The production of crude dauer pheromone results in a yellow liquid (Figure 1) that is both heat and cold stable. Aliquots of crude pheromone are stored at -20 °C indefinitely with no obvious loss in activity. Following the production of pheromone, a single dose-response bioassay is sufficient for a rough estimate of potency. However, for most experiments it is necessary to repeat the bioassay 2 – 3 times and take an average from each experiment. Representative results from two pheromone bioassays are given in Figure 3. From these data an EC50 and EC90 can be calculated.
In addition to resistance to sodium dodecyl sulfate2, dauers can be recognized by several morphological features including an occluded stoma, lateral alae, refractile bodies in the anterior hypodermis and a shrunken pharynx (Figure 2 and Figure 4A for L2 larvae). Dauers also have several distinctive behavioral characteristics including: a propensity to remain behaviorally quiescent, the presence of an occasional dispersal strategy called nictation, suppression of head foraging movements, suppression of pharyngeal pumping and the induction of excretory duct pulsing2, 5, 23. The behavioral characteristics show variability among individuals.
The molt into dauer takes approximately 12 hr from the end of the pre-dauer L2 stage (L2d)4. This period corresponds with stereotypical growth of the IL2Q arbors. The first noticeable event of the molt into dauer is the suppression of pharyngeal pumping. Suppression of pumping is common to all molts in C. elegans. However, suppression of pharyngeal pumping continues throughout the dauer stage and ends approximately 4 hr following a return of the animal to favorable environmental conditions. During the initial period of pharyngeal pumping suppression, a layer of cuticle forms over the stoma. These changes occur during the first hour following the onset of the molt into dauer. During this initial period there is frequently the formation of an additional neurite process extending from the IL2Q cell bodies. However, this initial additional process usually retracts within 2 hr following the onset of the molt into dauer12.
During hours 2 – 5 following the onset of the molt into dauer the pharynx begins to remodel showing a gradual reduction in size (Figure 4B). Concomitantly, short puncta form and retract along the IL2Q primary dendrite (Figure 5). During hours 4 – 8 following the onset of the molt, the body undergoes a gradual radial shrinkage resulting in the pulling away of the nematode from the L2d cuticle (Figure 4C). This period is correlated with outgrowth of 2° dendrites from the primary dendrites and the establishment of additional dendrites from the IL2Q cell bodies (dauer-specific primary dendrites—1d°). The dendritic growth is not constant, but rather characterized by both the dynamic formation and retraction of processes as illustrated in Figure 5. The 2° and 1d° dendrites generally extend to the ventral (for IL2Vs) or dorsal (for IL2Ds) midlines. Once the dauer cuticle is completely separated from the L2d cuticle, the application of fluorescent light causes the animal to rapidly spin on its longitudinal axis, making subsequent imaging challenging. Attempts to remove the developing animal from the slide, mechanically remove the L2D cuticle, and mount the animal on a new slide while still maintaining dauer development were not successful.

Figure 1. Crude dauer pheromone is a yellow liquid that can be stored indefinitely at -20 °C. Please click here to view a larger version of this figure.
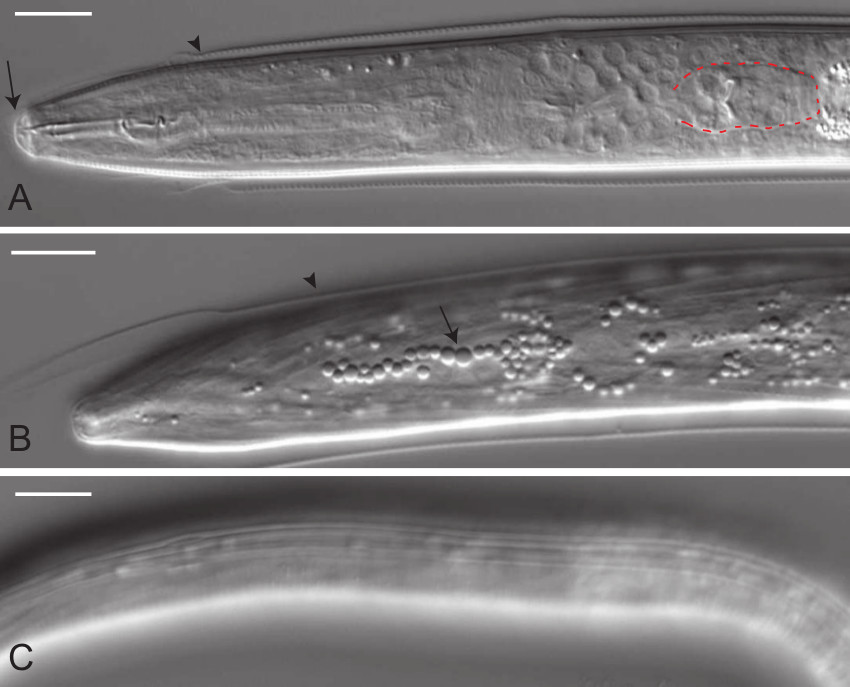
Figure 2. DIC micrographs of C. elegans dauers with typical dauer morphological features. (A) Dorsal mid-body view of a dauer demonstrating an occluded stoma (arrow) and a shrunken pharynx with rectangular terminal bulb (outlined in red, compare with L2 larva shown in Figure 4A). Newly formed dauers will frequently retain part of the L2d cuticle (arrowhead). (B) Dorsal view of hypodermis showing highly refractile bodies (arrow) typical of dauers. Again the L2d cuticle is evident (arrowhead). (C) Lateral cuticle view showing lateral alae, a set of four prominent longitudinal ridges that run from the anus to anterior of the nerve ring. Scale bars, 10 µm. Please click here to view a larger version of this figure.

Figure 3. Dose response assay to crude dauer pheromone. L1 animals exposed to increasing levels of dauer pheromone incorporated into modified NGM media are more likely to form dauers. Data were collected and pooled from two separate experiments except for the following concentrations, which were only tested once: 0.05%, 2%, and 4%. Data were fit to a sigmoidal dose response curve. The R2 and EC values were calculated using logistic regression. The number of animals examined for each concentration is as follows: 0% n=299, 0.05% n=81, 0.1% n=352, 0.5% n=368, 1% n=241, 2% n=125, 4% n=155. Error bars represent 95% confidence intervals.
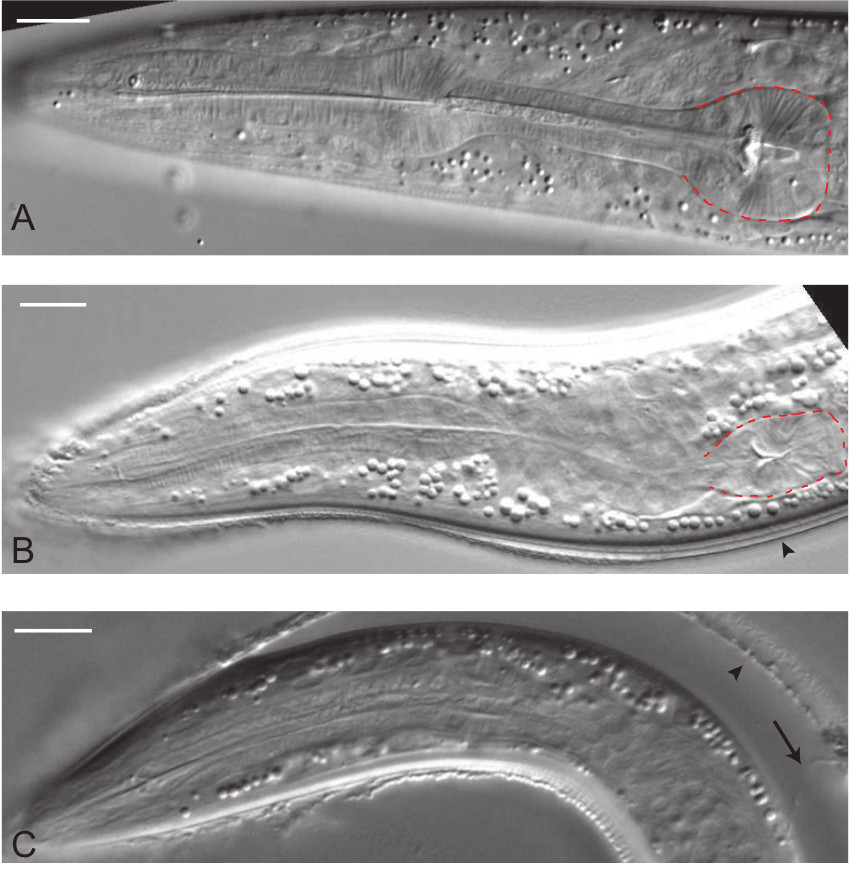
Figure 4. DIC lateral right view micrographs of C. elegans during different stages of the molt into dauer. (A) L2 animal with typical rounded terminal pharyngeal bulb (outlined in red). (B) Approximately 3 hr following the molt into dauer, the terminal bulb is shrinking and the L2d cuticle is beginning to detach from the newly formed dauer cuticle (arrowhead). (C) Approximately 10 hr following the onset of the dauer molt, the animal has undergone extensive radial shrinkage and detachment from the L2d cuticle (arrowhead). Occasionally, the cuticle lined excretory duct of the L2d stage (arrow) can be seen still attached to the developing dauer. Scale bars, 10 µm. Please click here to view a larger version of this figure.
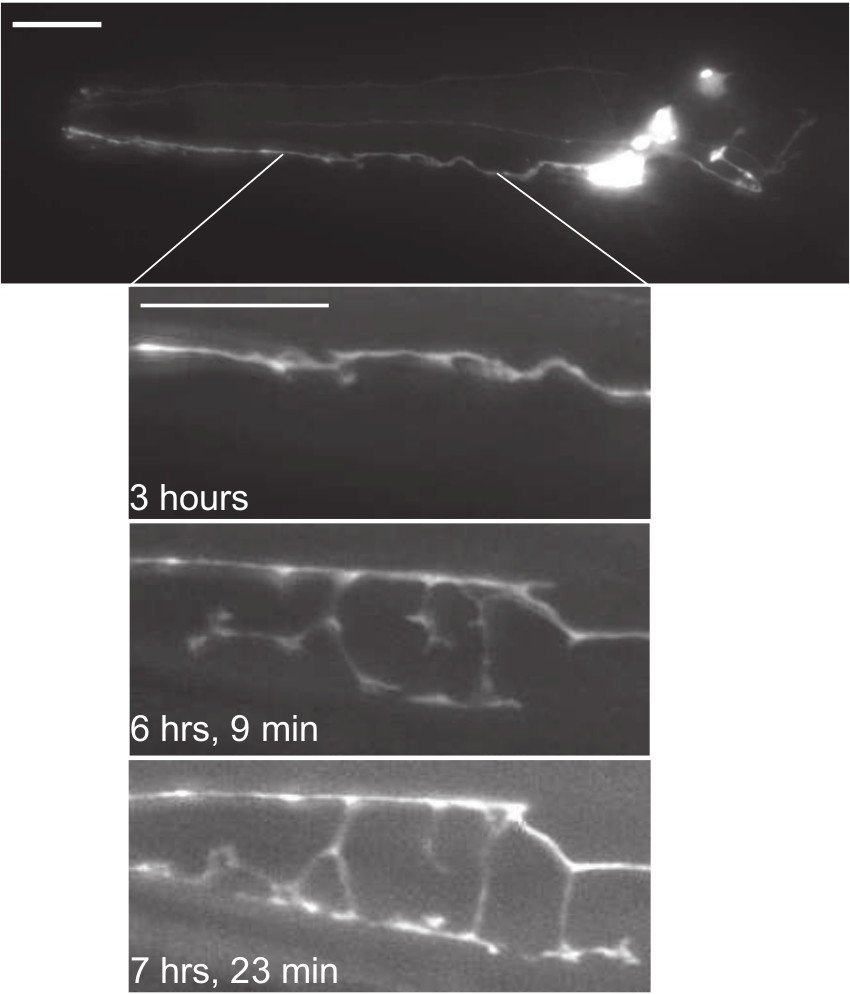
Figure 5. Lateral view epifluorescent micrographs of a single animal expressing the IL2 reporter Pklp-6::tdTomato. Top, full-length image of IL2 neurons approximately 3 hr following the onset of the molt into dauer. Inset shows portion of IL2Q dendrite at various time-points following the onset of the molt into dauer. A rapid extension of the IL2Q branches occurs starting approximately 5 hr following the onset of the molt into dauer. Scale bars, 10 µm. Please click here to view a larger version of this figure.
