Rapid Analysis of Chromosome Aberrations in Mouse B Lymphocytes by PNA-FISH
Summary
DNA repair pathways are essential for maintenance of genomic integrity and preventing mutation and cancer. The goal of this protocol is to quantify genomic instability by direct observation of chromosome aberrations in metaphase spreads from mouse B cells using fluorescent in situ hybridization (FISH) for telomeric DNA repeats.
Abstract
Defective DNA repair leads to increased genomic instability, which is the root cause of mutations that lead to tumorigenesis. Analysis of the frequency and type of chromosome aberrations in different cell types allows defects in DNA repair pathways to be elucidated. Understanding mammalian DNA repair biology has been greatly helped by the production of mice with knockouts in specific genes. The goal of this protocol is to quantify genomic instability in mouse B lymphocytes. Labeling of the telomeres using PNA-FISH probes (peptide nucleic acid – fluorescent in situ hybridization) facilitates the rapid analysis of genomic instability in metaphase chromosome spreads. B cells have specific advantages relative to fibroblasts, because they have normal ploidy and a higher mitotic index. Short-term culture of B cells therefore enables precise measurement of genomic instability in a primary cell population which is likely to have fewer secondary genetic mutations than what is typically found in transformed fibroblasts or patient cell lines.
Introduction
Cancer is caused by the accumulation of mutations affecting genes that regulate normal cell growth. Mutation is a consequence of changes to the structure and sequence of the genome caused by damage to DNA. DNA damage can occur through a variety of process, including exogenous agents such as ionizing radiation, and as a by-product of normal cellular metabolism, such as spontaneous deamination of nucleotide bases or damage occurring by contact with reactive oxygen species1.
Although mammalian cells possess a range of repair activities which can reverse DNA damage or restore the sequence at break sites, mutations nonetheless accumulate throughout the lifetime of a cell. DNA damage can furthermore contribute to senescence and loss of potency of stem cells, two processes which are associated with aging-associated disease2. Understanding the repair of DNA damage is therefore of central importance in addressing two significant issues in public health. Increasing evidence suggests that mammalian DNA repair pathways can contribute to the evolution of the cancer cell genome3,4, making it even more imperative to understand the processes involved in suppressing mutation at the molecular level.
Direct visualization of chromosome aberrations is a powerful and quantitative means of determining the extent of genomic instability in a particular cell type. Condensed chromosomes from cells at metaphase can be isolated and inspected using light or fluorescent microscopy. Such cytogenetic approaches have been in practice for several decades and can be used to demonstrate the appearance of translocations or specific types of chromosome aberrations associated with loss of DNA repair activities. The protocol lends itself to several potential extensions: chromosomes can be labeled with probes for spectral karyotyping (SKY) or multicolor fluorescent in situ hybridization (mFISH) to identify translocations5,6. These techniques also enable the frequency of chromosome translocations and the structure of complex chromosome translocations to be determined, which provides additional information beyond what is possible with this protocol. Alternatively, sequence-specific probes can be generated and used to test the frequency of DNA breakage at selected genomic sites7.
In this protocol, we describe preparation of metaphase chromosome spreads from B lymphocytes. A fluorescently-labeled peptide nucleic acid (PNA) probe for telomeric repeats is used, which efficiently marks telomeres in metaphase chromosome spreads This protocol has several advantages. B cells can be induced to grow at high mitotic index so that high-quality spreads can consistently be produced. B cells from genetically-modified mice are also much less likely to contain secondary genetic mutations that can confound the analysis of the contribution of specific genes to genomic integrity. The PNA-FISH approach can be completed in one day, and allows more accurate scoring of chromosome breaks. By using this approach, particularly in combination with specific equipment described in this protocol, it is possible to produce very consistent, high-quality spreads and rapidly analyze the rate and type of genomic instability.
Protocol
This procedure was approved by the Institutional Animal Care and Use Committee at Rutgers, the State University of New Jersey. Mice were treated in accordance with the NIH Guide for the Care and Use of Laboratory Animals. Scientists should consult their national and institutional animal organizations for established and approved guidelines.
Before beginning, prepare the solutions listed in Table 1.
1. B Cell Isolation and Activation
Euthanize a mouse using CO2 followed by cervical dislocation. Position the mouse so the left side of the body is up and spray the mouse with 70% ethanol until the fur is damp. Dissect the spleen and place in a 35 mm tissue culture dish with 2 ml wash buffer. In a laminar flow hood, gently break up the spleen in the tissue culture dish using the flat end of a 5 ml syringe.
- Insert a 70 μm nylon mesh into a 50 ml tube and transfer the spleen sample in 2 ml wash buffer into the nylon mesh. Gently disrupt the spleen in the mesh using the flat end of the syringe.
- Rinse the back of the syringe and the 35 mm plate with 8 ml of wash buffer and filter through the 70 μm nylon mesh. Centrifuge at 300 x g for 10 min at 4 °C.
- Aspirate and discard the supernatant and resuspend the pellet in 3 ml of ACK lysis buffer. Pipette up and down briefly to disrupt clumps and incubate for 5 min. Add 10 ml of wash buffer and centrifuge at 300 x g for 10 min at 4 °C.
- Aspirate and discard the supernatant and resuspend the pellet by tapping the bottom of the tube then by pipetting in 1 ml of wash buffer. Add 50 μl of anti-CD43 MACS micro-beads and incubate on ice for 30 min. Add 10 ml of wash buffer, mix gently, and centrifuge at 300 x g for 10 min at 4 °C. Add 10 ml of wash buffer, mix gently, and centrifuge at 300 x g for 10 min at 4 °C.
- Set up a Mini-MACS column by placing a column on the MACS magnet. Position a labeled 15 ml conical tube below the column. Add 1 ml of wash buffer to column and allow to run through to the 15 ml tube.
- Aspirate the supernatant and resuspend in 1 ml of wash buffer. Load the sample in the column after the wash buffer has run through.
- After the sample has run through the column, add 1 ml wash buffer to wash the column. Add 7 ml of wash buffer directly to the collected sample in the 15 ml tube.
- Count the cells using a hemocytometer. Transfer the required volume of cells needed for 5,000,000 cells/well into a 50 ml tube. Centrifuge at 300 x g for 10 min at 4 °C. Supplement B cell medium with LPS 1:500 and IL4 1:1,000.
- Aspirate the supernatant and add the appropriate volume of supplemented B cell medium needed for final concentration of 1,000,000 cells/ml.
- Plate cells in 6-well plates with 5 ml of cells at 1,000,000 cell/ml and place in a humidified cell culture incubator at 37 °C, 5% CO2.
2. B Cell Fixation
- Add 50 μl of colcemid to the well containing 5 ml of B cells (for a final concentration of 100 ng/ml) and incubate 1 hr at 37 °C. Prewarm 75 mM KCl solution in a 37 °C water bath. Transfer the B cells to a labeled 15 ml tube. Cover the label with tape and centrifuge at 300 x g for 10 min at 4 °C.
- Aspirate the supernatant leaving 1 ml in the tube and resuspend the pellet by pipetting. Add 5 ml of the prewarmed KCl drop by drop while tapping or pulsing on a vortex.
- Add 10 ml of KCl and mix by inverting. Incubate in a 37 °C water bath for 15 min. Add 5 drops of fresh fixative and mix by inverting. Centrifuge at 300 x g for 10 min at 4 °C. Aspirate the supernatant leaving 1 ml in the tube and resuspend pellet by pipetting up and down.
- Add 5 ml of fresh fixative drop by drop while tapping or pulsing on a vortex. Add an additional 10 ml of fixative and incubate for 30 min at room temperature.
- Centrifuge at 300 x g for 10 min at 4 °C. Aspirate the supernatant leaving 1 ml in tube and resuspend by pipetting. Add 14 ml of fresh fixative and centrifuge at 300 x g for 10 min at 4 °C.
- Repeat step 2.7 twice more.
- Add 14 ml of fresh fixative. Seal the caps with Parafilm and store overnight at -20 °C.
3. Preparation of Metaphase Chromosome Spreads
- Set a regulated environmental chamber to 22.9 °C and 52% humidity.
- Centrifuge the fixed B cells at 300 x g for 10 min at 4 °C. Aspirate fixative leaving 70 μl in the tube and resuspend by pipetting.
- Place labeled slides in the humidity chamber at a 35° angle. Drop 35 μl of the resuspended B cells onto a slide and drop two slides per sample. Allow the slides to dry in the humidity chamber for 30 min then store the slides in a slide box at 37 °C.
4. Telomere PNA FISH
- Preparation of Probe
- Pre-warm deionized formamide to 37 °C. Mix 2 μl of telomere PNA probe with 7 μl deionized pre-warmed formamide and incubate at 37 °C with shaking for 1 hr.
- Pre-warm FISH master mix to 37 °C. Add 7 μl pre-warmed FISH master mix to the mixture from step 4.1.1 and incubate at 37 °C for 1-3 hr.
- Denature the probe mixture for 8 min at 80 °C. Pre-anneal the probe for 1 hr at 37 °C.
- Pretreatment of Chromosome Slides
- Pre-warm a glass slide-staining jar containing 50 ml of 0.01 M HCl to 37 °C in a water bath.
- Place slides in a different glass slide-staining jar containing 2x SSC for 5 min at room temperature.
- Add 2 μl of pepsin stock to the pre-warmed glass slide-staining jar and mix by inverting. Transfer the slides to this glass slide-staining jar and incubate 90 sec at 37 °C.
- Wash the slides 2x in a different glass slide-staining jar containing 1x PBS for 5 min each at room temperature, shaking. Then wash the slides for 5 min in 1x PBS/MgCl2 at room temperature, shaking.
- Transfer the slides to a glass slide-staining jar containing freshly-prepared 1% formaldehyde/1x PBS/MgCl2 for 10 min at room temperature.
- Wash the slides for 5 min in a different glass slide-staining jar containing 1x PBS at room temperature, shaking.
- Prepare an ethanol dehydration series by having separate glass slide-staining jars containing 70%, 90%, and 100% ethanol.
- Transfer the slides to each jar for 3 min, starting with 70% ethanol, then 90% ethanol, and then 100% ethanol. Place ethanol jars at -20 °C when finished.
- Allow the slides to air dry by tapping excess ethanol onto a paper towel and propping slides at a 35° angle. Once dry, mark an 18 x 18 mm area for hybridization on the back of the slides using a diamond pen.
- Denaturation of Slides for FISH
- Apply 120 μl 70% deionized formamide/2X SSC to a 24 x 60 mm coverslip. Touch the slide to the coverslip. Denature the slide at 80 °C on a hot plate for 90 sec.
- Quickly and carefully slide off the coverslip and place the slide in ice cold 70% ethanol for 3 min, followed by 90% ethanol, and 100% ethanol each for 3 min. Allow the slides to air dry.
- Prepare a humid chamber by lining a plastic box with a tight lid and wet tissue paper. The paper should be damp, but not saturated with water.
- In a humid chamber, apply the pre-annealed probe mix from step 4.1.3 to the marked area on the slide, cover with an 18 x 18 mm cover slip and incubate at 37 °C for 1 hr.
- Post-incubation Washes
- Pre-warm 50% formamide/2x SSC, 1x SSC, and 4x SSC/0.1% Tween-20 in a 45 °C water bath.
- Carefully remove the coverslips and wash the slides in pre-warmed 50% formamide/2x SSC 3xfor 5 min each in the dark and shaking. Wash the slides in pre-warmed 1x SSC 3xfor 5 min each, shaking. Finally, wash the slides in pre-warmed 4x SSC/0.1% Tween-20 3xfor 5 min each.
- Stain the slides for 3 min in DAPI in a light protected glass slide-staining jar.
- Wash the slides for 5 min in 2x SSC, shaking. Apply 35 μl of Mowiol mounting medium, cover with 24 x 60 mm coverslips and store the slides at 4 °C.
5. Microscopic Analysis of Chromosomes
- Analyze the metaphase slides using a standard epi-fluorescence microscope. Use an imaging platform with an automated stage such as the MetaSystems Metafer platform to obtain best results. For each metaphase spread, collect one image for DAPI fluorescence to visualize chromosomes, and one image for the fluorescent signal from the telomere probe. (e.g., Cy3- other fluors are available and work equally well.)
- Overlay the images with an image analysis program and analyze.
Representative Results
Metaphase chromosomes derived from one cell should form a discrete cluster containing 40 chromosomes (if using mouse cells) (Figure 1A). Each chromosome has a centromere at one end, which is visible as a light blue sphere. In normal chromosomes, two telomere signals are seen at the end of the chromatids and two signals by each centromere. Interphase nuclei will be present on the slide as well. These will be visible as blue spheres, with red telomere signals, but no condensed chromosomes (see for example Figure 1B). The interphase nuclei can be ignored for the purposes of quantifying genomic instability.
A number of different types of chromosome aberrations can be observed. Chromatid breaks are breaks in one of the two sister chromatids that make up each metaphase chromosome. They can be scored if the investigator sees a discontinuity in one chromatid, or loss of a single telomere signal from one end of a chromosome (see examples in Figure 1B). Chromosome breaks are identified by looking for loss of telomere signals from one end of a chromosome (example in Figure 1C). In some cases, the broken end of the chromosome can be observed as a fragment with telomere signals in the same metaphase spread (as is the case in Figure 1C). In other cases, the chromosome end may not be present.
Several types of chromosome rearrangements may be observed. Radial chromosomes take a number of forms, characterized by complex rearrangements involving two chromosomes (three examples are shown with red arrows in Figure 1D). Radial chromosomes also include more complex rearrangements, involving three or more chromosomes. Chromosomes may also become joined to form dicentric chromosomes, which appear as an end-to-end fusion of chromosomes with centromeres at both ends (shown with a white arrow in Figure 1D). Robertsonian translocations are a type of translocation between the centromeres of two chromosomes, which appear as four sister chromatid arms attached to a single centromere (seen in Figure 1E).
Figure 1F shows a metaphase that does not have telomere signals present. Absence of telomere signals can occur from errors in the probe preparation or air bubbles in the coverslip during hybridization.
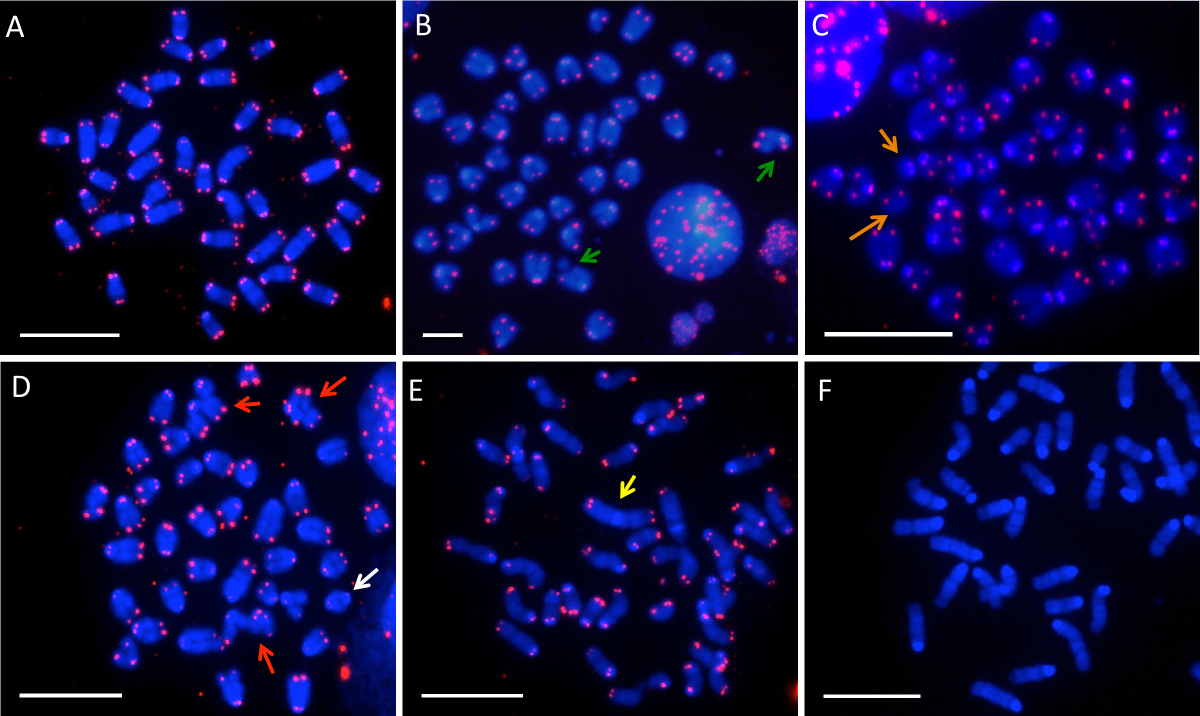
Figure 1. Chromosome aberrations in mouse B cells as visualized by PNA telomere FISH. A) The chromosome structure of a normal mouse B cell at metaphase. 40 chromosomes are visible, with two telomere signals at either end. B) A cell displaying two chromatid breaks (green arrows). C) A metaphase spread with one chromosome break (orange arrows). D) A cell displaying 3 radial chromosomes (red arrows) and one dicentric chromosome (white arrow). E) A metaphase spread with one chromosome with a Robertsonian translocation (yellow arrow). F) An example of a failed hybridization, showing chromosomes without telomere signals. The scale bar represents 10 μm in each image. Please click here to view a larger version of this figure.
Discussion
Whereas activated B lymphocytes are particularly suited to preparation of mitotic chromosome spreads, other cell types can also be used. T lymphocytes share many of the advantages of B cells, as they can be purified from the spleen or lymph nodes, and have a high mitotic index when stimulated by growth in medium containing appropriate mitogens8. Embryonic Stem cells (ES cells) are also suited for metaphase chromosome analysis9. If these are not available, fibroblast cells can be used, although a longer treatment with colcemid (e.g., 16 hr with colcemid at a final concentration of 10 ng/ml) is recommended to trap more cells in metaphase before fixation.
We find that the consistency of preparation of metaphase spreads is greatly facilitated by dropping the fixed chromosomes in a controlled humidity chamber such as the Thermotron CDS-5 Cytogenetic Drying Chamber, but it is possible to obtain good results while working at a bench depending on the humidity and temperature of the lab environment. To obtain reliable results, it is advisable to score chromosome aberrations from at least 100 metaphases per sample. As the process of manually imaging each metaphase in each channel can be laborious, we acquire metaphases using a Metasystems Metafer4 automated microscopy system, which can quickly find and accurately scan a large number of metaphase chromosomes.
Analysis of metaphase chromosomes labeled with telomere probes is ideal for measuring chromosome and chromatid breaks, and the telomere signals also aid the identification of complex chromosome rearrangements such as fusions and radial structures. Staining of fixed metaphase chromosomes with Giemsa’s solution is also suitable for quantifying chromosome aberrations, and offers the advantage of not requiring fluorescent microscopy10. Labeling telomeres with a pantelomeric PNA probe extends the range of chromosome aberrations that can be quantified, by allowing the identification of telomere fusions, which arise, for example, in cells from DNAPKcs-knockout mice11. PNA-FISH can also be used to measure the frequencies of telomere abnormalities, including chromosome telomere duplications and chromatid telomere loss12,13. Loss of a telomere signal can be caused by telomere shortening as well as by chromosome breakage. PNA-FISH can be used to sensitively measure telomere length using Q-FISH (quantitative FISH)14.
Cells lacking different DNA repair activities tend to accumulate different types of chromosome aberrations. For example, deficiency in the DNA damage response factor 53BP1, leads to accumulation of chromosome breaks15. By contrast, a high proportion of complex radial chromosome structures are observed in cells lacking BRCA14. These chromosome rearrangements are varied in their exact structure, but always involve fusion of material from two or more chromosomes. When cells have very high levels of genomic instability, as for example occurs after treatment with high doses of agents that cause DNA breaks, it becomes increasingly difficult to score individual chromosome aberrations, which puts a limit on the dynamic range of the experiment.
Quantification of chromosome breaks and rearrangements in metaphase spreads is useful for testing whether specific genes have a role in DNA repair. One potential issue in using metaphase chromosome analysis for this purpose is that cells which lack DNA repair activities may not grow normally. If repair-deficient cells do not reach metaphase, the importance of a gene for mediating DNA repair may be underestimated. For example, RNF8-/- cells, which are subject to p53-dependent senescence, show a modest level of chromosome instability, whereas RNF8-/-p53-/- double-knockout cells show many more chromosome aberrations, because deletion of p53 allows better cell growth16. It is therefore important to pair measurements of chromosome instability with a cell cycle assay, to show that knockout cell populations can grow normally and contain a meaningful proportion of mitotic cells.
The observation of genomic instability does not by itself give insight into the exact function of a repair activity provided by a specific gene, but merely acts as a starting point for more mechanistic studies. Finally, although this approach is useful for measuring chromosome breaks and complex rearrangements, the use of mFISH or SKY is recommended for quantifying chromosome translocations. These techniques also begin with metaphase chromosome spreads, but use a cocktail of probes that ‘paint’ each chromosome with a specific combination of fluorophores. Either of these techniques is recommended for measuring the full spectrum of chromosome instability in repair-deficient cells.
Disclosures
The authors have nothing to disclose.
Acknowledgements
This work is supported by NIH grant R00 CA160574 (SFB) and by a fellowship of the Rutgers Biotechnology Training Program (to SMM).
Materials
| Name | Company | Catalog Number | Comments |
| 50% Dextran sulfate | Intergen | S4030 | |
| Deionized formamide | Ambion | 9342 | |
| Pepsin (5g) | Sigma | P 6887 | |
| FCS | Gemini | 100-106 | |
| LPS (100mg) | Sigma | L2630 | |
| IL-4 (5μg) | Sigma | I1020 | |
| PNA Telomere Probe | PNA Bio Inc | F1002 | (CCCTAACCCTAACCCTAA) |
| Colcemid | Roche | 295892 | |
| Mowiol 4-88 | Sigma | 81381-50g | |
| CD43 (ly-48_) micro-beads | Miltenyl Biotec. | 130-049-801 | |
| DAPI | Sigma | 32670-5MG | |
| Eclipse E800 | Nikon | ||
| AxioImager.Z2 | Zeiss | ||
| CDS-5 Cytogenetic Drying Chamber | Thermotron |
References
- Sancar, A., Lindsey-Boltz, L. A., Unsal-Kacmaz, K., Linn, S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Ann Rev Biochem. 73, 39-85 (2004).
- Sperka, T., Wang, J., Rudolph, K. L. DNA damage checkpoints in stem cells, ageing and cancer. Nature reviews Mol Cell Biol. 13 (9), 579-590 (2012).
- Alexandrov, L. B., et al. Signatures of mutational processes in human cancer. Nature. 500 (7463), 415-421 (2013).
- Bunting, S. F., et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 141 (2), 243-254 (2010).
- Padilla-Nash, H. M., Barenboim-Stapleton, L., Difilippantonio, M. J., Ried, T. Spectral karyotyping analysis of human and mouse chromosomes. Nat Protoc. 1 (6), 3129-3142 (2006).
- Callen, E., et al. Chimeric IgH-TCRalpha/delta translocations in T lymphocytes mediated by RAG. Cell Cycle. 8 (15), 2408-2412 (2009).
- Boboila, C., et al. Alternative end-joining catalyzes robust IgH locus deletions and translocations in the combined absence of ligase 4 and Ku70. Natl Acad Sci USA. 107 (7), 3034-3039 (2010).
- Benn, P., Delach, J. Human lymphocyte culture and chromosome analysis. Cold Spring Harb Protoc. 2008, (2008).
- Nguyen, H. N., Reijo Pera, R. A. Metaphase spreads and spectral karyotyping of human embryonic stem cells. Cold Spring Harb Protoc. 2008, (2008).
- Schreck, R. R., Disteche, C. M. Chapter 4. Chromosome banding techniques. Unit 2, (2001).
- Gilley, D., et al. DNA-PKcs is critical for telomere capping. Proc Natl Acad Sci USA. 98 (26), 15084-15088 (2001).
- Bolzan, A. D. Chromosomal aberrations involving telomeres and interstitial telomeric sequences. Mutagenesis. 27 (1), 1-15 (2012).
- Bolzan, A. D., Telomeres Bianchi, M. S. interstitial telomeric repeat sequences and chromosomal aberrations. Mutat Res. 612 (3), 189-214 (2006).
- Poon, S. S., Lansdorp, P. M. Chapter 18. Quantitative fluorescence in situ hybridization (Q-FISH). Unit 18 14, (2001).
- Morales, J. C., et al. 53BP1 and p53 synergize to suppress genomic instability and lymphomagenesis. Proc Natl Acad Sci USA. 103 (9), 3310-3315 (2006).
- Halaby, M. J., et al. Synergistic interaction of Rnf8 and p53 in the protection against genomic instability and tumorigenesis. PLoS Genet. 9 (1), e1003259 (2013).
