For a successful E. histolytica culture, two important conditions must be fulfilled: growth in axenic conditions and harvest in logarithmic phase. Previously, cultures of E. histolytica were readily established in association with certain species of bacteria or trypanosomatids22. However, nowadays it is common to have axenic cultivation of this parasite meaning an indefinite subcultivation of amoebae in an environment free of metabolizing bacteria, fungi, protozoa, or metazoan cells. Additionally, harvesting the trophozoites during the late logarithmic to early stationary phase of growth is crucial to have viable and proliferating cells23. Therefore, it is important to distinguish this phase by quantification of trophozoites along several days and also by examining their intact morphology and rapid locomotion (Figure 1).
For interaction studies, epithelial cells need to be in a confluent monolayer to represent physiological conditions that amoebas would normally encounter in the body. Several epithelial cell lines, easy to cultivate, are available. While a more physiological cell model system would derive from the intestines such as Caco-2 or T84, these cell lines grow rather slowly. A faster growing cell line that has extensively been studied are MDCK cells24. These epithelial cells are of kidney origin and are characterized by high TER, strong TJ and easy cultivation. This cell line has been used for decades to study cell biology of junctions and TJ composition as well as mechanisms of TJ assembly and disassembly. For these reasons, MDCK cells are often chosen by researchers when studying TJ functions. MDCK cells have also been widely used as a model for the study of mechanisms induced during amoebiasis5,8,10,25-28. In a recent study, we reported that interaction of strain I MDCK cells and intestinal epithelial Caco-2 cells with live amoebas, lysates and amoeba products caused similar effects on TJ composition and TER10. Thus, MDCK cells are a suitable model to study mechanisms of amoeba-epithelia interactions. Furthermore, the use of several E. histolytica products (intact trophozoites, trophozoite total lysates or secreted products harvested as trophozoites culture supernatant) is crucial to provide additional and relevant information about molecules involved in these interactions, such as availability, mode of action, secretion, participation of other molecules and triggering of signaling pathways.
While reaching confluency, epithelial cells form contacts that are stabilized by TJ and AJ. These structures are composed of certain molecules that can be visualized by using specific antibodies in immunofluorescence stainings. For example, in Figure 3, cell contacts are visualized by an antibody against the TJ marker ZO-1 and a red fluorescently labeled secondary antibody. As it can be seen, cells gather in close proximity to form a dense monolayer without any hole. In this figure, the amoeba-derived complex EhCPADH112 has been stained using a monoclonal antibody that is detected by a green fluorescently labeled secondary antibody to study the interaction of this complex with the epithelial surface. Three different sources of this complex have been applied to the epithelial monolayer: live trophozoites (T), trophozoite total lysates (TL) and secreted products (SP). Strikingly, in all cases, co-localization of EhCPADH112 with ZO-1 can be observed (Figure 3, arrows), indicating that this complex could be required to facilitate E. histolytica penetration into the epithelial monolayer.
Although this interaction can already be observed as early as 2 min after amoeba-epithelial cell contact, the cell layer is not yet affected by this interaction since the ZO-1 staining still looks continuous. This completely changes after longer incubation times as shown in Figure 4. Here, images were taken after 30 min of exposure to the three different EhCPADH112 sources. A clear disruption of TJ is visualized by a discontinuous ZO-1 staining at cell borders (Figure 4, arrows), with the most evident effect occurring in MDCK cell in contact with T or TL. By contrast, ZO-1 gets internalized and is seen in intracellular vesicles. Exact co-localization with either TJ or AJ along the lateral cell membranes cannot be distinguished by just looking on top of the monolayer but rather by obtaining a “side”-view of cell contacts29. Confocal laser microscopy allows such a view along the xz-axis as demonstrated in Figures 3 and 4 below each xy view. These images show a clear statement whether proteins co-localize with TJ at the most apical portion of the lateral membrane or if they are rather located below TJ or above TJ at the apical membrane. In Figure 3 (arrowheads), an exact co-localization of the EhCPADH112 complex with ZO-1 can be observed, clearly revealing TJ as the place of action for this virulence factor. In contrast, as E. histolytica invasion progressed (30 min of interaction) into the epithelia, EhCPADH112 penetrated towards the intercellular space as the staining along the lateral membrane showed (Figure 4, arrowheads). In our recent paper, this method allowed us to distinguish between TJ and AJ because this complex only co-localized with the TJ markers occludin and claudin-1 but not with the AJ marker β–catenin at 2 min of interaction10.
Internalization of TJ components, as can be seen in the immunofluorescence stainings in Figure 4, is usually accompanied by a loss of barrier function that can be measured by transepithelial electrical resistance (TER). TER reflects the ion flow across the epithelial monolayer and can be measured using electrodes as shown in Figure 2B. When the monolayer is not yet completely formed or disrupted due to extracellular cues, a free flow of ions across the cell layer is indicated by a low TER. TER was measured of a control confluent monolayer that has not been in contact with amoeba products or monolayers in contact with trophozoites (Figure 5). Parasite contact disrupted epithelial barrier function indicated by a TER drop of 90% in comparison to the control monolayers. To investigate the mechanisms by which trophozoites affect barrier function, we pre-incubated the trophozoites with an antibody against EhCPADH112 to prevent adhesion of this complex or with protease inhibitors, to block the proteolytic activity of this complex and other proteases important for target cell damage. Both treatments led to an almost complete reversal of TER drop, suggesting that both proteolytic activity and adhesive properties are important virulence factors of this complex during E. histolytica invasion.
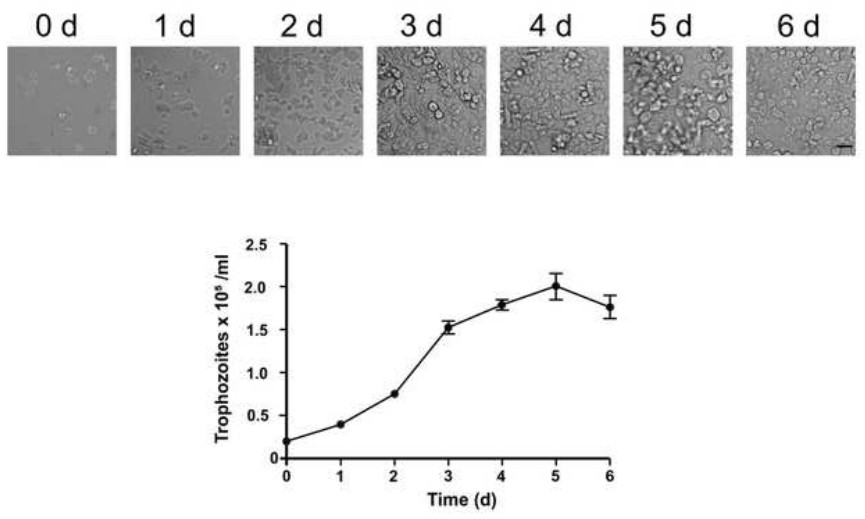
Figure 1. Typical growth curve of axenically cultivated E. histolytica trophozoites. Inocula of 2 x 105 trophozoites of E. histolytica HMI-IMSS clone A were grown in 6-well dishes with TYI-S-33 medium and after each 24 hr cell numbers were determined using a hematocytomer. Cellular growth was monitored by light microscopy and morphology of growing cells is depicted in the upper panel. The amount of trophozoites was monitored for 6 days (d) and values were plotted in the chart below. Data represent the mean and standard error of the mean of three independent measurements. Bar = 10 µm. Please click here to view a larger version of this figure.

Figure 2. Schematic representation of interactions among MDCK cells and different conditions of E. histolytica. A) Different conditions of E. histolytica are shown: live trophozoites (T), total trophozoites lysates (TL) and molecules secreted by trophozoites into the medium (SP). Any experimental condition was assayed for 2 and 30 min of incubation with confluent MDCK cells. After incubation, MDCK cells were abundantly washed to remove trophozoites or unbound parasitic molecules. Then samples were processed for immunofluorescence assays, employing mαEhCPADH112 and pαZO-1 antibodies to co-localize the parasitic complex EhCPADH112 and the TJ marker ZO-1, respectively. Later, species-specific secondary antibodies coupled to different fluorochromes were used to detect both proteins by confocal microscopy. B) Addition of trophozoites only or trophozoites pre-incubated with mαEhCPADH112 or protease inhibitors for 20 min at 4 °C in the upper compartment of transwells containing confluent MDCK cells. TER of epithelial cells was monitored using a STX2 electrode connected to an EVOM voltohmeter, during 30 min. Please click here to view a larger version of this figure.
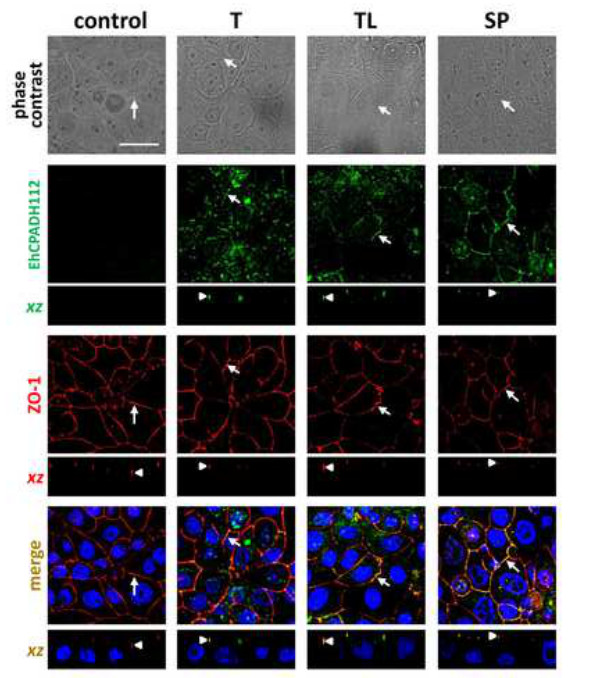
Figure 3. Localization of EhCPADH112 at TJ of MDCK monolayers. MDCK cells were incubated with live trophozoites (T), total trophozoites lysates (TL) and molecules secreted by trophozoites into the medium (SP) for 2 min. Upper panel: phase contrast images of MDCK cells. EhCPADH112 and ZO-1 proteins were identified by mαEhCPADH112 and pαZO-1 antibodies and then with FITC- and TRITC-secondary antibodies, respectively. Nuclei were stained with DAPI. Arrows: protein localization at cell borders. Arrowheads in xz-planes: protein localization at TJ. Bar = 10 μm. Please click here to view a larger version of this figure.
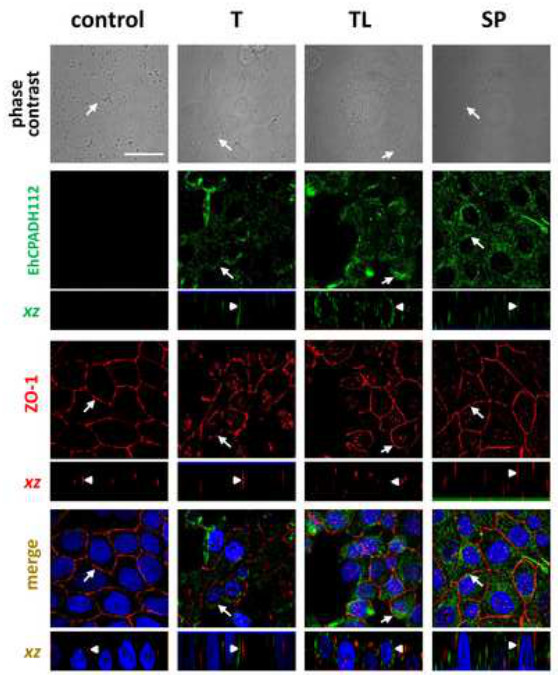
Figure 4. Internalization of EhCPADH112 into MDCK cells. MDCK cells were incubated with live trophozoites (T), total trophozoites lysates (TL) and molecules secreted by trophozoites into the medium (SP) for 30 min. Upper panel: phase contrast images of MDCK cells. EhCPADH112 and ZO-1 proteins were identified by mαEhCPADH112 and pαZO-1 antibodies and then with FITC- and TRITC-secondary antibodies, respectively. Nuclei were stained with DAPI. Arrows: protein localization at cell borders. Arrowheads in xz-planes: protein localization at lateral membrane (arrowheads). Bar = 10 μm. Please click here to view a larger version of this figure.

Figure 5. E. histolytica via EhCPADH112 induces epithelial barrier disruption. MDCK monolayers were incubated with live trophozoites (T) or T pre-incubated with protease inhibitors (PI) or mαEhCPADH112 antibody (α) for 30 min and TER was evaluated at indicated times. TER was normalized according to the initial value for each transwell (~3,200 Ω·cm2). Means and standard errors of the means are represented for each time point. Statistical analysis was performed with GraphPad Prism 5 software using one-way ANOVA test. *** P < 0.001.
