

ERRATUM NOTICE
Important: There has been an erratum issued for this article. Read more …
Summary
Den klustrade regelbundet mellanliggande kort palindromic repetitioner/CRISPR associerade protein 9 (CRISPR/Cas9) systemet ger ett lovande verktyg för genteknik och öppnar upp möjligheten att riktade integration av transgener. Vi beskriver en homologi-medierad slutet att gå (HMEJ)-baserat strategi för effektiv DNA riktade integration i vivo och riktade genterapi med hjälp av CRISPR/Cas9.
Abstract
Som en lovande genomet redigering plattform har CRISPR/Cas9 systemet stor potential för effektiv genmanipulation, särskilt för riktade integration av transgener. På grund av den låga verkningsgraden för homolog rekombination (HR) och olika ditsatta mutationer av icke-homolog slutet att gå (NHEJ)-baserade strategier i icke-dela celler, i vivo genomet redigering fortfarande en stor utmaning. Här, vi beskriver en homologi-medierad slutet att gå (HMEJ)-baserade CRISPR/Cas9-system för effektiv i vivo exakt riktade integration. I detta system, riktade genomet och givaren vektor innehållande homologi armar (~ 800 bp) flankerad av samma guide RNA (sgRNA) mål sekvenser klyvs av CRISPR/Cas9. Denna HMEJ-baserade strategi uppnår effektiva transgenens integration i mus zygoter, liksom i hepatocyter i vivo. Dessutom en HMEJ-baserad strategi erbjuder en effektiv metod för korrigering av fumarylacetoacetathydrolas hydrolas (Fah) mutation i hepatocyterna och räddar Fah-brist inducerad leversvikt möss. Sammantaget med fokus på riktade integration, denna HMEJ-baserade strategi ger ett lovande verktyg för en mängd applikationer, inklusive generationen genmodifierade djurmodeller och riktade genterapi.
Introduction
Exakt, riktade gen editering krävs ofta för att producera genetiskt modifierade djurmodeller och kliniska behandlingar. Mycket arbete har gjorts för att utveckla olika strategier för effektiv riktad genomet redigering, såsom zink finger nukleotid (ZFN), transkription aktivator-liknande effektor nukleaser (TALENs), och CRISPR/Cas9-system. Dessa strategier skapa riktade DNA dubbel-strand raster (DSB) i genomet och utnyttja inneboende DNA reparation system, såsom homolog rekombination (HR)1,2, microhomology-medierad slutet att gå (MMEJ)3 , 4 , 5, och icke-homolog slutet att gå (NHEJ)6,7,8 för att inducera riktade integration av transgener1,9. HR-baserade strategin är för närvarande den vanligaste genomet redigering tillvägagångssätt som är mycket effektiv i cellinjer, men inte lättillgängliga för icke-dela celler på grund av dess begränsade förekomst i den sena S/G2-fasen. HR-baserade strategin gäller således inte för i vivo gen editering. Nyligen, den NHEJ-baserade strategin utvecklades för effektiv gen knock-in i musen vävnader8. Metoden NHEJ-baserade införs dock vanligtvis indels knytpunkter, vilket gör det svårt att generera exakt gen editering, särskilt när man försöker konstruera i-frame fusion gener8. MMEJ-baserade riktade integration kan exakt gen editering. Men ökar det bara blygsamt riktade integration effektiviteten i tidigare rapporter5. Förbättring av exakt riktade integration i vivo effektivitet behövs därför snarast för breda terapeutiska tillämpningar3.
I ett nyligen publicerat arbete, vi visat en homologi-medierad slutet att gå (HMEJ)-baserat strategi, som visade den högsta riktade integration effektiviteten i alla rapporterade strategier både in vitro- och in-vivo10. Här beskriver vi ett protokoll för etableringen av det HMEJ systemet, och även byggandet av singel-guide RNA (sgRNA) vektorerna inriktning genen av intresse och givaren vektorer hysande sgRNA inriktade på områden och ~ 800 bp homologi vapen (figur 1) . I detta protokoll beskriver vi också de detaljerade anvisningarna för generering av DNA inpressning möss och korta steg för riktade integration i vävnader i vivo. Dessutom visat en proof-of-concept studie av HMEJ-baserade strategi sin förmåga att korrigera Fah mutation och rädda Fah- / - leversjukdom misslyckande möss, vilket ytterligare avslöjade dess terapeutiska potential.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
Alla förfaranden inklusive animaliska ämnen har godkänts av den biomedicinska forskningsetisk kommitté på Shanghai institut för biologisk vetenskap (CAS).
1. design av givare plasmider
-
Urval av sgRNA
- Använda online CRISPR design-verktyg för att förutsäga sgRNAs på mål regionen11,12,13,14,15. För det Cdx2 locus, designa sex olika sgRNAs (Cdx2-sgRNA1 ~Cdx2-sgRNA6) runt stoppet codon med högre rang och lägre potential off-mål (figur 1A)16.
- Linjär 2 µg av Cas9-CMV-andra uttryck vektorer och sgRNA av BbsI matsmältning (1 µL av BbsI för 2 h vid 37 ° C med en slutlig koncentration på 1 U/µL i en volym av 20 µL). Sedan rena produkten genom gel rening kit med en 1% agarosgel i 1 × TAE buffert.
- Blanda ett par sgRNA oligonukleotider i 10 µL 1 × T4 DNA ligase buffert till en slutlig koncentration på 50 µM. Inkubera oligo lösningen med en temperaturgradient från 95 ° C till 25 ° C med en temperatur förändring av 5 ° C 5 min (95 ° C i 5 min sedan 90 ° C i 5 min, 85 ° C i 5 min, etc.), som kommer att glödga oligos.
- Blanda 4 µL glödgas produkt, 2 µL av linearized vektorn med 1 µL T4 DNA-ligase i 10 µL 1 × T4 DNA-ligase buffert, och sedan ligera vid 22 ° C i 1-2 h (figur 1B).
-
Surveyor nuclease haltbestämning av sgRNA
Obs: Den sgRNA som används för inpressning experimentet inriktning effektivitet utvärderas av surveyor nuclease assay (även känd som T7 Amiiiiin jag (T7EI) assay)17. Välj sgRNA med hög DNA klyvning effektivitet och låg avstånd mellan sgRNA skärande webbplats och den stop kodon.- Transfect Cas9-sgRNA-andra uttryck vektorer in N2a cellinjer odlade i DMEM kompletteras med 10% fetalt bovint serum, 1% PSG och 1% icke-essentiell aminosyra av transfection kit (se Tabell för material). Inkubera vid 37 ° C i 5% CO2transfekterade cellerna.
- Efter 48 h för inkubation, samla in 5 000 transfekterade celler (GFP+) av fluorescens-aktiverad cell sortering (FACS) med icke-transfekterade celler som en kontroll.
- Smälta de insamlade cellerna i 2-5 µL oflysis buffert (0,1% Triton x-100, 0,1% Tween 20 och 100 µg/mL proteinas K) på 56 ° C i 30 min och sedan värme inaktivera proteinas K vid 95 ° C i 10 min.
- Förstärka provet med nästlade PCR (tabell 1) med tillverkarens protokollet. Storleken på PCR-produkter är inställd på 300-500 bp.
- Blanda 1 µL av Lys produkt med DNA-polymeras och ett par yttre primers erkänner sekvensen runt målplatsen sgRNA (0.1 µM, slutlig koncentration) (tabell 1), och utföra den primära PCR i en volym av 20 µL.
- Aktivera DNA-polymeras vid 95 ° C i 5 min och utföra den primära PCR för 30 cykler vid 95 ° C under 30 s, 60 ° C under 30 s och 72 ° C under 24 s (1 min/1 kb), med en sista förlängning vid 72 ° C i 5 min.
- Utföra den sekundära PCR använder 1 µL av PCR primärprodukt och ett par av kapslade inre primers.
- Denaturera och åter glödga 300-600 ng av renat PCR-produkt i 20 µL av 1 × T7EI reaktion buffert (50 mM NaCl, 10 mM Tris-HCl, 10 mM MgCl2, 1 mM DTT pH 7,9) med en gradient av temperatur från 95 ° C till 25 ° C med en hastighet av 5 ° C 5 min.
- Lägg till 1 µL T7EI enzym i glödgad PCR-produkter och smälta vid 37 ° C i 2 h. Kör sedan matsmältningen produkten på 2% agarosgel i 1 × TAE buffert vid 120 V för 40 min tills fragmenten separeras (se Tabell för material).
- Använda ImageJ för att avgöra bandet stödnivåerna cut och uncut DNA. Beräkna den ditsatta frekvensen med metoder som tidigare rapporterats9 (figur 1C).
-
Byggandet av givare vektor
Obs: För att generera HMEJ givare vektorer för Cdx2 gen, konstruera en donator DNA (800 bp HAL-p2A-mCherry-800 bp HAR) flankerad med 23 nt Cdx2-sgRNAs inriktning sekvens i båda ändar (figur 1D och figur 1E). PAM mål sekvens var intill slutet av den homologa arm. Gibson montering rekommenderas för HMEJ givare kloning.- Förstärka 800 bp vänster homologi armen (HAL) med forward primer-1F (innehållande 15-20 nt överlappning sekvens från vector, 23 nt Cdx2-sgRNA inriktning sekvens, och cirka 20 nt sekvens från HAL) och reverse primer-1R (innehållande 15-20 bp överlappning sekvens från p2A-mCherry och cirka 20 nt sekvens från HAL) på 0,1 µM slutlig koncentration använda musen genomiskt DNA på 200 ng/µL (figur 1D, tabell 1).
- Förstärk p2A-mCherry införande fragmentet med forward primer-2F (innehållande 15-20 nt överlappning sekvens från HAL och ca 20 nt sekvens från insättningspunkten fragment) och reverse primer-2R (innehållande 15-20 nt överlappning sekvens från HAR och ca 20 nt sekvens från insättningspunkten fragmentet) på 0.1 µM slutlig koncentration med genomiskt DNA eller plasmid med reporter sekvenser på 100 ng/µL eller 30 ng/µL (figur 1D, tabell 1).
- Förstärka 800 bp rätt homologi armen (HAR) med forward primer-3F (innehållande 15-20 nt överlappning sekvens från vector, 23 nt Cdx2-sgRNA inriktning sekvens, och cirka 20 nt sekvens från HAR) och reverse primer-3R (innehållande 15-20 nt överlappning sekvens från p2A-mCherry och cirka 20 nt sekvens från HAR) på 0,1 µM slutlig koncentration använda musen genomiskt DNA på 200 ng/µL (figur 1D, tabell 1).
- Kör alla PCR-produkter på 1% agarosgel i 1 × TAE buffert och rena PCR-produkter av förväntad storlek genom gel utvinning kit, enligt tillverkarens instruktioner (tabell 1).
- Smälta 50-100 ng av en konstruktion vektor med KpnI och XbaI. Blanda 2 µL av linearized vektor på 30-40 ng/µL med tre PCR amplifieras fragment (1 µL för varje, 100-200 ng/µL) 2 x Gibson mix. Lägg till H2O justera den slutliga volymen till 10 µL.Incubate mixen vid 50 ° C i 60 min.
- Omvandla behöriga E. coli celler med alla sammansatta produkt och extrakt plasmiden konstruktioner av DNA extraktion kit enligt tillverkarens anvisningar. Verifiera HMEJ givaren genom DNA-sekvensering.
2. gen editering i musembryon som använder metoden HMEJ-baserade
-
Produktion av Cas9 mRNA
- För Cas9 mRNA förberedelse, lägga till T7 promotorn sekvensen till Cas9 kodande regionen av PCR-amplifiering med lämplig primer paret anges i tabell 1. Lägg till primern Cas9 F/R på en slutlig koncentration på 0,1 µM och 20 ng av Cas9 uttrycker vektor till 1 × HiFi DNA-polymeras mix. Anpassa den slutliga volymen till 50 µL med H2O.
- Aktivera DNA-polymeras vid 95 ° C i 5 min och utför PCR för 36 cykler vid 95 ° C under 30 s, 60 ° C under 30 s och 68 ° C i 4 min (1 min/1 kb), med en sista förlängning vid 68 ° C i 10 min.
- Rena T7-Cas9 PCR-produkt för in vitro- transkription (IVT) och sedan transkribera 0,5-1 µg DNA av mRNA transcription kit vid 37 ° C i 8 h i en total volym på 20 µL, enligt tillverkarens anvisningar (se Tabell för material).
- Tillsätt 1 µL DNAS i blandningen för att ta bort mallen DNA vid 37 ° C i 15 min. Lägg en poly-A svans för 45 min vid 37 ° C och Återställ Cas9 mRNA av RNA rening kit, enligt tillverkarens anvisningar (se Tabell för material).
-
Produktion av sgRNA
- Skapa mallen sgRNA drivs av en T7 promotorn med HiFi DNA-polymeras som ovan. Välja en sgRNA byggnadsställning som innehåller vektor som mall. Primers används listas i tabell 1.
- Rena T7-sgRNA PCR-produkten och använder 0,5-1 µg DNA som mall för in vitro- transkription av sgRNA med en kort RNA transkriptionen kit vid 37 ° C för 6 h i en total volym på 20 µL, enligt tillverkarens anvisningar (se tabellen av material < / c11 >).
- Tillsätt 1 µL av DNAS till blandningen och fortsätta inkubationen vid 37 ° C i 15 min att ta bort mallen DNA. Rena sgRNAs av RNA rening kit, som ovan (se Tabell för material).
- Späd sgRNA till 500 ng/µL i RNase-gratis vatten och lagra proverna vid −80 ° C i upp till 3 månader.
Obs: CRISPR ribonucleoproteins (RNPs) är en alternativ ersättning med en bättre skära effektivitet18,19,20.
-
Embryo insamling, Mikroskop och in vitro- kultur
- Superovulate B6D2F1 (C57BL/6 × DBA2J) honmöss (7-8 veckor gamla) av gravida mare serum gonadotropin (PMSG), följt av humant koriongonadotropin (hCG) 48 h senare. Efter hCG-injektionen, hus honor med B6D2F1 hanar över natten.
- Offra honorna av CO2 anestesi, 24 h efter hCG-injektionen. Samla de befruktade embryona från deras oviducts (med 30-50 embryon för varje kvinna) i M2 medium.
- Plats de befruktade embryona (ca 300 ägg en dag injektionsvätska) till KSOM medium (5,55 g/L NaCl, 0,19 g/L KCl, 0,05 g/L KH2PO4, 0,05 g/L MgSO4•7H2O, 0.04 g/L glukos, 1,12 g/L natrium laktat, 2,1 g/L NaHCO3 , 0,02 g/L natrium pyruvat, 0,25 g/L CaCl2•2H2O, 0,004 HB EDTA, 0.146 HB L-glutamin och 1 g/L bovint serum albumin) vid 37 ° C i en inkubator med 5% CO2.
- Mix-Cas9 mRNA (100 ng/µL), sgRNA (50 ng/µL), HMEJ givare vektor (100 ng/µL) och lägga till H2O justera den slutliga volymen till 10 µL. Lägg blandningen på is.
- Dra kapillär nålar (yttre diameter 1,0 mm, innerdiameter 0.78 mm med glödtråd) med hjälp av en mikropipett avdragare (parametrar: värme, hastighet, 80; 74, tidsfördröjning, 200; dra, 60; tryck, 300. Se tabell material). Kommersiella nålar skulle vara en alternativ ersättning för Mikroskop.
- Injicera en trolig volymen av blandningen i cytoplasman i zygoter med väldefinierade pronuclei i en droplet HEPES-CZB medium innehållande 5 µg/mL cytochalasin B med en microinjector med konstant flöde inställningar (figur 2A) (se En tabell av)21.
Obs: Varje grupp av zygoter ska injiceras inom 20-30 min. Cytochalasin B kunde öka lönsamheten för mus zygot efter injektionen. Alternativt, Mikroskop kan drivas med piezo-systemet, som tidigare beskrivits22. - Kultur de injicerade zygoter i KSOM medium vid 37 ° C under 5% CO2 tills blastocystan stage efter 3,5 dagar för fluorescens observation (siffror 2B och 2C).
-
Embryotransfer och generering av möss
- Mate estrous ICR honmöss med vasektomerade ICR hanmöss samma dag som injektion.
- Kultur de injicerade zygoter in i 2-cellstadie vid 37 ° C under 5% CO2, och överföring 25-30 2-cells embryon till oviducts av pseudopregnant ICR honor vid 0,5 dag post intravenösa (dpc). Mottagarens mödrar leverera valpar på 19,5 dpc.
-
Mus genotypning
- Extrahera mus genomiskt DNA från tå eller svans prover med ett DNA extraktion kit, enligt tillverkarens anvisningar (se Tabell för material).
- Identifiera 5' och 3' korsningen av inpressning händelser med 200-400 ng av genomisk DNA mätt med UV/vis spektrometri som en mall för att utföra en PCR-förstärkning.
- Aktivera DNA-polymeras vid 95 ° C i 5 min och utför PCR för 38 cykler vid 95 ° C under 30 s, 60 ° C under 30 s och 72 ° C i 1 min (1 min/1 kb), med en sista förlängning vid 72 ° C i 10 min. För 5' korsningen, Använd framåt primer på uppströms av HAL, med den omvända för inpressning fragmentet (p2A-mCherry). Om 3' junction, Använd framåt primer på inpressning fragmentet (p2A-mCherry), med den omvända för nedströms av HAR (tabell 1).
- Kör 6 µL av PCR-produkten på 1% agarosgel i 1 × TAE buffert och kontrollera för beräknade fragment storlek. Kontrollera dem genom DNA sekvensering (figur 2D).
3. HMEJ-baserade In Vivo genomet redigering i hepatocyter
- Placera mottagaren C57BL/6J mus (8 veckor) i en återhållande enhet och sätta svansen genom skåran.
- Blanda HMEJ givare vektorer (30 µg) och spCas9 uttryck vektorer (30 µg) i 2 mL koksaltlösning. För kontroll experimentet, upphäva HMEJ givare vektorer (30 µg) i 2 mL koksaltlösning (figur 3A).
- Rengöra musen svansen med 70% etanol. Infoga nålen in svansen ven och injicera plasmid DNA lösningen inom 5-7 s. ut nålen och släpp musen från besöksförbud enheten.
- Offra möss av CO2 anestesi efter 5-9 dagar efter injektion. BEGJUTA möss transcardially med 0,9% koksaltlösning, följt av 4% PARAFORMALDEHYD använder en Peristaltisk pump och åtgärda levern under natten vid 4 ° C.
- Torkar ut vävnaden med 30% sackaros över natten, tills det sjunker till botten av röret.
- Avsnitt frysta vävnaden med en tjocklek på 10 µm för lever prover.
- Skölj i avsnitt tre gånger i 0,1 M fosfatbuffrad (PB) och inkubera dem med primär antikropp: kanin anti-mCherry (utspätt i 5% NGS) över natten vid 4 ° C.
- Tvätta sektioner tre gånger i PB, och sedan ruva dem med sekundär antikropp: Cy3-AffiniPure get anti-kanin IgG för 2 h i rumstemperatur i orbitalskak.
- Motfärg avsnitten med DAPI i 20 min och montera med glycerin på glasskivor för ytterligare fluorescens observation (figur 3B).
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
HMEJ-baserade gen editering i musembryon: För att definiera inpressning effektiviteten i metoden HMEJ-baserat i mus zygoter, levererat vi Cas9 mRNA, sgRNA inriktning Cdx2 genen och HMEJ givaren in i musen zygoter, som var avsedd att smälta en p2A-mCherry reporter gen till den sista kodon av Cdx2 gen (figur 2A). De injicerade zygoter utvecklats till blastocyster i kulturen. Utvärdera effektiviteten inpressning, analyserade vi de mCherry fluorescensen med fluorescerande Mikroskop, och vi fann att 12,9% av de blastocyster emot HMEJ givare var positivt för mCherry, som var strikt uttryckt i trophectoderm (siffror 2B 2 C). Genom sekvensering av PCR-positiva möss, fann vi också att alla undersökta integration händelser var exakt i-frame integrationer i både 5' och 3' korsningar (figur 2D).
HMEJ-baserade gen editering i vuxna vävnader och HMEJ-medierad genterapi: För att undersöka huruvida HMEJ-baserade gen editering kunde tillämpas i vuxna vävnader, infogade vi mCherry kassett precis innan den stop kodon av Actb gen av transducing Actb- HMEJ konstruktioner till C57/B6J mus lever av svans-ven hydrodynamiska injektion (figur 3A). Efter 7 dagar av injektioner fann vi att nästan hälften av de transfekterade hepatocyterna uttryckta mCherry målat på levern delar (figur 3B).
För att undersöka möjligheten att använda en HMEJ-baserad strategi för genterapi, vi anställt fumarylacetoacetathydrolas hydrolas (Fah)-bristfällig möss. Fah- / - musen är en väletablerad hereditär tyrosinemi typ I (HTI) musmodell, som hamnar ett införande fragment i exon 5 av Fah genen orsakar frameshift mutationer i den följande sekvens23. För att bibehålla Fah- / - möss, behandlat vi Fah- / - möss med en hämmare av uppströms tyrosin katabola utbildningsavsnitt, 2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione (NTBC)24. Här har vi satt för att se huruvida MMEJ - och HMEJ-medierad gen korrigering kunde rädda Fah mutation i Fah- / - mus. Vi injiceras hydrodynamiskt Cas9 konstruera tillsammans med Fah- MMEJ eller Fah- HMEJ konstruktioner, utformad för att infoga Fah cDNA av exon 5 till 14 i intron 4 av Fah gen, Fah - / - mus () lever Figur 3 C). en vecka efter injektionen, NTBC drogs för att inducera leverskada (figur 3C). Efter återkallande av NTBC, Fah-korrigerade hepatocyter Fah- / - mössens mottagande Fah- HMEJ och Cas9 konstruktioner visade effektivare spridning än MMEJ-baserade metod (figur 3D ).

Figur 1 : HMEJ-medierad riktade integration i vitro.
(A) experimentella system för urval av sgRNAs: sex olika sgRNAs (Cdx2-sgRNA1 ~Cdx2-sgRNA6) runt den stop kodon av den Cdx2 locus med högre rang och off-target potential var väljs baserat på online CRISPR-design verktyg. Protospacer angränsande motiv (PAM) sekvensen är i rött. (B) experimentell design: The Cas9-CMV-GFP uttryck plasmider uttrycker sgRNA, Cas9 och god Jordbrukarsed infördes i N2a celler. GFP+ celler sorterades på dag 3 för surveyor assay. (C) Surveyor assay för Cdx2 inriktning: 6 olika sgRNAs utformades för surveyor assay. Normal N2a cell genomiskt DNA fungerar som kontroll. *, den sgRNA som används för Cdx2-2A-mCherry inpressning experiment. (D) Schematisk översikt av byggandet av HMEJ givare med Gibson församling. (E) Schematisk översikt över HMEJ-medierad genmodifiering strategi på Cdx2 locus. HAL/HAR, vänster/höger homologi arm; trianglar, sgRNA mål platser; AV / OR, yttre framåt/bakåt primer; Om / IR, inre framåt/bakåt primer. Figur modifierad från föregående rapport10. Klicka här för att se en större version av denna siffra.

Figur 2 : Gen editering i musembryon via HMEJ-medierad riktade integration
(A) experimentella system av Mikroskop: en blandning av Cas9 mRNA (100 ng/µL), sgRNA (50 ng/µL), och givaren plasmider (100 ng/µL) skulle injiceras i mus zygoter. (B) representativa fluorescens bilder av musembryon som redigeras av HMEJ strategi. Bar, 20 µm. (C) inpressning effektivitet anges av andelen mCherry+ blastocyster. Numret ovanför varje stapel, totala blastocyster räknas. (D) sekvens analys av gen-redigeras möss vid Cdx2 locus. PCR-produkter som amplifierats från 5' och 3' junction platser var sekvenserade. Övre, homologi arm; lila, p2A; röd, mCherry; HAR eller HAL, höger eller vänster homologa arm. Streckade linjer markerar regionen utelämnas för tydlighet. Figur modifierad från föregående rapport10. Klicka här för att se en större version av denna siffra.

Figur 3 : HMEJ-medierad riktade integration i vivo.
(A) Schematisk översikt av hydrodynamisk svans ven injektion. En blandning av plasmider uttrycker givare sekvens och sgRNA och plasmider uttrycker spCas9 levererades till levern via hydrodynamiska svans ven injektion. (B) representativa immunofluorescens bilder av hepatocyter. Avsnitten levern var insamlade 7 dagar efter injektionen. Skalstapeln, 50 µm. GFP, transfekterade celler. (C) plasmider antingen MMEJ - eller HMEJ-medierad gen ersättare strategi avsedd att infoga Fah cDNA av exon 5 till 14 i intron 4 Fah gen levererades till Fah- / - mus lever genom hydrodynamisk injektion. NTBC på: Fah- / - möss bibehölls på NTBC vatten; NTBC off: tillbakadragande av NTBC vatten (den första dagen av NTBC uttag definierades som dag 0, som är den 7: e dagen efter injektionen). (D) Fah immunohistokemi färgning av levern sektioner från Fah- / - möss som injicerats med MMEJ eller HMEJ plasmider. Skalstapeln, 100 µm. figur modifierad från föregående rapporter5,10. Klicka här för att se en större version av denna siffra.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
De mest kritiska steg i byggandet av HMEJ givare plasmider är: (1) val av sgRNA med hög DNA klyvning effektivitet och låga avståndet mellan sgRNA skärande webbplats och stop kodon, och (2) korrekt uppförande av HMEJ givare. CRISPR/Cas9-medierad klyvning på båda transgenens givare vektor (innehållande sgRNA inriktade på områden och ~ 800 bp homologi armar) och riktade genomet är nödvändiga för effektiv och exakt riktade integration invivo. De mest kritiska steg av generation av inpressning möss med HMEJ-baserade metoden är: (1) utarbetandet av högkvalitativa Cas9 mRNA och sgRNA (inga degeneration finns i Cas9 mRNA och sgRNA) och (2) utarbetandet av högkvalitativa HMEJ givare Plasmiden. Plasmiden visar inga toxiska effekter på fosterutvecklingen.
Nyligen, en NHEJ-baserad metod hade har också rapporterats för effektiv i vivo genomet redigering8. Olika typer av ditsatta mutationer förmåddes emellertid vanligtvis knytpunkter, enligt beskrivningen i tidigare rapporter8, vilket gör det svårt att uppnå exakt vilka integrationsåtgärder. Här, visade i HMEJ-baserade strategi som vi beskrivit ovan exakt riktade integration med knappast någon ditsatta mutationer. Således kunde en HMEJ-baserad strategi vara en idealisk plattform för att ersätta en muterad sekvens (till exempel en punktmutation) med den rätta, som inte är tillämplig för NHEJ-baserad metod.
Mosaicism är ett stort problem för genen redigering i embryon. Injektion av Cas9 protein istället för mRNA vid ett tidigare embryonala stadium kan uppnå transgenens inpressning en cellstadie utan mosaicism. För kliniska tillämpningar är leverans av de CRISPR/Cas9-system till vuxna vävnader fortfarande utmanande.
I området i närheten finns det många framtida potentiella användningsområden för HMEJ-baserade gen editering. Det kan användas för att generera genetiskt modifierade djurmodeller. Med tanke på dess inpressning högeffektiv i embryon, denna metod kan avsevärt minska antalet djur behövs för att skapa genetiskt modifierade djurmodeller, och öppnar särskilt upp möjligheten att generera icke-mänskliga primater genetiska modeller. HMEJ-baserade gen editering kan härstamning spåra enskilda celltyper i vuxna vävnader, vilket är särskilt användbart för djurmodeller, eftersom det finns en brist på tillgängliga djurmodeller, såsom icke-mänskliga primater. Det kan användas för riktade genterapi: mest attraktiva tillämpningen av en HMEJ-baserad strategi är genterapi för kliniken använder. I denna studie vi korrigerat Fah mutationen av hereditär tyrosinemi typ jag möss genom hydrodynamisk injektion indikerade vektorer. Leverans av CRISPR/Cas9 systemet till vuxna vävnader är dock fortfarande en stor teknisk utmaning för klinisk användning, eftersom hydrodynamiska injektion sannolikt utföras hos patienter. För närvarande ytterligare behövs förbättring av leverans strategi omgående innan att översätta denna HMEJ-baserade metod till kliniken.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Författarna har något att avslöja.
Acknowledgments
Detta arbete stöds av CAS strategiska prioriterade Research Program (XDB02050007, XDA01010409), den nationella Hightech R & D Program (863 Program; 2015AA020307), den nationella naturvetenskap Foundation i Kina (NSFC beviljar 31522037, 31500825, 31571509, 31522038), Kina Youth tusen talanger Program (att HY), Break genom projektet av kinesiska vetenskapsakademin, projektet Shanghai City kommittén för vetenskap och teknik (16JC1420202 till HY), ministeriet för vetenskap och teknik i Kina (de flesta; 2016YFA0100500).
Materials
| Name | Company | Catalog Number | Comments |
| pX330 | Addgene | 42230 | |
| pAAV vector | Addgene | 37083 | |
| pX260 | Addgene | 42229 | |
| AAV_Efs_hSpCas9_NLS_FLAG-SV40 | Addgene | 97307 | AAV vector for encoding a human codon-optimized SpCas9 driven by EFs promoter |
| AAV_Actb HMEJ donor_U6_sgRNA_EF1a_GFP_polyA | Addgene | 97308 | HMEJ donor for fusing a p2A-mCherry reporter to mouse Actb. EGFP driven by EF1a promoter and U6-driven sgRNAs targeting Actb. AAV backbone. |
| AAV_Cdx2 HMEJ donor | Addgene | 97319 | HMEJ donor for fusing a p2A-mCherry reporter to mouse Cdx2. |
| Lipofectamine 3000 Transfection Reagent | Life Technology | L3000015 | |
| Nuclease-Free Water | Life Technologies | AM9930 | |
| Bbs I | New England Biolabs | R0539S | |
| NEB Buffer 2 | New England Biolabs | B7002S | |
| T7 endonuclease I | New England Biolabs | M0302L | |
| NEBuilder HiFi DNA Assembly Master Mix | New England Biolabs | E2621L | |
| Plasmid EndoFree-Midi Kit | Qiagen | 12143 | |
| MMESSAGE MMACHINE T7 ULTRA | Life Technologies | AM1345 | |
| MEGACLEAR KIT 20 RXNS | Life Technologies | AM1908 | |
| MEGASHORTSCRIPT T7 KIT 25 RXNS | Life Technologies | AM1354 | |
| Flaming/Brown Micropipette Puller | Sutter Instrument | P-97 | Micropipette Puller (parameters: heat, 74; pull, 60; velocity, 80; time/delay, 200; pressure, 300) |
| Borosilicate glass | Sutter Instrument | B100-78-10 | type of capillaries (outer diameter 1.0 mm, inner diameter 0.78 mm with filament) |
| FemtoJet microinjector | Eppendorf | ||
| Freezing microtome | Leica | CM1950-Cryostat | thickness of 40 μm for brain, 10 μm for liver |
| Rabbit anti-mCherry | GeneTex | ||
| Cy3-AffiniPure Goat Anti-Rabbit IgG | Jackson Immunoresearch | ||
| DMEM | Gibco | 11965092 | |
| FBS | Gibco | 10099141 | |
| NEAA | Gibco | 11140050 | |
| Pen,Strep,Glutamine | Gibco | 10378016 | |
| Gel Extraction Kit | Omega | D2500-02 | |
| FACS | BD AriaII | ||
| PMSG | Ningbo Sansheng Medicine | S141004 | |
| HCG | Ningbo Sansheng Medicine | B141002 | |
| Cytochalasin B | Sigma | CAT#C6762 | |
| KSOM+AA with D-Glucose and Phenol Red | Millipore | CAT#MR-106-D | |
| M2 Medium with Phenol Red | Millipore | CAT#MR-015-D | |
| Mineral oil | Sigma |
References
- Yang, H., et al. Generation of Mice Carrying Reporter and Conditional Alleles by CRISPR/Cas-Mediated Genome Engineering. Cell. 154 (6), 1370-1379 (2013).
- Hockemeyer, D., et al. Genetic engineering of human pluripotent cells using TALE nucleases. Nature Biotechnology. 29 (8), 731-734 (2011).
- Nakade, S., et al. Microhomology-mediated end-joining-dependent integration of donor DNA in cells and animals using TALENs and CRISPR/Cas9. Nature Communications. 5, 5560 (2014).
- Hisano, Y., et al. Precise in-frame integration of exogenous DNA mediated by CRISPR/Cas9 system in zebrafish. Scientific reports. 5, 8841 (2015).
- Yao, X., et al. Cas9 - Mediated Precise Targeted Integration In Vivo Using a Double Cut Donor with Short Homology Arms. EBioMedicine. , (2017).
- Auer, T. O., Duroure, K., De Cian, A., Concordet, J. P., Del Bene, F. Highly efficient CRISPR/Cas9-mediated knock-in in zebrafish by homology-independent DNA repair. Genome research. 24 (1), 142-153 (2014).
- Maresca, M., Lin, V. G., Guo, N., Yang, Y. Obligate ligation-gated recombination (ObLiGaRe): custom-designed nuclease-mediated targeted integration through nonhomologous end joining. Genome Research. 23 (3), 539-546 (2013).
- Suzuki, K., et al. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature. 540 (7631), 144-149 (2016).
- Cong, L., et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 339 (6121), 819-823 (2013).
- Yao, X., et al. Homology-mediated end joining-based targeted integration using CRISPR/Cas9. Cell Research. 27 (6), 801-814 (2017).
- Han, D. W., et al. Direct reprogramming of fibroblasts into epiblast stem cells. Nature Cell Biology. 13 (1), 66-71 (2011).
- Han, D. W., et al. Direct Reprogramming of Fibroblasts into Neural Stem Cells by Defined Factors. Cell Stem Cell. , (2012).
- Ambasudhan, R., et al. Direct reprogramming of adult human fibroblasts to functional neurons under defined conditions. Cell Stem Cell. 9 (2), 113-118 (2011).
- Sparman, M., et al. Epigenetic reprogramming by somatic cell nuclear transfer in primates. Stem Cells. 27 (6), 1255-1264 (2009).
- Schatten, G., Mitalipov, S. Developmental biology: Transgenic primate offspring. Nature. 459 (7246), 515-516 (2009).
- Hsu, P. D., et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nature Biotechnology. 31 (9), 827-832 (2013).
- Cong, L., et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science. 339 (6121), 819-823 (2013).
- Quadros, R. M., et al. Easi-CRISPR: a robust method for one-step generation of mice carrying conditional and insertion alleles using long ssDNA donors and CRISPR ribonucleoproteins. Genome Biology. 18 (1), 92 (2017).
- Park, K. E., et al. Targeted Gene Knockin in Porcine Somatic Cells Using CRISPR/Cas Ribonucleoproteins. International journal of molecular sciences. 217 (6), (2016).
- Woo, J. W., et al. DNA-free genome editing in plants with preassembled CRISPR-Cas9 ribonucleoproteins. Nature biotechnology. 33 (11), 1162-1164 (2015).
- Harms, D. W., et al. Mouse Genome Editing Using the CRISPR/Cas System. Current protocols in human genetics. 83, 11-27 (2014).
- Yang, H., Wang, H., Jaenisch, R. Generating genetically modified mice using CRISPR/Cas-mediated genome engineering. Nature protocols. 9 (8), 1956-1968 (2014).
- Grompe, M., et al. Loss of Fumarylacetoacetate Hydrolase Is Responsible for the Neonatal Hepatic-Dysfunction Phenotype of Lethal Albino Mice. Genes & development. 7 (12), 2298-2307 (1993).
- Paulk, N. K., et al. Adeno-associated virus gene repair corrects a mouse model of hereditary tyrosinemia in vivo. Hepatology. 51 (4), 1200-1208 (2010).
Tags
Genetik fråga 133 CRISPR/Cas9 riktade Integration homologi-medierad slutet att gå In Vivo Embryo modifierade genetiskt möss hydrodynamisk injektionErratum
Formal Correction: Erratum: CRISPR/Cas9-mediated Targeted Integration In Vivo Using a Homology-mediated End Joining-based Strategy
Posted by JoVE Editors on 03/10/2021.
Citeable Link.
An erratum was issued for: Studying TGF-β Signaling and TGF-β-induced Epithelial-to-mesenchymal Transition in Breast Cancer and Normal Cells. The phrases "surveyor assay" and "Surveyor Nuclease" have been updated to "T7E1 assay" to " T7 endonuclease I" respectively.
Step 1.2 in the Protocol has been updated from:
- Surveyor nuclease assay of sgRNA
NOTE: The targeting efficiency of the sgRNA used for the knock-in experiment is evaluated by surveyor nuclease assay (also known as T7 endonuclease I (T7EI) assay)17. Select the sgRNA with high DNA cleavage efficiency and a low distance between the sgRNA cutting site and the stop codon.
to:
- T7 endonuclease assay of sgRNA
NOTE: The targeting efficiency of the sgRNA used for the knock-in experiment is evaluated by T7 endonuclease (T7EI) assay17. Select the sgRNA with high DNA cleavage efficiency and a low distance between the sgRNA cutting site and the stop codon.
Figure 1 in the Representative Results has been updated from:
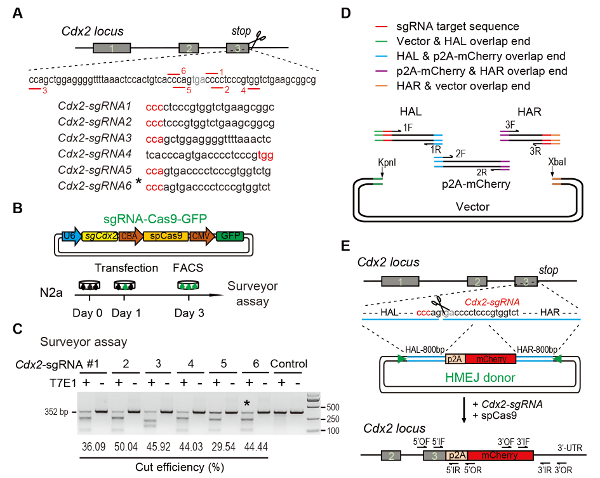
Figure 1: HMEJ-mediated targeted integration in vitro.
(A) Experimental scheme for selection of sgRNAs: Six different sgRNAs (Cdx2-sgRNA1~Cdx2-sgRNA6) around the stop codon of the Cdx2 locus with a higher rank and off-target potential were chosen based on online CRISPR design tool. The protospacer adjacent motif (PAM) sequence is in red. (B) Experimental design: The Cas9-CMV-GFP expression plasmids expressing sgRNA, Cas9, and GFP were introduced into N2a cells. GFP+ cells were sorted at day 3 for surveyor assay. (C) Surveyor assay for Cdx2 targeting: 6 different sgRNAs were designed for surveyor assay. Normal N2a cell genomic DNA serves as control. *, the sgRNA used for Cdx2-2A-mCherry knock-in experiment. (D) Schematic overview of construction of HMEJ donors using Gibson assembly. (E) Schematic overview of HMEJ-mediated gene targeting strategy at Cdx2 locus. HAL/HAR, left/right homology arm; triangles, sgRNA target sites; OF/OR, outer forward/reverse primer; IF/IR, inner forward/reverse primer. Figure modified from previous report10. Please click here to view a larger version of this figure.
to:
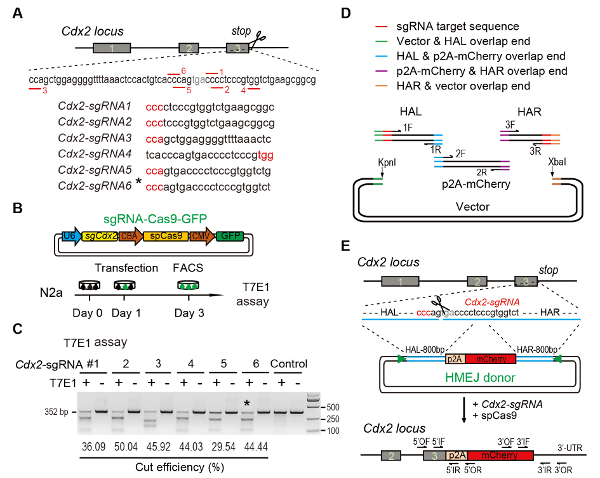
Figure 1: HMEJ-mediated targeted integration in vitro.
(A) Experimental scheme for selection of sgRNAs: Six different sgRNAs (Cdx2-sgRNA1~Cdx2-sgRNA6) around the stop codon of the Cdx2 locus with a higher rank and off-target potential were chosen based on online CRISPR design tool. The protospacer adjacent motif (PAM) sequence is in red. (B) Experimental design: The Cas9-CMV-GFP expression plasmids expressing sgRNA, Cas9, and GFP were introduced into N2a cells. GFP+ cells were sorted at day 3 for T7EI assay. (C) T7EI assay for Cdx2 targeting: 6 different sgRNAs were designed for T7EI assay. Normal N2a cell genomic DNA serves as control. *, the sgRNA used for Cdx2-2A-mCherry knock-in experiment. (D) Schematic overview of construction of HMEJ donors using Gibson assembly. (E) Schematic overview of HMEJ-mediated gene targeting strategy at Cdx2 locus. HAL/HAR, left/right homology arm; triangles, sgRNA target sites; OF/OR, outer forward/reverse primer; IF/IR, inner forward/reverse primer. Figure modified from previous report10. Please click here to view a larger version of this figure.
