Using this protocol for transfection of THP-1 macrophages with siRNA we usually achieve transfection rates of above 90% without significant reduction of cell vitality. Figure 1 shows representative data characterizing the state of the cells 24 hr after transfection with fluorescently labeled siRNA versus an untransfected control, which were not treated with Nucleofection reagents and pulse or siRNA but received all changes of culture media as transfected samples. In Figure 1A the cellular morphology is shown according to flow cytometric measurement. Microscopic images are shown in Figure 1B. Figures 1C and 1D represent the low rates for apoptosis (control: 1.5%; Nucleofection: 5.4%) and necrosis (control: 2.0%; Nucleofection: 3.2%) indicating unaffected cell vitality. Necrosis and apoptosis are slightly higher for the transfected sample as transfected THP-1 macrophages are more difficult to detach than untransfected cells, thus the probability of cell damage during detachment increases. Finally transfection efficiency is presented in Figure 1E. As the entire transfected population is shifted against the control, this indicates that all cells are transfected which is confirmed by fluorescence microscopic images (Figure 2B). Both flow cytometric data as well as fluorescence microscopic images indicate that all cells are consistently transfected with homogenous distribution of siRNA among cells as well as within cells. This is in contrast to many chemical transfection reagents, for comparison Figures 2A and 2B show the respective flow cytometric data and fluorescence microscopic images for a chemical transfection agent using a lipid based approach. These figures show that two distinct populations of transfected cells can be detected. The first population is similar to the transfected cells following Nucleofection. They are characterized by low overall fluorescence and homogenous distribution of siRNA within the cells. The second population is marked by a more intense fluorescence which originates from very bright intracellular agglomerates of siRNA. The cellular morphology remains unaffected for both transfection approaches as shown by differential interference contrast in Figure 2C. Transfections using plasmid DNA yield effectively similar results, for examples please refer to Robenek et al. (2009)9 or Xie et al. (2006)7.
The choice of the cell culture medium after transfection is of great relevance; therefore different cell culture media were tested in comparison. The selected media are all suitable for cultivation of THP-1 cells and were chosen according to recommendations of Lonza as applicable media for post-Nucleofection cultivation. Figure 3 represents the siRNA mediated knockdown efficiency using a siRNA directed against IL10RB (interleukin 10 receptor β chain) mRNA. For all tested media the expression of IL10RB was significantly reduced to about 10 to 20% of the control level (Figure 3). However the transfected cells differ depending on the culture medium in their potential for polarization. THP-1 macrophages transfected with either unspecific control siRNA or IL10RB-specific siRNA were cultivated in different media after transfection and treated with IL10. Subsequently the expression levels of the IL10-induced gene SOCS3 (suppressor of cytokine signaling 3) on mRNA level were measured by RT-qPCR. Differences between culture media occur in regard to the extent of the induction of SOCS3 mRNA expression (Figure 4), the inductions for each of the tested media Mouse T Cell Nucleofector Medium, LGM3, X-VIVO 20 and IMDM were 19.5 [17.7-21.5], 11.5 [8.5-15.7], 8.0 [4.6-14.2] and 7.2 [5.6-9.5] fold, respectively. Therefore application of the Mouse T Cell Nucleofector medium is vital. In addition, differences in the effect of the knockdown of IL10RB on SOCS3 expression after IL10 treatment were observable, expression levels of SOCS3 mRNA were reduced to 9.5 [8.8-10.3] fold in Mouse T Cell Nucleofector medium, 2.1 [1.1-4.1] fold in LGM3, 4.8 [3.2-7.2] fold in X-VIVO 20 and 3.3 [2.7-3.9] fold in IMDM. These values correspond to reductions of SOCS3 expression in the different media to 49%, 18%, 60%, and 45%, respectively. Reduction of IL-10-induced SOCS3 expression after transfection of IL10RB siRNA confirms the successful downregulation of IL10RB by the transfection.
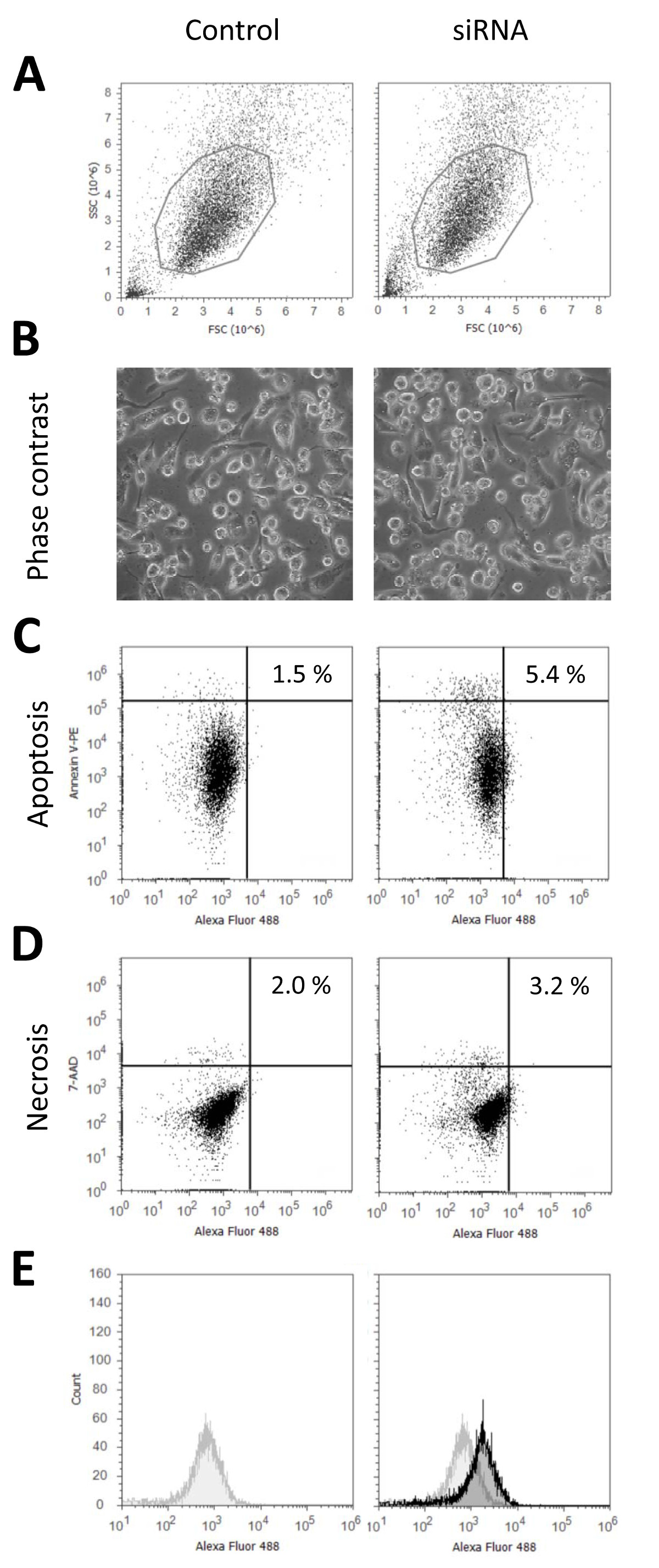
Figure 1. Characterization of transfected THP-1 macrophages. The characteristic status of the cells is unaffected as revealed by light microscopic and flow cytometric analyses of untransfected control cells versus THP-1 macrophages transfected according to the presented protocol. THP-1 macrophages were differentiated with 10 ng/ml PMA for 48 hr and transfected with fluorescently labeled (Alexa 488) unspecific control siRNA. 24 hr after transfection either images of live cells were taken (B), or the cells were detached by Accutase I treatment and analyzed by flow cytometry (A). Apoptosis and necrosis staining was performed using Annexin V-phycoerythrin (PE) (C) and 7-aminoactinomycin (7-AAD) (D). Transfection efficiency (E) was determined by flow cytometry using the fluorescence label attached to the siRNA. The fluorescence signal of the transfected cells (black) is shown against the control signal (grey). Please click here to view a larger version of this figure.

Figure 2. Comparison of intracellular distribution of siRNA after Nucleofection versus lipofection. THP-1 cells were differentiated for 48 hr according to this protocol and then transfected either by Nucleofection or by lipofection. The presented lipofection results were obtained using the Xtremegene siRNA kit according to the manufacturer’s recommendations with a charge ratio of reagent to siRNA of 4:1. 24 hr after transfection cells were either detached with Accutase I for flow cytometric analysis or evaluated microscopically with live cells. (A) Transfection efficiency and distribution of siRNA per cell within the population was determined by flow cytometry using the fluorescence label attached to the siRNA, controls transfected without siRNA are shown in grey, samples transfected with siRNA are shown in black. (B) Fluorescence microscopy images showing intracellular distribution of fluorescently labeled siRNA (Alexa Fluor 488); the scale bar represents 40 µm. (C) Differential interference contrast image corresponding to the fluorescence image; the scale bar represents 40 µm. Please click here to view a larger version of this figure.
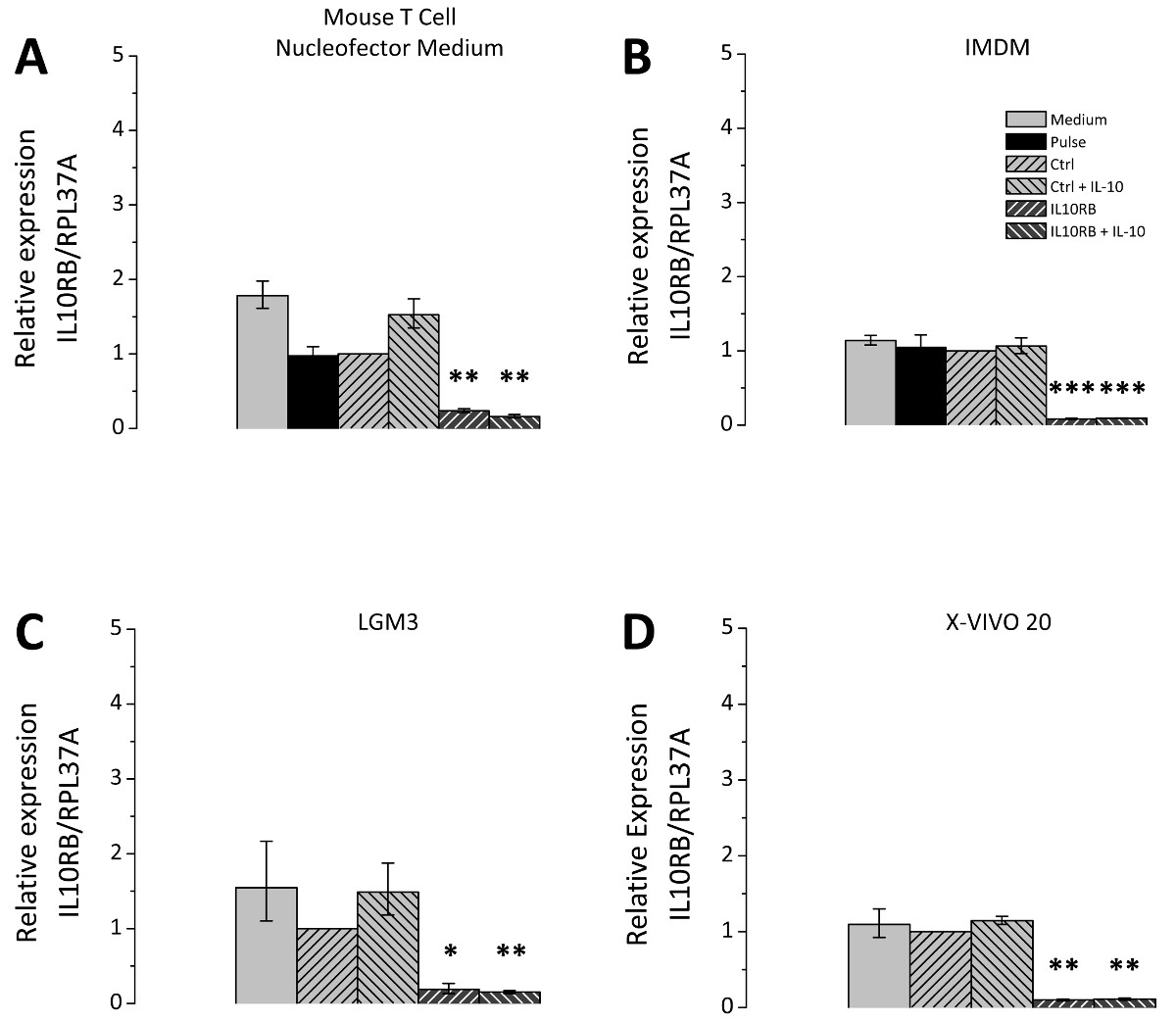
Figure 3. siRNA-mediated knockdown of IL10RB in transfected THP-1 macrophages. THP-1 macrophages were transfected according to the outlined protocol. After transfection the cells were cultivated in four different culture media, namely Mouse T Cell Nucleofector medium (A), IMDM (B), LGM3 (C), and X-VIVO 20 (D), supplemented as described in the Protocol section. Cells were either transfected with unspecific control siRNA (Ctrl) or IL10RB-specific siRNA (IL10RB). As additional controls the following samples were included: Pulse control, i.e. cells which underwent transfection in the absence of siRNA (Pulse), and a medium control, i.e. cells that received only the changes of culture media but remained otherwise untreated. 24 hr after transfection the cells were incubated in serum-free medium with or without 50 ng/ml IL10 for further 24 hr. IL10RB expression was measured by RT-qPCR; diagrams show the mean of three independent experiments, error bars represent the standard error of the mean; * p <0.05; ** p <0.01; *** p <0.001 vs. Ctrl. Please click here to view a larger version of this figure.
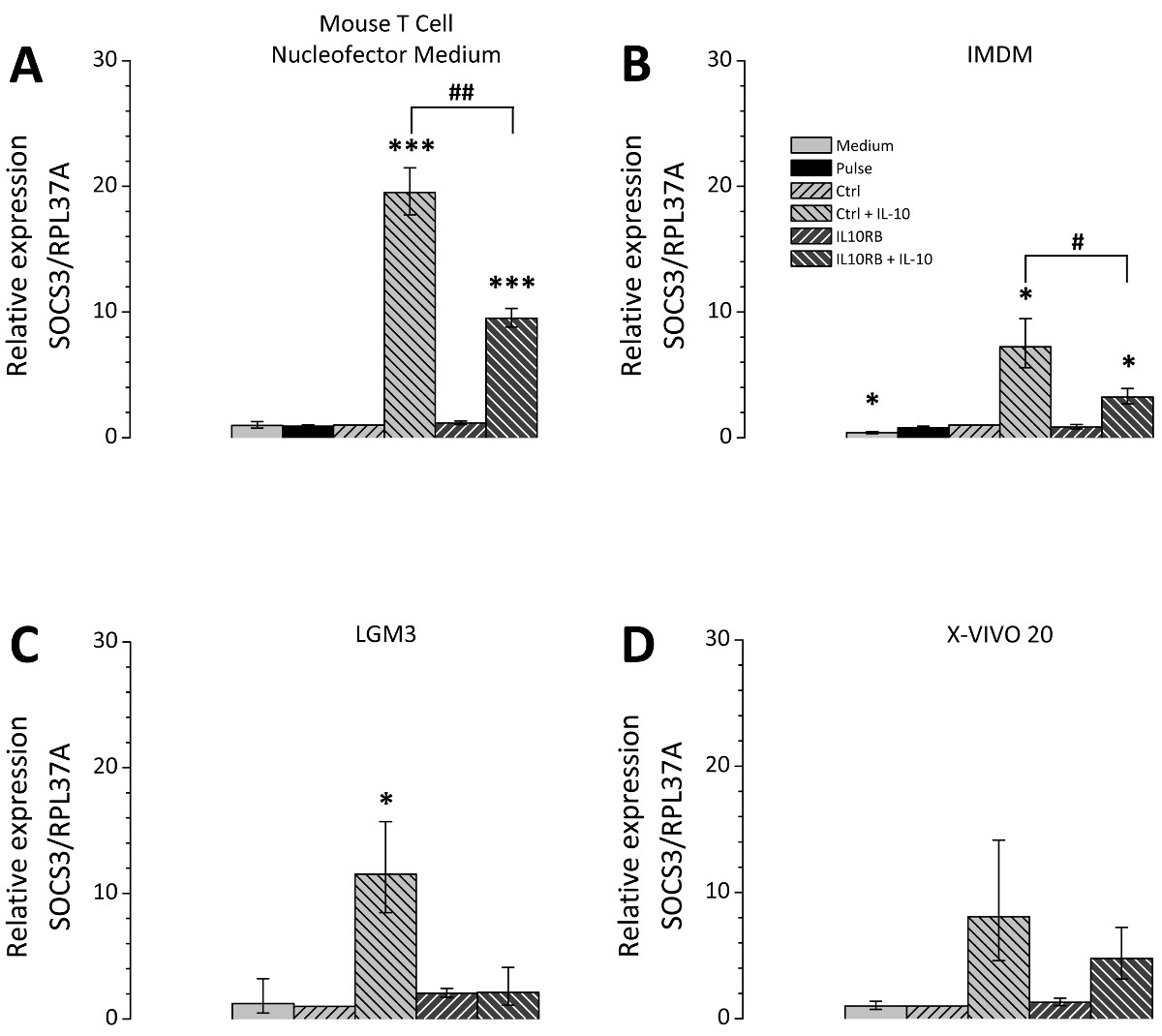
Figure 4. IL10-dependent regulation of SOCS3 after knockdown of IL10RB in transfected THP-1 macrophages. THP-1 macrophages were transfected according to the described protocol. After transfection the cells were cultivated in four different culture media Mouse T Cell Nucleofector medium (A), IMDM (B), LGM3 (C) and X-VIVO 20 (D) supplemented as described in the Protocol section. Cells were either transfected with unspecific control siRNA (Ctrl) or IL10RB-specific siRNA (IL10RB). As additional controls the following samples were included: Pulse control, i.e. cells which underwent transfection in the absence of siNRA (Pulse), and a medium control, i.e. cells that received only the changes of culture media but remained otherwise untreated. 24 hr after transfection the cells were incubated in serum free medium with or without 50 ng/ml IL10 for further 24 hr. SOCS3 expression was measured by RT-qPCR; diagrams show the mean of three independent experiments, error bars represent the standard error of the mean; *, # p <0.05; **, ## p <0.01; ***, ### p <0.001 vs. Ctrl and vs. Ctrl + IL-10, respectively. Please click here to view a larger version of this figure.
