

Summary
RNA-sequencing en bioinformatica analyses werden significant verschillend tot expressie en transcriptiefactoren in Lin-CD34 + en CD34-Lin subpopulaties van muis EMLcells identificeren. Deze transcriptiefactoren kunnen een belangrijke rol spelen bij het bepalen van de switch tussen zelfvernieuwende Lin-CD34 + en gedeeltelijk gedifferentieerd Lin-CD34-cellen.
Abstract
Hematopoietische stamcellen (HSC) worden klinisch gebruikt voor transplantatie behandeling hematopoëtische systeem van een patiënt bouwen bij vele ziekten zoals leukemie en lymfoom. Ophelderen van de mechanismen die HSCs zelfvernieuwing en differentiatie is belangrijk voor de toepassing van HSCs voor onderzoek en klinische toepassingen. Het is echter niet mogelijk om grote hoeveelheden van HSCs hetzij vanwege hun onvermogen tot prolifereren in vitro. Om deze hindernis te overwinnen, hebben we gebruik gemaakt van een muis beenmerg afgeleide cellijn, de EML (erythroïde, myeloïde en lymfatische) cellijn, als een modelsysteem voor de studie.
RNA-sequencing (RNA-Seq) wordt in toenemende mate gebruikt om microarray voor genexpressie studies vervangen. Wij rapporteren hier een gedetailleerde methode waarbij RNA-Seq technologie het potentieel belangrijke factoren bij de regulering van EML cel zelfvernieuwing en differentiatie te onderzoeken. Het protocol in dit papier is verdeeld in drie delen. De eerste part wordt uitgelegd hoe u de cultuur EML cellen en aparte Lin-CD34 + en Lin-CD34-cellen. Het tweede deel van het protocol biedt gedetailleerde procedures voor de totale RNA-bereiding en de daaropvolgende bibliotheek constructie voor high-throughput sequencing. Het laatste deel beschrijft de methode voor het RNA-Seq data-analyse en wordt uitgelegd hoe u de gegevens gebruiken om differentieel tot expressie transcriptiefactoren tussen Lin-CD34 + en Lin-CD34 cellen te identificeren. Het meest significant verschillend tot expressie transcriptiefactoren werden geïdentificeerd aan de potentiële sleutel regulatoren controleren van EML cel zelfvernieuwing en differentiatie zijn. In de discussie sectie van dit document, hebben we aandacht voor de belangrijkste stappen voor een succesvolle uitvoering van dit experiment.
Kortom, dit document biedt een methode waarbij RNA-Seq technologie mogelijke regulatoren van zelfvernieuwing en differentiatie in EML cellen te identificeren. De vastgestelde sleutelfactoren zijn onderworpen aan downstream functionele analyse in vitro en in vivo.
Introduction
Hematopoietische stamcellen zijn zeldzaam bloedcellen die vooral in de volwassen beenmerg niche wonen. Zij zijn verantwoordelijk voor de productie van cellen nodig is om de bloed vullen en het immuunsysteem 1. Als een soort van stamcellen, HSCs kunnen zowel zelfvernieuwing en differentiatie. Ophelderen van de mechanismen die het lot beslissing van HSC's te beheersen, in de richting van ofwel zelf-vernieuwing of differentiatie, zal een waardevolle leidraad bieden aan de manipulatie van HSC voor bloedziekte onderzoeken en klinische gebruik 2. Een probleem van de onderzoekers dat HSCs kunnen worden gehandhaafd en geëxpandeerd in vitro in zeer beperkte mate; de meeste afstammelingen gedeeltelijk gedifferentieerd in kweek 2.
Om de belangrijkste toezichthouders dat de processen van zelf-vernieuwing en differentiatie te controleren op een genoom-brede schaal te identificeren, hebben we gebruik gemaakt van een muis primitieve hematopoietische voorlopercellen cellijn EML als modelsysteem. Thwordt cellijn werd afgeleid van muizen beenmerg 3,4. Wanneer gevoed met verschillende groeifactoren kunnen EML cellen differentiëren tot erythroïde, myeloïde en lymfoïde cellen in vitro 5. Belangrijk is, kan deze cellijn worden gekweekt in grote hoeveelheid in kweekmedium met stamcel factor (SCF) en het behoud van hun multipotentiality. EML cellen kunnen worden gescheiden in subpopulaties van zelf-vernieuwing Lin-SCA + CD34 + en gedeeltelijk gedifferentieerde Lin-SCA-CD34- cellen op basis van oppervlakte markers CD34 en SCA 6. Net als bij de korte termijn HSC, SCA + CD34 + cellen zijn in staat van zelf-vernieuwing. Bij behandeling met SCF, Lin-SCA + CD34 + cellen snel kan regenereren een gemengde populatie van Lin-SCA + CD34 + en Lin-SCA-CD34- cellen en blijven prolifereren 6. De twee populaties zijn vergelijkbaar in morfologie en hebben vergelijkbare niveaus van c-kit mRNA en eiwit 6. Lin-SCA-CD34- cellen kunnen voortplanten in medium dat IL-3 in plaats van SCF 3. Unveiling de belangrijkste toezichthouders in de EML Celgedrag zal beter begrip van de moleculaire en cellulaire mechanismen bieden in vroege ontwikkelingsstoornissen overgang tijdens hematopoiesis.
Om de onderliggende moleculaire verschillen tussen de zelf-vernieuwing Lin-SCA + CD34 + en gedeeltelijk gedifferentieerde Lin-SCA-CD34- cellen te onderzoeken, gebruikten we RNA-Seq differentieel tot expressie gebrachte genen te identificeren. In het bijzonder richten we ons op transcriptiefactoren, zoals transcriptiefactoren zijn cruciaal bij het bepalen lot van de cel. RNA-Seq is een recent ontwikkelde aanpak die de mogelijkheden van de next-generation sequencing gebruikt (NGS) technologieën te profileren en te kwantificeren RNA getranscribeerd uit genoom 7,8. In het kort, totaal RNA poly-A geselecteerd en gefragmenteerd met de eerste template.The RNA matrijs wordt vervolgens omgezet in cDNA met behulp van reverse transcriptase. Om in kaart volledige lengte RNA transcripten, met intacte, niet-afgebroken RNA voor het construeren van cDNA-bibliotheek is belangrijk. Voor de purstelt van sequencing worden specifieke adapter sequenties toegevoegd aan beide uiteinden van cDNA. Vervolgens, in de meeste gevallen cDNA moleculen worden geamplificeerd door PCR en sequentie in een high-throughput manier.
Na sequencing, het resulterende leest kan worden afgestemd om een referentie genoom en een transcriptome database. Het aantal staat dat de kaart aan de referentie-gen wordt geteld en deze informatie kan worden gebruikt om genexpressie kan worden gemeten. De leest kan worden gemonteerd zonder de novo referentie genoom, waardoor de studie van transcriptomes in niet-modelorganismen 9. RNA-seq technologie is ook gebruikt voor splice isovormen 10-12, nieuwe transcripten 13 en genfusies 14 detecteren. Naast de detectie van eiwit-coderende genen, kunnen RNA-Seq ook worden gebruikt om nieuwe transcriptieniveau van niet-coderende RNA's, zoals detecteren en analyseren van lange niet-coderende RNA 15,16, microRNA 17, siRNA etc. 18. Vanwege thij nauwkeurigheid van deze methode, is gebruikt voor de detectie van enkelvoudige nucleotide variaties 19,20.
Vóór de komst van de RNA-Seq-technologie, microarray was de belangrijkste methode die wordt gebruikt voor de analyse van genexpressie-profiel. Vooraf ontworpen probes worden gesynthetiseerd en vervolgens bevestigd aan een vast oppervlak op een microarray dia 21 vormen. mRNA wordt geëxtraheerd en omgezet in cDNA. Tijdens het reverse transcriptie proces worden fluorescent gelabelde nucleotiden opgenomen in het cDNA en de cDNA kan worden gehybridiseerd op de microarray dia. De intensiteit van het vanuit een specifieke plek signaal afhankelijk van de hoeveelheid cDNA binding aan de specifieke probe op die plek 21. In vergelijking met RNA-Seq-technologie, micro-array heeft een aantal beperkingen. Eerste microarray gebaseerd op de reeds bestaande kennis van gen annotatie, terwijl RNA-Seq technologie kan nieuwe transcripten detecteren relatief hoog achtergrondniveau, die het gebruik ervan bij beperkt gene uitdrukking niveau is laag. Bovendien, de RNA-Seq technologie veel hoger dynamisch bereik van detectie (8000 voudig) 7, dat, als gevolg van achtergrondlicht en verzadiging van de signalen, de nauwkeurigheid van microarray beperkt zowel sterk en bescheiden expressie gebrachte genen 7,22. Tenslotte microarray sondes verschillen in hun hybridisatie efficiëntie, waarin de resultaten minder betrouwbaar maken bij het vergelijken van de relatieve expressie niveaus van verschillende transcripts binnen één monster 23. Hoewel RNA-Seq veel voordelen heeft boven microarray, de data analyse complex. Dit is een van de redenen dat veel onderzoekers nog steeds gebruik van microarray in plaats van RNA-Seq. Verschillende bioinformatica nodig voor RNA-Seq data en analyse 24.
Onder verschillende next-generation sequencing (NGS) platforms, 454, Illumina, SOLID en Ion Torrent zijn de meest gebruikte plaatsen. 454 was de eerste commerciële NGS platform. In tegenstelling tot de andere sequencing platformszoals Illumina en SOLID, genereert de 454-platform meer lezen lengte (gemiddeld 700 base gelezen) 25. Langere leest zijn beter voor eerste karakterisering van transcriptiome vanwege de hogere efficiëntie monteren 25. Het belangrijkste nadeel van het platform 454 is de hoge kosten per megabase sequentie. De Illumina en SOLID platforms genereren leest met verhoogde aantallen en korte lengtes. De kosten per megabase sequentie is veel lager dan de 454 platform. Vanwege het grote aantal korte leest de Illumina SOLID platforms, gegevensanalyse veel computerintensief. De prijs van het instrument en reagentia voor sequencing voor de Ion Torrent platform is goedkoper en de sequencing is korter 25. Echter, het aantal fouten en de kosten per megabase van sequence zijn hoger in vergelijking met de Illumina en SOLID platforms. Verschillende platforms hebben hun eigen voordelen en nadelen en vereisen verschillende methoden voor gegevensanalyse. De plaTForm moet worden gekozen op basis van de sequencing doel en de beschikbaarheid van financiële middelen.
In dit artikel nemen we Illumina RNA-Seq platform als voorbeeld. We gebruikten EML cel als modelsysteem om de belangrijkste toezichthouders in EML cel zelfvernieuwing en differentiatie te onderzoeken, en op voorwaarde dat een gedetailleerde methoden voor RNA-Seq bibliotheek bouw en de data-analyse voor expressie niveau berekening en nieuwe transcript detectie. We hebben aangetoond in onze eerdere publicatie die RNA-seq studie EML modelsysteem 2, wanneer gekoppeld aan functionele test (bijvoorbeeld shRNA knockdown) een krachtige aanpak in het begrijpen van de moleculaire mechanismen van de vroege stadia van hematopoëtische differentiatie en kan dienen als een model voor analyse van cel zelfvernieuwing en differentiatie in het algemeen.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. EML celcultuur en Scheiding van Lin-CD34 + en Lin-CD34 cellen met behulp van Magnetic Cell Sorting System en Fluorescentie-Activated Cell Sorting Methode
- Voorbereiding van babyhamsternier (BHK) celcultuur medium voor stamcelfactor collectie:
- Cultuur BHK cellen in DMEM medium met 10% FBS in 25 cm2 fles (tabel 1) bij 37 ° C, 5% CO2 in een celkweek incubator.
- Wanneer cellen groeien tot 80-90% confluentie was cellen eenmaal met 10 ml PBS. Voeg 5 ml van 0,25% trypsine-EDTA oplossing van de monolaag en incubeer de cellen gedurende 1-5 min bij kamertemperatuur (RT) tot de cellen losgemaakt.
- Pipet de oplossing op en neer voorzichtig te breken klonten van cellen. Voeg 5 ml compleet DMEM aan de kolf aan trypsine-activiteit te stoppen. Verzamel cellen door centrifugatie bij 200 xg gedurende 5 minuten bij kamertemperatuur.
- Verwijder het medium en resuspendeer de celpellet in 10 ml verse BHK celkweekmedium. Overdracht 2 ml van de celsuspensie uit stap 1.1.4 een nieuwe 75 cm2 kolf en voeg 48 ml verse BHK celkweekmedium aan de kolf.
- Cultuur BHK cellen gedurende twee dagen en laat het kweekmedium. Passage het medium door een 0,45 urn filter. Bewaar het medium -20 ° C tot verder gebruik.
- EML celcultuur:
- Kweek EML cellen (in suspensie) in EML basisch medium dat BHK celkweek medium (tabel 1) bij 37 ° C, 5% CO2 in een celkweek incubator.
- Handhaaf de EML cellen bij lage celdichtheid (0,5-5 x 10 5 cellen / ml) met de piek dichtheid lager dan 6 x 10 5 cellen / ml. Splits de cellen om de 2-3 dagen in een verhouding van 1: 5. Passage EML cellen zachtjes en de cultuur gooi deze na passage van 10 generaties.
- Uitputting van lineage positieve cellen:
- Oogst de EML cellen door centrifugatie bij 200 xg for 5 min en was de cellen eenmaal met PBS. Verzamel de cellen door centrifugatie bij 200 xg gedurende 5 minuten.
- Resuspendeer de cellen met PBS en tel de cellen met een hemocytometer. Bepaal het antilichaam concentratie in de daaropvolgende celscheiding stap naar het getal van de cellen (zie de instructies die door de aanbieder van de cel isolatie-systeem).
- Isoleer het geslacht negatieve (Lin) cellen middels stam antilichaam cocktail (cocktail van biotine-geconjugeerde monoklonale antilichamen CD5, CD45R (B220), CD11b, Anti-Gr-1 (Ly-6G / C), 7-4 en Ter-119 ) en een magnetisch geactiveerde celsortering volgens instructies van de fabrikant.
- Scheiding van Lin-CD34 + en Lin-CD34 cellen:
- Spin down de Lin cellen uit stap 1.3.3 bij 200 xg gedurende 5 minuten. Resuspendeer de celpellet met PBS en tel de cellen met een hemocytometer.
- Was de cellen tweemaal met FACS-buffer en pellet de cellen bij 200 xggedurende 5 min.
- Label vijf 1,5 ml microcentrifuge buizen met respectievelijk 1, 2, 3, 4, 5. Resuspendeer de cellen met 100 ul FACS buffer per 10 6 cellen (10 6 cellen per buis).
- Voeg 1 ug van anti-muis CD34 FITC antilichaam aan buis 1 en buis 2 en meng de buizen voorzichtig.
- Incubeer alle buizen bij 4 ° C gedurende 1 uur in het donker.
- Voeg 0,25 ug Anti-SCA1 antilichaam PE-geconjugeerde en 20 pl APC geconjugeerde Lineage Cocktail antilichamen buis 1, 0,25 ug van PE-geconjugeerd anti-SCA1 antilichaam buis 3, en 20 pl APC-geconjugeerde antilichamen Lineage Cocktail buis 4.
- Meng alle buizen zachtjes en incubeer de cellen bij 4 ° C gedurende 30 min in het donker.
- Voeg 300 ul FACS buffer om de cellen en de cellen af te draaien bij 200 xg gedurende 5 minuten.
- Was de cellen met 500 pi FACS buffer driemaal.
- Resuspendeer de celpellet in 500 ui FACS buffer.
- Gebruik de cellen in tubes 2, 3, 4 en 5 voor het opzetten compensatie. Isoleer Lin-SCA + CD34 + en Lin-SCA-CD34 cellen in buis 1 met behulp van FACS Aria.
2. RNA Voorbereiding en Bibliotheek Bouw voor High-throughput sequencing
- Isolatie, kwaliteit analyse en kwantificering van RNA:
- Extract totaal RNA van Lin-CD34 + en Lin-CD34 cellen respectievelijk behulp TRIzol na het produceert 'protocol.
- Verwijder de verontreinigde DNA met desoxyribonuclease I (DNase I) volgens het protocol van de fabrikant. Eventueel bewaar het RNA bij -80 ° C bij deze stap voor verder gebruik.
- Beoordeel de kwaliteit totaal RNA met Bioanalyzer volgens de instructies van de aanbieder. Gebruik RNA monster met RNA Integrity Number (RIN) lager dan 9.
- Bibliotheek Bouw en high-throughput sequencing:
OPMERKING: Dit protocol beschrijft RNA-Seq gebruik van Illumina platform. Voorandere sequencing platforms, zijn andere bibliotheek bereidingswijzen vereist.- Gebruik 0,1-4 ug van hoge kwaliteit totaal RNA per monster voor bibliotheek voorbereiding. Normaal 2 ug totaal RNA kan worden geëxtraheerd uit 10 5 EML cellen.
- Gebruik een RNA-sequencing monstervoorbereiding systeem voor RNA zuivering en fragmentatie, eerste en tweede streng cDNA synthese, einde reparatie, 3'-uiteinden adenylering, adapter ligatie en PCR-amplificatie, volgens de gedetailleerde standaardprocedures van de instructies van de provider.
- Positief selecteren PolyA mRNA met behulp van oligo-dT magnetische korrels en versnippering van de mRNA.
- Voer reverse transcriptie met behulp van willekeurige primers met het cDNA te verkrijgen en vervolgens synthetiseren de tweede streng cDNA dubbelstrengs cDNA te genereren.
- Verwijder de 3 'overhangen en vul de 5' overhang door DNA polymerase. Adenylaat 3 'uiteinden te voorkomen cDNA fragmenten uit ligeren elkaar.
- Voeg multiplex indexering adapters beide uiteinden van de dscDNA. Voer PCR voor de verrijking van DNA-fragmenten.
- Meet de A260 / A280 informatie over de concentratie van de bibliotheek met behulp van een spectrofotometer verkrijgen.
- Beoordeel de bibliotheek kwaliteit en meet de orde van grootte van de DNA-fragmenten met een Bioanalyzer.
3. Data Analysis
Ter referentie van de software die wordt gebruikt in dit deel, zie (tabel 2).
- Gegevensbestand verwerking voor downstream-analyse:
- Omzetten .bcl (base call-bestand) bestand naar bestand .fastq behulp Casava software (Illumina, versie 1.8.2).
- Vuur van de 'Terminal' in Linux-systeem. Ga naar de map met gegevens die het gegevensbestand van een Illumina HiSeq2000 sequencing machine bevat. Stel dat de map resultaat is 'NASboy1 / JiaqianLabData / HiSeq_RUN / 2013_07_11 / 130627_SN860_0309_A_2013-166_H0PW9ADXX /', het typein de opdracht in figuur S1A, en voer de map data.
- Installeer Casava 1.8.2 in het Linux-systeem. Stel dat de outputfolder is 'Niet afgestemd', gebruikt u de opdracht in figuur S1B het configuratie bestand voor te bereiden voor het omzetten. Gebruik de optie --fastq-cluster-count 0 te verzekeren slechts één .fastq bestand wordt aangemaakt voor elk monster. De gegenereerde .fastq bestand is in .gz formaat. Unzip het voor downstream-analyse (Figuur S1B).
- Na de 'Niet afgestemd' map is aangemaakt, gaat u naar de 'Niet afgestemd' map (Figuur S1C).
- Gebruik de opdracht in figuur S1D om de omzetten te starten. De parameter '-j' levert het cpu nummer dat wordt gebruikt.
- Nadat het systeem klaar met het omzetten van proces, ga naar de map resultaat onder 'Niet afgestemd' map (Figuur S1E).
- Gebruik de opdracht in figuur S1F </ Strong> om de .fastq.gz bestand uitpakken in .fastq bestand onder elke map monster.
- Omzetten .bcl (base call-bestand) bestand naar bestand .fastq behulp Casava software (Illumina, versie 1.8.2).
- Detecteren roman transcripties en de expressie niveau te evalueren met behulp van Tuxedo Suite 26:
- Wijs de gepaarde-end RNA-Seq leest met de referentie muis genoom (UCSC versie MM9, verkregen uit http://cufflinks.cbcb.umd.edu/igenomes.html ) met behulp van Tophat software (versie 1.3.3) 27, die gebruik maakt van de Bowtie lezen mapper (versie 0.12.7) 28. Tophat wordt geleverd met "-geen-roman-Juncs" optie om de schatting nauwkeurigheid van meningsuiting niveau te verbeteren.
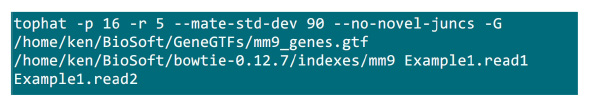
- Doe de .fastq bestanden in een map waar de mapping zal worden uitgevoerd. Stel dat er 2 .fastq bestanden (hernoemen naar Example1.read1, Example1.read2) voor een gepaarde-end sequencing sample, gebruikt u de opdracht in figuur S2 om de mapping (pas de parameters volgens de instelling systeem) doen.De parameter "-p" levert het cpu nummer dat wordt gebruikt. De "-r" en "-mate-std-dev" parameters kunnen worden verkregen bij de bibliotheek QC of is afgeleid van een subset van uitgelijnde leest (figuur S2).
- Monteer de in kaart gebrachte leest in RNA-transcripten met behulp van de manchetknopen software (versie 1.3.0) 29. Ren Manchetknopen met behulp van de annotatie-bestand van bekende genen (zelfde .gtf bestand dat wordt gebruikt door Tophat) en .bam bestand geproduceerd door Tophat.
- Na klaar Tophat hardlopen, in dezelfde map, gebruikt u de opdracht in figuur S3A om manchetknopen lopen om transcriptome en schatting transcript expressie niveau te construeren. De 'mm9_repeatMasker.gtf' en genoomsequentie bestanden in de map 'GenomeSeqMM9' kan worden verkregen bij UCSC Genome Browser.
- De resulterende genes.expr en transcripts.expr bestanden bevatten de uitdrukking waarde van genen en transcripties (isovormen). Kopiëren en plakkende inhoud van het bestand naar een Excel-bestand en te manipuleren met spreadsheetprogramma (Figuur S3B).
- Gebruik de opdracht in figuur S3C om het resulterende 'transcripts.gtf' bestand naar de referentie 'mm9_genes.gtf' bestand te vergelijken met het oog op nieuwe transcripties identificeren.
- Het resulterende .tmap bevat het vergelijkingsresultaat. Kopieer en plak de inhoud van een bestand naar een Excel-bestand en te manipuleren met een spreadsheetprogramma. Afschriften met klasse code 'u' kan worden beschouwd als 'nieuwe' in vergelijking met de referentie-.gtf bestand verstrekt (figuur S3D).
OPMERKING: Voor downstream analyse gemak, stel de FPKM waarden tot 0,1 als de waarden onder 0.1.
OPMERKING: Stap 3.2.3 - 3.2.6 is optioneel voor degenen die wensen om de nauwkeurigheid van meningsuiting schatting roman transcripts 'te verbeteren. Dit zal een veel langere tijd in beslag nemen, omdat het in kaart brengen en transcriptome constructie moeten run meer dan eens.
- Run Tophat behulp standaard parameters en voer manchetknopen tot gegenereerd .gtf bestand met de opdracht in figuur S3E.
- Vergelijk de resulterende .gtf bestand met de referentie genoom .gtf bestand met de opdracht in figuur S3F.
- Ontleden het resulteerde .tmap bestand zoals beschreven in de stap 3.2.2.4. Kopieer en plak de inhoud van een bestand naar een Excel-bestand en te manipuleren met een spreadsheetprogramma. Afschriften met klasse code 'u' kan worden beschouwd als 'nieuwe' in vergelijking met de referentie-.gtf bestand aangeboden.
- Na de stap 3.2.5, er een .combined.gtf in de map die kan worden gebruikt als referentie .gtf bestand. Een tweede run van Tophat en manchetknopen kan worden uitgevoerd zoals beschreven in de stap 3.2.1 en 3.2.2 tot een nauwkeuriger FPKM schatting van nieuwe transcripties te verkrijgen.
- Wijs de gepaarde-end RNA-Seq leest met de referentie muis genoom (UCSC versie MM9, verkregen uit http://cufflinks.cbcb.umd.edu/igenomes.html ) met behulp van Tophat software (versie 1.3.3) 27, die gebruik maakt van de Bowtie lezen mapper (versie 0.12.7) 28. Tophat wordt geleverd met "-geen-roman-Juncs" optie om de schatting nauwkeurigheid van meningsuiting niveau te verbeteren.
- Detect differentially uitgedrukt genen met behulp DESeq pakket 30.
- De input van DESeq is een ruwe tafel read telt. Om een dergelijke tafel te krijgen, gebruik dan de htseq-count script verspreid met de HTSeq Python pakket dat kan worden gedownload van HTSeq website ( http://www-huber.embl.de/users/anders/HTSeq/doc/count.html ) .
- Zorg ervoor dat samtools, python, en htseq-count programsare geïnstalleerd in het systeem. Verkrijgen rauwe LEES AANTAL nummers van tophat uitgang met behulp van het commando in figuur S4A.
- Bereid 'Raw_Count_Table.txt', 'ExperimentDesign.txt' bestanden met behulp van Excel. Kopieer en de inhoud in .txt formaat voor de DESeq R pakket (Figuur S4B) op te slaan.
- Installeer R-programma in het systeem. In de terminal, type 'R' en druk ENTER.A screen melding appearas toonde in figuur S4C.
- Lees 'Raw_Count_Table.txt ',' ExperimentDesign.txt 'in R met behulp van het commando in figuur S4D.
- Laad DESeq pakket met de opdracht in figuur S4E.
- In factoren omstandigheden in R (figuur S4F).
- Gebruik de opdracht in figuur S4G naar negatief binominale test uit te voeren op de genormaliseerde tabel met aantallen.
- Gebruik de opdracht in figuur S4H output significant differentiële expressie gebrachte genen in een CSV-bestand.
- De input van DESeq is een ruwe tafel read telt. Om een dergelijke tafel te krijgen, gebruik dan de htseq-count script verspreid met de HTSeq Python pakket dat kan worden gedownload van HTSeq website ( http://www-huber.embl.de/users/anders/HTSeq/doc/count.html ) .
- Lookup transcriptiefactoren '(TFS) FPKM waarden over monsters met behulp van Excel. Intersect DE gen tafel en TFs tafel. Genen behoren zowel tabel worden differentieel tot expressie transcriptiefactoren.
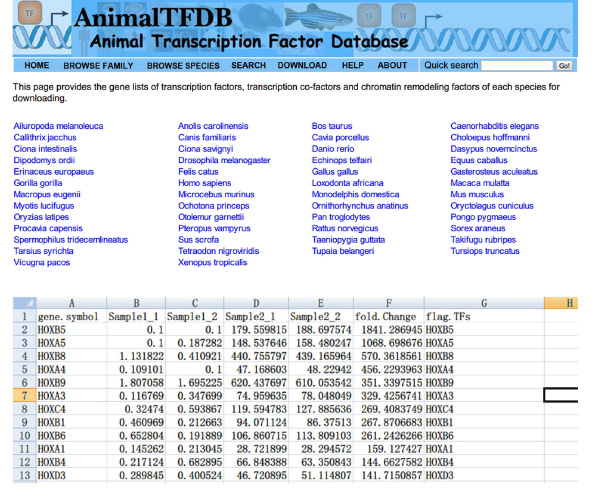
- Ga naar de website http://www.bioguo.org/AnimalTFDB/download.php en download de transcriptiefactoren. Opzoeken dan het voorste transcriptiefactoren in de Excel (< strong> Figuur S5).
- Het genereren .bigwig bestand voor UCSC genoom browser visualisatie.
- Download 'bedtools' software pakket van de website https://github.com/arq5x/bedtools2 en installeer de software in het systeem 31. Download de UCSC gereedschappen 'bedGraphToBigWig' van de website http://hgdownload.cse.ucsc.edu/admin/exe/ en installeer de software in het systeem.
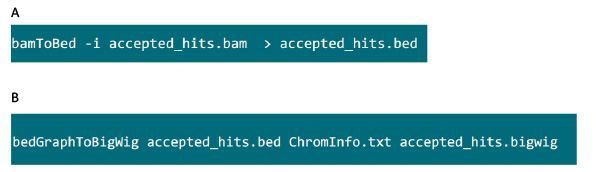
- In de map met de .bam bestand, gebruikt u de opdracht in figuur S6A om .bam bestand gegenereerd door tophat in .bed bestand te converteren.
- Na het .bed bestand wordt geproduceerd, gebruikt u de opdracht in figuur S6B om .bigwig bestand te genereren. Het bestand 'ChromInfo.txt' is te verkrijgen bij onderstaande url:e onerzoeken = "_blank"> http://hgdownload.cse.ucsc.edu/goldenPath/mm9/database/chromInfo.txt.gz.
- Observeer een aangepaste track op UCSC Genome Browser. Raadpleeg de website http://genome.ucsc.edu/goldenPath/help/customTrack.html over hoe je een aangepaste track met UCSC genoom browser weer te geven.

Figuur S1: Het omzetten .bcl bestand naar bestand .fastq behulp Casava software.

Figuur S2: Mapping leest om te verwijzen naar het genoom met behulp van Tophat.
Figuur S3: Detectie van nieuwe transcripties en expressie niveau schatting.

Figuur S4: Calling differentieel uitgedrukt gen met behulp DESeq pakket.

Figuur S5: Identificatie van differentieel tot expressie transcriptiefactoren.

Figuur S6: Het omzetten van mapping resultaat voor data visualisatie.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Om differentieel tot expressie gebrachte genen in Lin-CD34 + en CD34-Lin EML cellen analyseren gebruikten we RNA-Seq techniek. Figuur 1 toont de werkstroom van de procedures. Na isolatie van lineage negatieve cellen door magnetische celsortering, we gescheiden Lin-SCA + CD34 + en Lin-SCA-CD34 cellen met behulp van FACS Aria. Lin-verrijkte EML cellen werden gekleurd met anti-CD34, anti-SCA1 afstamde cocktail antilichamen. Alleen Lin- cellen werden afgesloten voor analyse van SCA1 en CD34 expressie. Twee populaties (SCA + CD34 + en CD34- EML SCA-cellen) worden waargenomen door FACS-analyse (Figuur 2) 6.
Na scheiden, we totaal RNA geëxtraheerd van CD34 + en CD34- cellen respectievelijk de kwaliteit van RNA geanalyseerd. De nauwkeurigheid van RNA-Seq gegevens grotendeels op de kwaliteit van RNA-Seq bibliotheek en de kwaliteit van het totale RNA is essentieel voor het bereiden van een hoge kwaliteit bibliotheek. Hoge kwaliteit RNA monster moet een OD 260/280 waarde tussen 1 hebben.8 en 2.0. Naast het gebruik van de spectrofotometer, werd RNA kwaliteit verder beoordeeld nauwkeuriger door Bioanalyzer. Figuur 3 toont een resultaat van een hoge kwaliteit RNA monster met de RIN gelijk aan 9.4. Alleen hoge kwaliteit totaal RNA monster met RIN waarde groter dan 9 werd gebruikt voor mRNA-extractie en de daaropvolgende bibliotheek bouw procedures.
Ribosomaal RNA is de meest voorkomende vorm van RNA in cellen. Momenteel zijn er twee belangrijke strategieën, uitputting van rRNA of positief selectie van gepolyadenyleerde mRNA (poly-A mRNA), worden gebruikt voor de verrijking van target RNA voordat bibliotheek constructie. Niet gepolyadenyleerd RNA soorten worden verloren tijdens de selectie van poly A-mRNA. Daarentegen kan rRNA uitputting werkwijzen zoals RiboMinus niet gepolyadenyleerd RNA species behouden. Het doel van deze studie is het zoeken naar differentieel tot expressie coderende genen in twee celtypen, waardoor gebruikten we de poly-A mRNA selectiemethode voor verrijking van target RNA's voor bibliotheek constructie. Bij bibliotheekconstructie klaar was, de grootte van DNA fragmenten in de bibliotheek werd gecontroleerd voor sequencing met Bioanalyzer. Figuur 4 toont een goede kwaliteit bibliotheek met de fragmentgrootte pieken bij ongeveer 300 bp.
In de volgende stap werd de bibliotheek onderworpen aan high-throughput sequencing. In principe zal het langer lezen lengte nuttig voor lezen mapping zijn. Het kan de waarschijnlijkheid verminderen dat de gelezen is toegewezen aan meerdere locaties door gelijkenis tussen dubbele genen of gen-familie. Aangezien het paar-end sequencing sequenties van beide uiteinden van de fragmenten moet de gelezen gekozen lengte minder dan de helft van de gemiddelde lengte fragmenten. Als het belangrijkste doel van het experiment is om de expressie in plaats van de aanleg van transcript structuur te meten, single-end lezen (75 of 100 bp) kan de kosten verlagen zonder verlies van te veel informatie. Gepaarde-end sequencing is nuttiger voor transcript structuur bouw en korterlees lengte kan worden gebruikt om de kostprijs te verlagen. Zeker, wanneer er voldoende middelen beschikbaar zijn, wordt meer gelezen lengte voorkeur.
Voor differentiële expressie analyse, zijn er vele andere dan DESeq alternatieve algoritmen. Er is ook één in manchetknopen pakket met de naam cuffdiff 32. DESeq is een van de meest gebruikte telling gebaseerd DE genanalyse algoritmen. DESeq methode is gebaseerd op een goed gekarakteriseerde statistieken model - negatieve binomiale verdeling. In onze ervaring, DESeq is stabieler vergelijken cuffdiff. Vroege versies van cuffdiff geven vaak sterk verschillende aantallen DE genen. Daarom gebruikten we DESeq voor DE analyse hier.
Omdat transcriptiefactoren zijn cruciaal voor het lot van de cel bepalen, hebben we ons gericht op het significant verschillend tot expressie transcriptiefactoren 33. De TFs gewijzigd> 1,5 maal tussen Lin-CD34 + en Lin-CD34 werden gevonden en worden getoond op de heatmap (Figur5 e) 2. Met name de relatieve expressieniveau van Tcf7 in Lin-CD34 + cellen meer dan 100 maal hoger dan die in Lin-CD34- cellen. Zo werd Tcf7 gekozen voor verdere ChIP-Sequencing (chromatine Immunoprecipitatie en sequencing) analyse en de functionele test functie Tcf7 's in de regulering van EML cel zelfvernieuwing en differentiatie 2 bevestigen.

Figuur 1:. Workflow van de procedures Lin-CD34 + en CD34- Lin-cellen werden door magnetische celscheiding systeem en fluorescentie-geactiveerde celsortering methode. Totaal RNA werd geëxtraheerd mRNA gevolgd door zuivering en bibliotheekconstructie. Na analyse van de bibliotheek kwaliteit, werden monsters onderworpen aan high throughput sequencing. De gegevens werden geanalyseerd en differentieel tot expressie transcriptiefactoren werden geïdentificeerd.

Figuur 2: Scheiding van Lin-CD34 + en Lin-CD34 EML cellen 6 Lin- EML cellen werden verrijkt door magnetische celsortering.. Lin- cellen werden gekleurd met anti-CD34, anti-SCA1 afstamde mengsel antilichamen. Lin- cellen werden gated voor expressie van CD34 en SCA1. Lin-CD34 + SCA + en Lin-CD34-schaalniveaus EML celpopulaties werden opgelost.

Figuur 3. Een vertegenwoordiger van hoogwaardige totale RNA monsters De kwaliteit totaal RNA werd beoordeeld door Bioanalyzer. Het RNA Integriteit Number is 9,4 (FU, Fluorescentie Units).

Figuur 4:. Fragmenten van grootte van Gekoppelde-End bibliotheek De DNA grootteverdeling van de bibliotheek werd geanalyseerd middels Bioanalyzer. De meeste fragmenten zijn in de orde van grootte van 250-500 bp.

Figuur 5:. Differentieel tot expressie transcriptiefactoren (> 1,5-voudig) tussen Lin-CD34 + cellen en Lin-CD34- cellen 2 Voor elk celtype, twee onafhankelijke experimenten werden uitgevoerd. Up-gereguleerde genen worden aangeduid als rode kleur en omlaag gereguleerde genen worden aangeduid als groene kleur.
Tabel 1: Buffers en Celcultuur mediums.
| Software | Gebruik | Verwijzing | |||
| Bowtie 1.2.7 | Gebruikt door Tophat voor mapping | [28] | |||
| Tophat 1.3.3 | Mapping leest terug naar referentie-genoom | [27] | |||
| Manchetknopen 1.3.0 | Afschriften constructie en expressie niveau schatting | [29] | |||
| DESeq 1.16.0 | Differentiële expressie analyse | [30] | Bedtools 2.18 | Omzetten .bam bestand in .bed bestand | [31] |
| bedGraphToBigWig | Omzetten .bed bestand naar bestand .bigwig | http://genome.ucsc.edu/ |
Tabel 2: Lijst van software voor gegevensanalyse.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Zoogdieren transcriptome is zeer complex 34-38. RNA-Seq-technologie speelt een steeds belangrijkere rol in de studies van transcriptoomanalyse, roman transcripties detectie en single nucleotide variatie discovery enz. Het heeft vele voordelen ten opzichte van andere methoden voor de analyse van genexpressie. Zoals vermeld in de inleiding, overwint de hybridisatie artefacten van microarray en kan worden gebruikt om nieuwe transcripten de novo identificeren. Een beperking van RNA-sequencing relatief korte lengte gelezen vergelijking met Sanger sequentiebepaling. Echter, met de snelle verbetering van sequencing technologie, lees lengte voortdurend toe. In dit artikel geven we gedetailleerde methoden voor het gebruik van deze technologie om potentiële belangrijke regulatoren in de muis EML cel zelfvernieuwing en differentiatie te identificeren.
De eerste belangrijke stap voor dit protocol is EML celcultuur. Hoewel EML is een hematopoietische voorloper cellijn en het kan zijngepropageerd in grote hoeveelheid met SCF. Het kweken conditie van EML cellen vereist meer aandacht dan de gebruikelijke onsterfelijk gemaakte cellijnen. De cellen moeten worden gevoed en gepasseerd op regelmatige basis met zachte werking; anders de cellen kunnen veranderen in de eigenschappen van zelfvernieuwing en differentiatie ondergaan celdood. Als eerste stap na het verzamelen van genoeg cellen die we lineage negatieve cellen met een magnetische celsortering systeem. Vervolgens afgescheiden we CD34 + en CD34- cellen met fluorescentie-geactiveerde celsortering. De EML cellen worden normaal gepasseerd minder dan 10 generaties voordat u voor RNA-extractie en de aantallen CD34 + en CD34 cellen moeten vergelijkbaar na scheiding zijn. Als de twee populaties variëren sterk in aantal cellen, is het raadzaam om de cultuur ontdoen en opnieuw ontdooien andere buis cel stock cultuur.
Na scheiding van CD34 + en CD34 cel werd totaal RNA-extractie uitgevoerd, opnieuw een belangrijke stap voor deze stUdy. Hoogwaardige RNA is de basis voor de bouw van een hoge kwaliteit bibliotheek, die de nauwkeurigheid van de sequentie data belooft. In deze kritieke stap dienen de contacten met RNase vermeden. Alle reagentia moeten gratis worden RNase. Het is belangrijk om handschoenen te allen tijde tijdens het hanteren RNA. Hoge kwaliteit RNA monster een OD 260/280 waarde tussen 1,8 en 2,0. Bij het verzamelen van de waterige fase die RNA, wees voorzichtig om geen organische fase voeren met de RNA-monster. Eventuele resterende organische oplosmiddelen zoals fenol of chloroform in RNA zal leiden tot een OD260 / 280 waarde van minder dan 1,65. Wanneer de OD260 / 280 lager is dan 1,65, precipiteren RNA opnieuw met ethanol. Na het wassen met 75% ethanol, dat niet Kreukherstellend RNA pellet. Drogen RNA pellet volledig zal de oplosbaarheid van RNA beïnvloeden en leiden tot een lage opbrengst van RNA.
De volgende belangrijke stap voor dit protocol is bibliotheek voorbereiding. Na totaal RNA extractie, een stap van het gebruiken DNase voor het verwijderen van vervuild DNA is zeer aan te bevelen, omdat DNA contaminatie zou kunnen leiden tot verkeerde schatting van de totale hoeveelheid RNA gebruikt. Aanbevolen wordt de procedure onmiddellijk stroomafwaarts voeren na RNA-isolatie, aangezien na langdurige opslag en invriezen-ontdooien, zal RNA degraderen enigszins. Als de volgende stappen na RNA-isolatie niet onmiddellijk kan worden uitgevoerd, opgeslagen in de RNA -80 ° C. Voor totaal RNA wordt gebruikt voor mRNA zuivering en cDNA-synthese, moet de kwaliteit altijd gecontroleerd. Hoge kwaliteit RNA kan worden gebruikt voor bibliotheek bereiding. Met behulp van lage kwaliteit of gedegradeerde RNA zou kunnen leiden tot een oververtegenwoordiging van 3'-uiteinden. Voordat sequencing, werd bibliotheek kwaliteit beoordeeld om maximale sequencing efficiëntie te garanderen.
In de data-analyse deel, na het uitvoeren van een run van Manchetknopen zonder verwijzing transcriptome, combineerden we de roman transcripties met bekende transcripties van een referentie .gtf bestand en run Tophat en manchetknopen voor de tweede keer te vormen.Deze twee-run procedure wordt aanbevolen, omdat deze zorgen voor meer accurate FPKM schatting dan het draaien van slechts één keer. Na data-analyse, werden de differentieel tot expressie gebrachte genen geïdentificeerd. Downstream experimenten kunnen worden uitgevoerd om de functie van genen te valideren in vitro en in vivo. In onze vorige publicatie 2, kozen we het aanzienlijk verschillend tot expressie transcriptiefactoren en identificeerde het genoom bindingsplaats van deze factoren door het uitvoeren van chromatine immunoprecipitatie en sequencing (ChIP-Seq). Daarnaast, pasten wij shRNA knockdown assay het functionele effect van Tcf7 testen. We vonden dat in Tcf7 knockdown cellen opwaarts gereguleerd genen genen sterk verrijkt in CD34- cellen terwijl down-gereguleerde genen bleken significant verrijkt in CD34 + cellen. Daarom is de genexpressie profiel van Tcf7 knockdown cellen verschoven in de richting van een gedeeltelijk gedifferentieerde CD34 state.Overall, met behulp van EML cel als een modelsysteemgekoppeld aan RNA-Sequencing technologie en functionele testen, identificeerden we en bevestigd Tcf7 als een belangrijke regulator van EML cel zelfvernieuwing en differentiatie.
Subscription Required. Please recommend JoVE to your librarian.
Materials
| Name | Company | Catalog Number | Comments |
| Antibiotic-Antimycotic | Invitrogen | 15240-062 | BHK cell culture |
| Anti-Mouse CD34 FITC | eBioscience | 11-0341-81 | FACS sorting |
| Anti-Mouse Ly-6A/E (Sca-1) PE | eBioscience | 12-5981-81 | FACS sorting |
| APC Mouse Lineage Antibody Cocktail | BD Biosciences | 558074 | FACS sorting |
| BD FACSAria Cell Sorter | BD Biosciences | Special offer sysmtem | FACS sorting |
| Corning™ Cell Culture Treated Flasks 75 cm2 | Corning incorporated | 430641 | Cell culture |
| Corning™ Cell Culture Treated Flasks 25 cm2 | Corning incorporated | 430639 | Cell culture |
| Deoxyribonuclease I, Amplification Grade | Invitrogen | 18068-015 | Library preparation |
| DMEM | Invitrogen | 11965-092 | BHK cell culture |
| DPBS | Gibco | 14190 | Cell culture |
| HI FBS | Invitrogen | 16140071 | BHK cell culture |
| Horse Serum | Invitrogen | 16050-122 | EML cell culture |
| IMDM | HyClone | SH30228.02 | EML cell culture |
| L-Glutamine | Invitrogen | 25030-081 | Cell culture |
| Lineage Cell Depletion Kit, mouse | Miltenyi Biotec | 130-090-858 | Isolation of lineage negative cells |
| NanoVue Plus spectrophotometer | GE Healthcare | 28-9569-62 | Quality control |
| Thermo Scientific™ Napco™ 8000 Water-Jacketed CO2 Incubators | Thermo Scientific | 15-497-002 | Cell culture |
| Penicillin-Streptomycin | Invitrogen | 15140-122 | EML cell culture |
| TRIzol® Reagent | Invitrogen | 15596-018 | RNA exraction |
| TruSeq™ RNA Sample Prep Kit v2 -Set B (48 rxn) | Illumina | RS-122-2002 | Library preparation |
| 2100 Electrophoresis Bioanalyzer Instrument | Agilent | G2939AA | Quality control |
| 0.25% Trypsin-EDTA | Gibco | 25200 | Cell culture |
| 0.45 µm Syringe Filters | Nalgene | 190-2545 | Cell culture |
References
- Chambers, S. M., Goodell, M. A. Hematopoietic stem cell aging: wrinkles in stem cell potential. Stem Cell Rev. 3, 201-211 (2007).
- Wu, J. Q., et al. Tcf7 is an important regulator of the switch of self-renewal and differentiation in a multipotential hematopoietic cell line. PLoS genetics. 8, (2012).
- Ye, Z. J., et al. Complex interactions in EML cell stimulation by stem cell factor and IL-3. Proceedings of the National Academy of Sciences of the United States of America. 108, 4882-4887 (2011).
- Tsai, S., Bartelmez, S., Sitnicka, E., Collins, S. Lymphohematopoietic progenitors immortalized by a retroviral vector harboring a dominant-negative retinoic acid receptor can recapitulate lymphoid, myeloid, and erythroid development. Genes Dev. 8, 2831-2841 (1994).
- Weiler, S. R., et al. D3: a gene induced during myeloid cell differentiation of Linlo c-Kit+ Sca-1(+) progenitor cells. Blood. 93, 527-536 (1999).
- Ye, Z. J., Kluger, Y., Lian, Z., Weissman, S. M. Two types of precursor cells in a multipotential hematopoietic cell line. Proc Natl Acad Sci U S A. 102, 18461-18466 (2005).
- Wang, Z., Gerstein, M., Snyder, M. RNA-Seq: a revolutionary tool for transcriptomics. Nature reviews. Genetics. 10, 57-63 (2009).
- Chu, Y., Corey, D. R. RNA sequencing: platform selection, experimental design, and data interpretation. Nucleic acid therapeutics. 22, 271-274 (2012).
- Hornett, E. A., Wheat, C. W. Quantitative RNA-Seq analysis in non-model species: assessing transcriptome assemblies as a scaffold and the utility of evolutionary divergent genomic reference species. BMC genomics. 13, 361 (2012).
- Eswaran, J., et al. RNA sequencing of cancer reveals novel splicing alterations. Scientific reports. 3, 1689 (2013).
- Wang, E. T., et al. Alternative isoform regulation in human tissue transcriptomes. Nature. 456, 470-476 (2008).
- Wu, J. Q., et al. Dynamic transcriptomes during neural differentiation of human embryonic stem cells revealed by short, long, and paired-end sequencing. Proceedings of the National Academy of Sciences of the United States of America. 107, 5254-5259 (2010).
- Loraine, A. E., McCormick, S., Estrada, A., Patel, K., Qin, P. RNA-seq of Arabidopsis pollen uncovers novel transcription and alternative splicing. Plant physiology. 162, 1092-1109 (2013).
- Edgren, H., et al. Identification of fusion genes in breast cancer by paired-end RNA-sequencing. Genome biology. 12, 6 (2011).
- Ilott, N. E., Ponting, C. P. Predicting long non-coding RNAs using RNA sequencing. Methods. 63, 50-59 (2013).
- Sun, L., et al. Prediction of novel long non-coding RNAs based on RNA-Seq data of mouse Klf1 knockout study. BMC bioinformatics. 13, 331 (2012).
- Luo, S. MicroRNA expression analysis using the Illumina microRNA-Seq Platform. Methods in molecular biology. 822, 183-188 (2012).
- Bolduc, F., Hoareau, C., St-Pierre, P., Perreault, J. P. In-depth sequencing of the siRNAs associated with peach latent mosaic viroid infection. BMC molecular biology. 11, 16 (2010).
- Chepelev, I., Wei, G., Tang, Q., Zhao, K. Detection of single nucleotide variations in expressed exons of the human genome using RNA-Seq. Nucleic acids research. 37, 106 (2009).
- Djari, A., et al. Gene-based single nucleotide polymorphism discovery in bovine muscle using next-generation transcriptomic sequencing. BMC genomics. 14, 307 (2013).
- Murphy, D. Gene expression studies using microarrays: principles, problems, and prospects. Advances in physiology education. 26, 256-270 (2002).
- Chen, K., et al. RNA-seq characterization of spinal cord injury transcriptome in acute/subacute phases: a resource for understanding the pathology at the systems level. PLoS one. 8, 72567 (2013).
- Marioni, J. C., Mason, C. E., Mane, S. M., Stephens, M., Gilad, Y. RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome research. 18, 1509-1517 (2008).
- Ramskold, D., Kavak, E., Sandberg, R. How to analyze gene expression using RNA-sequencing data. Methods in molecular biology. 802, 259-274 (2012).
- Glenn, T. C. Field guide to next-generation DNA sequencers. Mol Ecol Resour. 11, 759-769 (2011).
- Trapnell, C., et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature protocols. 7, 562-578 (2012).
- Trapnell, C., Pachter, L., Salzberg, S. L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 25, 1105-1111 (2009).
- Langmead, B., Trapnell, C., Pop, M., Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome biology. 10, 25 (2009).
- Trapnell, C., et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature. 28, 511-515 (2010).
- Anders, S., Huber, W. Differential expression analysis for sequence count data. Genome biology. 11, 106 (2010).
- Quinlan, A. R., Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 26, 841-842 (2010).
- Cheranova, D., et al. RNA-seq analysis of transcriptomes in thrombin-treated and control human pulmonary microvascular endothelial cells. J Vis Exp. , (2013).
- Zhang, H. M., et al. AnimalTFDB: a comprehensive animal transcription factor database. Nucleic acids research. 40, 144-149 (2012).
- Wu, J. Q., et al. Systematic analysis of transcribed loci in ENCODE regions using RACE sequencing reveals extensive transcription in the human genome. Genome Biol. 9, 3 (2008).
- Wu, J. Q., et al. Large-scale RT-PCR recovery of full-length cDNA clones. Biotechniques. 36, 690-696 (2004).
- Wu, J. Q., Shteynberg, D., Arumugam, M., Gibbs, R. A., Brent, M. R. Identification of rat genes by TWINSCAN gene prediction, RT-PCR, and direct sequencing. Genome Res. 14, 665-671 (2004).
- Dewey, C., et al. Accurate identification of novel human genes through simultaneous gene prediction in human, mouse, and rat. Genome Res. 14, 661-664 (2004).
- Wu, J. Characterize Mammalian Transcriptome Complexity. , LAP Lambert Academic Publishing. (2011).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}