

ERRATUM NOTICE
Important: There has been an erratum issued for this article. Read more …
Summary
De grupperede regelmæssigt interspaced korte palindromiske gentager/CRISPR forbundet protein 9 (CRISPR/Cas9) system giver et lovende redskab for genteknologi, og åbner op for muligheden for målrettede integration af transgener. Vi beskriver en homologi-medieret ende sammenføjning (HMEJ)-baseret strategi for effektiv DNA målrettet integration i vivo og målrettet genterapier bruger CRISPR/Cas9.
Abstract
Som en lovende genom redigering platform har CRISPR/Cas9-systemet et stort potentiale for effektiv genetisk manipulation, især for målrettede integration af transgener. Men på grund af den lave effektivitet af homologe rekombination (HR) og forskellige indel mutationer af ikke-homologe ende sammenføjning (NHEJ)-baserede strategier i ikke-dividere celler, i vivo genom-redigering er fortsat en stor udfordring. Her, vi beskriver en homologi-medieret ende sammenføjning (HMEJ)-baseret CRISPR/Cas9 system til effektiv i vivo præcist målrettede integration. I dette system, de målrettede genom og donor vektor indeholdende homologi arme (~ 800 bp) flankeret af enkelt guide RNA (sgRNA) mål sekvenser er kløvet af CRISPR/Cas9. Denne HMEJ-baseret strategi opnår effektiv transgen integration i mus zygotes samt i hepatocytter i vivo. Desuden en HMEJ-baseret strategi tilbyder en effektiv tilgang til korrektion af fumarylacetoacetate hydrolase (Fah) mutation i hepatocytter og redder Fah-vitaminmangel induceret leversvigt mus. Taget sammen, med fokus på målrettet integration, denne HMEJ-baseret strategi giver et lovende redskab for en bred vifte af applikationer, herunder generation af genetisk modificerede dyremodeller og målrettede genterapier.
Introduction
Præcise, målrettede genom-redigering er ofte påkrævet for at producere genetisk modificerede dyremodeller og kliniske behandlinger. Stor indsats er gjort at udvikle forskellige strategier for effektiv målrettede genom-redigering, såsom zink nukleasen (ZFN), transkription aktivere-lignende effektor nukleaser (TALENs), og CRISPR/Cas9 systemer. Disse strategier skabe målrettede DNA dobbelt-strenget pauser (DSB) i genomet, og drage fordel af iboende DNA reparation systemer, såsom homologe rekombination (HR)1,2, microhomology-medieret ende sammenføjning (MMEJ)3 , 4 , 5, og ikke-homologe ende sammenføjning (NHEJ)6,7,8 for at fremkalde målrettede integration af transgener1,9. Den HR-baseret strategi er i øjeblikket mest almindeligt anvendte genom-redigering tilgang, som er meget effektiv i cellelinjer, men ikke let tilgængelige for ikke-dividere celler på grund af dens begrænsede forekomst i den sene S/G2 fase. Således, den HR-baseret strategi er ikke gældende for i vivo genom-redigering. For nylig, den NHEJ-baseret strategi er udviklet til effektiv gen banke i i mus væv8. Ikke desto mindre, den NHEJ-baseret metode normalt introducerer indels på vejkryds, hvilket gør det vanskeligt at generere præcis genom-redigering, især når de forsøger at konstruere i-frame fusion gener8. MMEJ-baserede målrettede integration er i stand til præcis genom-redigering. Det øger dog kun beskedent målrettede integration effektiviteten i tidligere rapporter5. Derfor, effektivisering af præcist målrettede integration i vivo er bydende nødvendigt for bred terapeutisk anvendelse3.
I en nyligt offentliggjort arbejde, vi viste en homologi-medieret ende sammenføjning (HMEJ)-baseret strategi, som viste den højeste målrettede integration effektivitet i alle rapporterede strategier både in vitro- og i vivo10. Her, vi beskriver en protokol om oprettelse af HMEJ systemet, og også opførelsen af single-guide RNA (sgRNA) vektorer målretning gen af interesse og donor vektorer husly sgRNA målwebsteder og ~ 800 bp homologi våben (figur 1) . I denne protokol beskriver vi også de detaljerede trin til generering af DNA knock-i mus og korte trin for målrettede integration i væv i vivo. Desuden viste en proof of concept undersøgelse af HMEJ-baseret strategien sin evne til at korrigere Fah mutation og redde Fah- / - leveren manglende mus, som yderligere afslørede dets terapeutiske potentiale.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
Alle procedurer, herunder animalske emner er blevet godkendt af den biomedicinske forskning etiske komité på Shanghai institutter for biologisk videnskab (CAS).
1. design af Donor plasmider
-
Valg af sgRNA
- Brug online CRISPR design værktøj til at forudsige sgRNAs på den indskyde region11,12,13,14,15. Til Cdx2 locus, designe seks forskellige sgRNAs (Cdx2-sgRNA1 ~Cdx2-sgRNA6) omkring en stop codon med højere rang og lavere potentiale ud-mål (fig. 1A)16.
- Linearize 2 µg for Cas9-CMV-EGFP udtryk vektorer og sgRNA af BbsI fordøjelse (1 µL af BbsI i 2 timer ved 37 ° C i en endelig koncentration på 1 U/µL i et volumen på 20 µL). Så rense produktet af gel rensning kit med en 1%-agarosegel i 1 × TAE buffer.
- Mix et par af sgRNA oligonukleotider i 10 µL af 1 × T4 DNA ligase buffer til en slutkoncentration på 50 µM. Inkuber oligo løsning ved hjælp af en temperaturgradient fra 95 ° C til 25 ° C med en temperatur ændring af 5 ° C/5 min (95 ° C i 5 min derefter 90 ° C i 5 min, 85 ° C i 5 min, osv.), som vil anneal oligos.
- Mix 4 µL af udglødet produkt, 2 µL af den lineariseret vektor med 1 µL af T4 DNA ligase i 10 µL af 1 × T4 DNA ligase buffer, og derefter ligate på 22 ° C i 1-2 h (figur 1B).
-
Landinspektør nukleasen analyse af sgRNA
Bemærk: Målretning effektiviteten af sgRNA anvendes til knock-i eksperimentet er evalueret af landinspektør nukleasen assay (også kendt som T7 liv1975 jeg (T7EI) assay)17. Vælg sgRNA med høj DNA kavalergang effektivitet og en lav afstand mellem sgRNA skæring websted og stop-codon.- Transfect Cas9-sgRNA-EGFP udtryk vektorer i N2a cellelinjer kulturperler i DMEM suppleret med 10% føtal bovint serum, 1% PSG, og 1% ikke-essentiel aminosyre af Transfektion kit (Se Tabel af materialer). Der inkuberes ved 37 ° C i 5% CO2transfected cellerne.
- Efter 48 h inkubation, indsamle 5.000 transfekteret celler (normal god landbrugspraksis+) ved fluorescens-aktiveret celle sortering (FACS) ved hjælp af ikke-transfekteret celler som kontrol.
- Fordøje de indsamlede celler i 2-5 µL oflysis buffer (0,1% Triton X-100, 0,1% Tween 20 og 100 µg/mL Proteinase K) ved 56 ° C i 30 min, og derefter varme inaktivere proteinase K ved 95 ° C i 10 min.
- Forstærke stikprøve af indlejrede PCR (tabel 1) ved hjælp af producentens protokol. Størrelsen af PCR produkter er indstillet til 300-500 bp.
- Mix 1 µL af lysis produkt med DNA polymerase og et par af ydre primere anerkende sekvens omkring sgRNA mål-webstedet (0,1 µM, endelige koncentration) (tabel 1), og udføre den primære PCR i et volumen på 20 µL.
- Aktivere DNA polymerase ved 95 ° C i 5 min, og udføre den primære PCR for 30 cykler ved 95 ° C til 30 s, 60 ° C i 30 s og 72 ° C i 24 s (1 min/1 kb), med en sidste udvidelse ved 72 ° C i 5 min.
- Udføre den sekundære PCR ved hjælp af 1 µL af primære PCR produkt og et par af indlejrede indre primere.
- Denaturere og re anneal 300-600 ng af renset PCR produkt i 20 µL 1 × T7EI reaktion buffer (50 mM NaCl, 10 mM Tris-HCl, 10 mM MgCl2, 1 mM DTT pH 7,9) ved hjælp af en gradient i temperatur fra 95 ° C til 25 ° C med en hastighed af 5 ° C/5 min.
- Tilføje 1 µL af T7EI enzym til udglødet PCR produkter og fordøje ved 37 ° C i 2 timer. Derefter køre fordøjelsen produkt på 2%-agarosegel i 1 × TAE buffer på 120 V i 40 min. indtil fragmenter adskilles (Se Tabel af materialer).
- Bruge ImageJ til at bestemme band intensiteterne af snit og uncut DNA. Beregne indel frekvens ved hjælp af metoder som tidligere rapporteret9 (figur 1C).
-
Opførelsen af donor vektor
Bemærk: For at generere HMEJ donor vektorer for Cdx2 gen, konstruere en donor DNA (800 bp HAL-p2A-mCherry-800 bp HAR) flankeret med 23 nt Cdx2-sgRNAs rettet mod sekvens i begge ender (fig. 1D og figur 1E). PAM af target sekvens var støder op til slutningen af den homologe arm. Gibson montage anbefales for HMEJ donor kloning.- Forstærke de 800 bp venstre homologi arm (HAL) med fremad primer-1F (der indeholder 15-20 nt overlapning sekvens fra vektor, 23 nt Cdx2-sgRNA målretning sekvens, og omkring 20 nt sekvens fra HAL) og vende primer-1R (der indeholder 15-20 bp overlapning sekvens fra p2A-mCherry og ca. 20 nt sekvens fra HAL) på 0,1 µM endelige koncentration ved hjælp af musen genomisk DNA på 200 ng/µL (figur 1D, tabel 1).
- Forstærke p2A-mCherry indsættelse fragment med fremad primer-2F (indeholdende 15-20 nt overlapning sekvens fra HAL og ca. 20 nt sekvens fra indsættelse fragment) og vende primer-2R (der indeholder 15-20 nt overlapning sekvens fra HAR og ca. 20 nt sekvens fra indsættelse fragment) på 0,1 µM endelige koncentration ved hjælp af genomisk DNA eller plasmid med reporter sekvenser på 100 ng/µL eller 30 ng/µL (figur 1D, tabel 1).
- Forstærke de 800 bp rigtige homologi arm (HAR) med fremad primer-3F (der indeholder 15-20 nt overlapning sekvens fra vektor, 23 nt Cdx2-sgRNA målretning sekvens, og omkring 20 nt sekvens fra HAR) og vende primer-3R (der indeholder 15-20 nt overlapning sekvens fra p2A-mCherry og ca. 20 nt sekvens fra HAR) på 0,1 µM endelige koncentration ved hjælp af musen genomisk DNA på 200 ng/µL (figur 1D, tabel 1).
- Køre alle PCR produkter på 1%-agarosegel i 1 × TAE buffer, og rense PCR-produkter af forventet størrelse af gel udvinding kit, ifølge producentens anvisninger (tabel 1).
- Fordøje 50-100 ng af en konstruktion vektor med KpnI og XbaI. Bland 2 µL af den lineariseret vektor på 30-40 ng/µL med tre PCR forstærket fragmenter (1 µL for hvert, 100-200 ng/µL) i 2 x Gibson mix. Tilføj H2O for at justere det endelige rumfang til 10 µL.Incubate mix ved 50 ° C til 60 min.
- Omdanne kompetente E. coli celler med alle den samlede vare og uddrag plasmidet konstruktioner af DNA udvinding kit ifølge producentens anvisninger. Kontroller HMEJ donor af DNA-sekventering.
2. genom-redigering i mus embryoner ved hjælp af metoden HMEJ-baserede
-
Produktion af Cas9 mRNA
- Cas9 mRNA forberedelse, tilføje T7 promotor sekvens til Cas9 kodende region af PCR-amplifikation ved hjælp af passende primer parret i tabel 1anførte. Tilføje primer Cas9 F/R på en endelig koncentration på 0,1 µM og 20 ng af Cas9 udtrykker vektor til 1 × high fidelity DNA polymerase mix. Juster det endelige rumfang på 50 µL med H2O.
- Aktivere DNA polymerase ved 95 ° C i 5 min, og udføre PCR for 36 cyklusser ved 95 ° C til 30 s, 60 ° C i 30 s og 68 ° C i 4 min (1 min/1 kb) med filtypenavnet endelige på 68 ° C i 10 min.
- Rense T7-Cas9 PCR produkt til in vitro- transskription (IVT), og derefter transskribere 0,5-1 µg DNA af mRNA transskription kit ved 37 ° C i 8 timer i en samlet maengde paa 20 µL, ifølge producentens anvisninger (Se Tabel af materialer).
- Tilføje 1 µL af DNase til blandingen for at fjerne skabelonen DNA ved 37 ° C i 15 min. Tilføj en poly-en hale i 45 min. ved 37 ° C og genoprette Cas9 mRNA af RNA oprensning kit, ifølge producentens anvisninger (Se Tabel af materialer).
-
Produktion af sgRNA
- Generere skabelonen sgRNA drevet af en T7 promotor med high fidelity DNA polymerase som ovenfor. Vælg en sgRNA stillads indeholdende vektor som skabelon. Primere bruges er angivet i tabel 1.
- Rense T7-sgRNA PCR produktet og bruge 0,5-1 µg DNA som skabelon for in vitro- transskription af sgRNA ved hjælp af et kort RNA transskription kit ved 37 ° C i 6 timer i en samlet maengde paa 20 µL, ifølge producentens anvisninger (Se tabel af materialer < / c11 >).
- Tilsæt 1 µL af DNase til blandingen og fortsætte inkubationen ved 37 ° C i 15 min at fjerne skabelonen DNA. Rense sgRNAs af RNA oprensning kit, som ovenfor (Se Tabel af materialer).
- Fortynd sgRNA til 500 ng/µL i RNase-fri vand og opbevare prøver på −80 ° C i op til 3 måneder.
Bemærk: CRISPR ribonucleoproteins (RNPs) er en alternativ substitution med en bedre skære effektivitet18,19,20.
-
Embryo indsamling, mikroinjektion og in vitro- kultur
- Superovulate B6D2F1 (C57BL/6 × DBA2J) hunmus (7-8 uger gamle) af gravide mare serum gonadotropin (PMSG), efterfulgt af humant choriongonadotropin (hCG) 48 timer senere. Efter hCG injektion, hus kvinder med B6D2F1 mænd natten over.
- Ofre hunnerne af CO2 anæstesi, 24 h efter hCG injektion. Indsamle de befrugtede embryoner fra deres æggelederne (med 30-50 embryoner for hver kvinde) i M2 medium.
- Sted de befrugtede embryoner (ca. 300 æg til endags-injektion) til KSOM medie (5,55 g/L NaCl, 0,19 g/L KCl, 0.05 g/L KH2PO4, 0.05 g/L MgSO4•7H2O, 0,04 g/L glukose, 1,12 g/L Sodium lactat, 2,1 g/L NaHCO3 , 0,02 g/L natrium pyruvat, 0,25 g/L CaCl2•2H2O, 0.004 g/L EDTA, 0.146 g/L L-glutamin og 1 g/L bovint serumalbumin) ved 37 ° C i en inkubator med 5% CO2.
- Mix Cas9 mRNA (100 ng/µL), sgRNA (50 ng/µL) og HMEJ donor, vektor (100 ng/µL), og tilføje H2O til at justere den endelige mængden til 10 µL. Sæt blandingen på køl.
- Trække kapillær nåle (ydre diameter 1.0 mm, indvendig diameter 0,78 mm med glødetråd) ved hjælp af en mikropipette Puller (parametre: varme, 74, pull, 60, hastighed, 80; tidsforsinkelse, 200, pres, 300. Se tabel over materialer). Kommercielle nåle ville være en alternativ substitution for mikroinjektion.
- Indsprøjte en sandsynlig volumen af blandingen i cytoplasma af zygotes med veldefinerede pronuclei i en dråbe af HEPES-CZB medium indeholdende 5 µg/mL cytochalasin B ved hjælp af et microinjector med konstant flow indstillinger (figur 2A) (Se Tabel af materialer)21.
Bemærk: Hver gruppe af zygotes bør sprøjtes inden for 20-30 min. Cytochalasin B kunne øge rentabiliteten af musen zygote efter injektion. Alternativt, mikroinjektion kan betjenes med piezo-systemet, som tidligere beskrevet22. - Kultur de injiceres zygotes i KSOM medium ved 37 ° C under 5% CO2 indtil blastocyst fase efter 3,5 dage til fluorescens observation (tal 2B og 2C).
-
Ægoplægning og generation af mus
- Mate duft ICR hunmus med vasectomized ICR mandlige mus på samme dag som injektion.
- Kultur de injiceres zygotes i de 2-celle stadium ved 37 ° C under 5% CO2og overførsel 25-30 2-celle embryoner i æggelederne på pseudopregnant ICR hunner på 0,5 dag post coitum (dpc). Modtagerens mødre levere unger på 19,5 dpc.
-
Musen genotypebestemmelse
- Uddrag mus genomisk DNA fra tå eller hale prøver ved hjælp af en DNA-ekstraktion kit, ifølge producentens anvisninger (Se Tabel af materialer).
- Identificere 5' og 3' krydset af knock-i events med 200-400 ng af genomisk DNA målt ved UV/vis SPEKTROMETRI som en skabelon til at udføre PCR amplification.
- Aktivere DNA polymerase ved 95 ° C i 5 min, og udføre PCR for 38 cyklusser ved 95 ° C til 30 s, 60 ° C i 30 s og 72 ° C i 1 min (1 min/1 kb), med en sidste udvidelse ved 72 ° C i 10 min. For 5' krydset, skal du bruge den fremskudte primer på upstream af HAL, med den omvendt på knock-i-fragment (p2A-mCherry). Om 3' junction, skal du bruge den fremad primer på knock-i-fragment (p2A-mCherry), hos den omvendte sig på downstream i HAR (tabel 1).
- Køre 6 µL PCR produkt på 1%-agarosegel i 1 × TAE buffer og kontrollere, om den forventede fragment størrelse. Derefter kontrollere dem ved DNA sekventering (figur 2D).
3. HMEJ-baserede In Vivo genom-redigering i hepatocytter
- Placere modtageren C57BL/6J mus (8 uger) i en fastholdende enhed og læg halen gennem spalten.
- Bland HMEJ donor vektorer (30 µg) og spCas9 udtryk vektorer (30 µg) i 2 mL af saltopløsning. For kontrol-eksperiment, at indstille HMEJ donor vektorer (30 µg) 2 mL af saltopløsning (fig. 3A).
- Ren mus hale med 70% ethanol. Indsætte nålen ind i halen vene og injicere plasmid DNA løsning inden for 5-7 s. fjerne nålen og slip musen fra fastholdende enheden.
- Ofre mus af CO2 anæstesi efter 5-9 dage efter injektion. Perfuse mus transcardially med 0,9% saltvand, efterfulgt af 4% PARAFORMALDEHYD ved hjælp af en peristaltisk pumpe og lave leveren natten over ved 4 ° C.
- Dehydrere væv med 30% saccharose natten over, indtil det synker til bunden af røret.
- Afsnit den frosne væv på en tykkelse på 10 µm til leveren prøver.
- Skyl sektioner tre gange i 0,1 M fosfatbufferet (PB) og inkuberes dem med primær antistof: kanin anti-mCherry (fortyndet i 5% NGS) natten over ved 4 ° C.
- Vaske sektioner tre gange i PB, og derefter inkuberes dem med sekundær antistof: Cy3-AffiniPure ged anti-kanin IgG i 2 timer ved stuetemperatur på en orbitalryster.
- Kontrastfarve sektioner med DAPI i 20 min. og mount med glycerin på glas dias for yderligere fluorescens observation (fig. 3B).
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
HMEJ-baseret genom-redigering i mus embryoner: Hvis du vil definere knock-i effektiviteten af HMEJ-baseret metode i mus zygotes, leverede vi Cas9 mRNA, sgRNA rettet mod Cdx2 -gen og HMEJ donor til musen zygotes, som var designet til at lunte en p2A-mCherry reporter gen til den sidste codon af Cdx2 gen (figur 2A). De injicerede zygotes udviklet sig til blastocyster i kulturen. For at evaluere overvågningsaktiviteternes effektivitet med knock-i, vi analyseret mCherry fluorescens med et fluorescerende mikroskop, og vi fandt at 12,9% af blastocyster modtager HMEJ donorer var positiv for mCherry, som var strengt udtrykt i trophectoderm (tal 2B 2 C). Ved sekventering af PCR positive musene, fandt vi også, at alle undersøgte integration begivenheder var præcis i-frame integrationer i både 5' og 3' vejkryds (figur 2D).
HMEJ-baseret genom-redigering i voksent væv og HMEJ-medieret genterapi: For at undersøge, om HMEJ-baseret genom-redigering kan anvendes i voksent væv, indsat vi mCherry kassetten lige før stop-codon af Actb gen af transducing Actb- HMEJ konstruktioner til C57/B6J mus lever af hale-vene Hydrodynamisk injektion (fig. 3A). Efter 7 dage af injektioner fandt vi, at næsten halvdelen af de transfected hepatocytter udtrykt mCherry, som farves på afsnittene leveren (fig. 3B).
Hvis du vil udforske muligheden af at anvende en HMEJ-baseret strategi for genterapi, vi ansat fumarylacetoacetate hydrolase (Fah)-mangelfuld mus. Fah- / - musen er en veletableret arvelige tyrosinemia skriver jeg (HTI) musemodel, som havne en indsættelse fragment i exon 5 af Fah -genet, forårsager frameshift mutationer i den følgende sekvens23. For at opretholde Fah- / - mus, behandlede vi Fah- / - mus med en hæmmer af upstream af tyrosin kataboliske pathway, 2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione (NTBC)24. Her satte vi sig for at se om MMEJ - og HMEJ-medieret gen korrektion kunne redde Fah mutation i Fah- / - musen. Vi hydrodynamically injiceres Cas9 konstruktion med Fah- MMEJ eller Fah- HMEJ konstruktioner, designet til at indsætte Fah cDNA af exon 5 til 14 i intron 4 af Fah gen, at Fah- / - mus () lever Figur 3 C). en uge efter injektion, NTBC blev trukket tilbage til at fremkalde leverskader (figur 3C). Efter tilbagetrækningen af NTBC, Fah-korrigerede hepatocytter Fah- / - musenes modtager Fah- HMEJ og Cas9 konstruktioner viste mere effektiv spredning end MMEJ-baseret metode (figur 3D ).

Figur 1 : HMEJ-medieret målrettet integration i vitro.
(A) eksperimentelle ordning for valg af sgRNAs: seks forskellige sgRNAs (Cdx2-sgRNA1 ~Cdx2-sgRNA6) omkring stop-codon af Cdx2 locus med en højere rang og off-target potentiale blev valgt baseret på online CRISPR design værktøj. Protospacer tilstødende motiv (PAM) sekvens er i rødt. (B) eksperimentelle design: The Cas9-CMV-NGL udtryk plasmider udtrykker sgRNA, Cas9 og normal god landbrugspraksis var indført i N2a celler. Normal god landbrugspraksis+ celler blev sorteret på dag 3 for landmåler assay. (C) landmåler assay for målretning af Cdx2 : 6 forskellige sgRNAs var designet til landinspektør assay. Normal N2a celle genomisk DNA fungerer som kontrol. *, sgRNA anvendes til Cdx2-2A-mCherry knock-i eksperimentet. (D) skematisk oversigt over opbygningen af HMEJ donorer med Gibson assemblyen. (E) skematisk oversigt over HMEJ-medieret gen målretning strategi på Cdx2 locus. HAL/HAR, venstre/højre homologi arm; trekanter, sgRNA målwebsteder; AF / OR, ydre frem/tilbage primer; Hvis / IR, indre frem/tilbage primer. Figur ændres fra forrige rapport10. Venligst klik her for at se en større version af dette tal.

Figur 2 : Genom-redigering i mus embryoner via HMEJ-medieret målrettet integration
(A) eksperimentelle ordning af mikroinjektion: en blanding af Cas9 mRNA (100 ng/µL), sgRNA (50 ng/µL) og donor plasmider (100 ng/µL) blev sprøjtet ind i mus zygotes. (B) repræsentative fluorescens billeder af musen embryoner redigeret af HMEJ strategi. Bar, 20 µm. (C) Knock-i effektivitet angivet ved procent af mCherry+ blastocyster. Tal over hver søjle, samlede blastocyster regnes. (D) sekvens analyse af gen-redigeret mus i Cdx2 locus. PCR produkter amplificeret fra 5' og 3' krydset steder blev sekventeret. Øvre, homologi arm; lilla, p2A; rød, mCherry; HAR eller HAL, højre eller venstre homologe arm. Stiplede linjer mark regionen udeladt for klarhed. Figur ændres fra forrige rapport10. Venligst klik her for at se en større version af dette tal.

Figur 3 : HMEJ-medieret målrettet integration i vivo.
(A) skematisk oversigt over hydrodynamiske hale vene injektion. En blanding af plasmider udtrykker donor sekvens og sgRNA og plasmider at udtrykke spCas9 blev leveret til leveren via hydrodynamiske hale vene injektion. (B) repræsentative immunofluorescens billeder af hepatocytter. Afsnittene leveren var indsamlet 7 dage post injektion. Skalalinjen, 50 µm. normal god landbrugspraksis, transfekteret celler. (C) plasmider af enten MMEJ - eller HMEJ-medieret gen udskiftning strategi designet til at indsætte Fah cDNA af exon 5 til 14 i intron 4 af Fah gen blev leveret i Fah- / - mus lever af hydrodynamiske injektion. NTBC på: Fah- / - mus blev opretholdt på NTBC vand; NTBC off: tilbagetrækning af NTBC vand (den første dag i NTBC tilbagetrækning blev defineret som dag 0, som er den 7 dag efter indsprøjtning). (D) Fah Immunhistokemi farvning af leveren sektioner fra Fah- / - musene injiceret med MMEJ eller HMEJ plasmider. Skalalinjen, 100 µm. figur ændres fra tidligere rapporter5,10. Venligst klik her for at se en større version af dette tal.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
De mest afgørende skridt i opbygningen af HMEJ donor plasmider er: (1) valg af sgRNA med høj DNA kavalergang effektivitet og lave afstand mellem sgRNA skæring websted og stop codon, og (2) en korrekt opførelse af HMEJ donor. CRISPR/Cas9-medieret kavalergang på begge transgen donor vektor (indeholdende sgRNA målwebsteder og ~ 800 bp homologi våben) og målrettede genom er nødvendige for effektivt og præcist målrettede integration in vivo. De mest kritiske trin i generation af knock-i mus ved hjælp af HMEJ-baseret metode er: (1) udarbejdelse af høj kvalitet af Cas9 mRNA og sgRNA (ingen degeneration findes i Cas9 mRNA og sgRNA), og (2) udarbejdelse af høj kvalitet HMEJ donor plasmid. Plasmidet viser ingen toksiske effekter på embryonale udvikling.
For nylig, var en NHEJ-baseret metode også rapporteret for effektiv i vivo genom-redigering8. Ikke desto mindre, forskellige typer af indel mutationer var normalt induceret i vejkryds, som beskrevet i tidligere rapporter8, hvilket gør det vanskeligt at opnå præcise integration. Her, viste den HMEJ-baseret strategi vi beskrevet ovenfor præcist målrettede integration med næsten ingen indel mutationer. Således, en HMEJ-baseret strategi kunne være en ideel platform til at erstatte en muteret sekvens (f.eks. et punkt mutation) med den korrekte, hvilket ikke gælder for NHEJ-baseret metode.
Mosaicisme er et stort problem for gen redigering i embryoner. Injektion af Cas9 protein i stedet for mRNA i en tidligere embryonale fase kan opnå transgen banke i på én celle stadium uden mosaicisme. Til kliniske applikationer er levering af CRISPR/Cas9-systemer i voksent væv stadig udfordrende.
Der er mange fremtidige potentielle anvendelser af HMEJ-baseret genom-redigering. Det kan bruges til at generere genetisk modificerede dyremodeller. I betragtning af dens høje banke-i effektivitet i embryoner, denne metode kunne reducere dyrs antallet behov for at skabe genetisk modificerede dyremodeller, og åbner især mulighed for at generere primat genetiske modeller. HMEJ-baseret genom-redigering kan afstamning sporingen enkelte celletyper i voksent væv, som er særligt nyttigt for dyremodeller, da der er mangel på tilgængelige dyremodeller, såsom ikke-menneskelige primater. Det kan anvendes til målrettede genterapier: den mest attraktive anvendelsen af en HMEJ-baseret strategi er genterapi for klinikken bruger. I denne undersøgelse, vi korrigeret Fah mutation af arvelige tyrosinemia type jeg mus ved hydrodynamiske injektion af de angivne vektorer. Levering af CRISPR/Cas9-systemet i voksent væv er dog stadig den største tekniske udfordring til klinisk brug, som hydrodynamiske injektion er usandsynligt, at være udført i patienter. I øjeblikket, er yderligere forbedring af levering strategi bydende nødvendigt før omsætte denne HMEJ-baseret metode til klinikken.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Forfatterne har ikke noget at oplyse.
Acknowledgments
Dette arbejde blev støttet af CAS strategiske prioritet Research Program (XDB02050007, XDA01010409), den nationale Hightech R & D Program (863 Program; 2015AA020307), National Natural Science Foundation of China (NSFC tilskud 31522037, 31500825, 31571509, 31522038), Kina ungdom tusinde talenter Program (til HY), bryde gennem projektet af kinesiske Academy of Sciences, projekt Shanghai City Udvalget for videnskab og teknologi (16JC1420202 til HY), Ministeriet for videnskab og teknologi i Kina (de fleste; 2016YFA0100500).
Materials
| Name | Company | Catalog Number | Comments |
| pX330 | Addgene | 42230 | |
| pAAV vector | Addgene | 37083 | |
| pX260 | Addgene | 42229 | |
| AAV_Efs_hSpCas9_NLS_FLAG-SV40 | Addgene | 97307 | AAV vector for encoding a human codon-optimized SpCas9 driven by EFs promoter |
| AAV_Actb HMEJ donor_U6_sgRNA_EF1a_GFP_polyA | Addgene | 97308 | HMEJ donor for fusing a p2A-mCherry reporter to mouse Actb. EGFP driven by EF1a promoter and U6-driven sgRNAs targeting Actb. AAV backbone. |
| AAV_Cdx2 HMEJ donor | Addgene | 97319 | HMEJ donor for fusing a p2A-mCherry reporter to mouse Cdx2. |
| Lipofectamine 3000 Transfection Reagent | Life Technology | L3000015 | |
| Nuclease-Free Water | Life Technologies | AM9930 | |
| Bbs I | New England Biolabs | R0539S | |
| NEB Buffer 2 | New England Biolabs | B7002S | |
| T7 endonuclease I | New England Biolabs | M0302L | |
| NEBuilder HiFi DNA Assembly Master Mix | New England Biolabs | E2621L | |
| Plasmid EndoFree-Midi Kit | Qiagen | 12143 | |
| MMESSAGE MMACHINE T7 ULTRA | Life Technologies | AM1345 | |
| MEGACLEAR KIT 20 RXNS | Life Technologies | AM1908 | |
| MEGASHORTSCRIPT T7 KIT 25 RXNS | Life Technologies | AM1354 | |
| Flaming/Brown Micropipette Puller | Sutter Instrument | P-97 | Micropipette Puller (parameters: heat, 74; pull, 60; velocity, 80; time/delay, 200; pressure, 300) |
| Borosilicate glass | Sutter Instrument | B100-78-10 | type of capillaries (outer diameter 1.0 mm, inner diameter 0.78 mm with filament) |
| FemtoJet microinjector | Eppendorf | ||
| Freezing microtome | Leica | CM1950-Cryostat | thickness of 40 μm for brain, 10 μm for liver |
| Rabbit anti-mCherry | GeneTex | ||
| Cy3-AffiniPure Goat Anti-Rabbit IgG | Jackson Immunoresearch | ||
| DMEM | Gibco | 11965092 | |
| FBS | Gibco | 10099141 | |
| NEAA | Gibco | 11140050 | |
| Pen,Strep,Glutamine | Gibco | 10378016 | |
| Gel Extraction Kit | Omega | D2500-02 | |
| FACS | BD AriaII | ||
| PMSG | Ningbo Sansheng Medicine | S141004 | |
| HCG | Ningbo Sansheng Medicine | B141002 | |
| Cytochalasin B | Sigma | CAT#C6762 | |
| KSOM+AA with D-Glucose and Phenol Red | Millipore | CAT#MR-106-D | |
| M2 Medium with Phenol Red | Millipore | CAT#MR-015-D | |
| Mineral oil | Sigma |
References
- Yang, H., et al. Generation of Mice Carrying Reporter and Conditional Alleles by CRISPR/Cas-Mediated Genome Engineering. Cell. 154 (6), 1370-1379 (2013).
- Hockemeyer, D., et al. Genetic engineering of human pluripotent cells using TALE nucleases. Nature Biotechnology. 29 (8), 731-734 (2011).
- Nakade, S., et al. Microhomology-mediated end-joining-dependent integration of donor DNA in cells and animals using TALENs and CRISPR/Cas9. Nature Communications. 5, 5560 (2014).
- Hisano, Y., et al. Precise in-frame integration of exogenous DNA mediated by CRISPR/Cas9 system in zebrafish. Scientific reports. 5, 8841 (2015).
- Yao, X., et al. Cas9 - Mediated Precise Targeted Integration In Vivo Using a Double Cut Donor with Short Homology Arms. EBioMedicine. , (2017).
- Auer, T. O., Duroure, K., De Cian, A., Concordet, J. P., Del Bene, F. Highly efficient CRISPR/Cas9-mediated knock-in in zebrafish by homology-independent DNA repair. Genome research. 24 (1), 142-153 (2014).
- Maresca, M., Lin, V. G., Guo, N., Yang, Y. Obligate ligation-gated recombination (ObLiGaRe): custom-designed nuclease-mediated targeted integration through nonhomologous end joining. Genome Research. 23 (3), 539-546 (2013).
- Suzuki, K., et al. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature. 540 (7631), 144-149 (2016).
- Cong, L., et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 339 (6121), 819-823 (2013).
- Yao, X., et al. Homology-mediated end joining-based targeted integration using CRISPR/Cas9. Cell Research. 27 (6), 801-814 (2017).
- Han, D. W., et al. Direct reprogramming of fibroblasts into epiblast stem cells. Nature Cell Biology. 13 (1), 66-71 (2011).
- Han, D. W., et al. Direct Reprogramming of Fibroblasts into Neural Stem Cells by Defined Factors. Cell Stem Cell. , (2012).
- Ambasudhan, R., et al. Direct reprogramming of adult human fibroblasts to functional neurons under defined conditions. Cell Stem Cell. 9 (2), 113-118 (2011).
- Sparman, M., et al. Epigenetic reprogramming by somatic cell nuclear transfer in primates. Stem Cells. 27 (6), 1255-1264 (2009).
- Schatten, G., Mitalipov, S. Developmental biology: Transgenic primate offspring. Nature. 459 (7246), 515-516 (2009).
- Hsu, P. D., et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nature Biotechnology. 31 (9), 827-832 (2013).
- Cong, L., et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science. 339 (6121), 819-823 (2013).
- Quadros, R. M., et al. Easi-CRISPR: a robust method for one-step generation of mice carrying conditional and insertion alleles using long ssDNA donors and CRISPR ribonucleoproteins. Genome Biology. 18 (1), 92 (2017).
- Park, K. E., et al. Targeted Gene Knockin in Porcine Somatic Cells Using CRISPR/Cas Ribonucleoproteins. International journal of molecular sciences. 217 (6), (2016).
- Woo, J. W., et al. DNA-free genome editing in plants with preassembled CRISPR-Cas9 ribonucleoproteins. Nature biotechnology. 33 (11), 1162-1164 (2015).
- Harms, D. W., et al. Mouse Genome Editing Using the CRISPR/Cas System. Current protocols in human genetics. 83, 11-27 (2014).
- Yang, H., Wang, H., Jaenisch, R. Generating genetically modified mice using CRISPR/Cas-mediated genome engineering. Nature protocols. 9 (8), 1956-1968 (2014).
- Grompe, M., et al. Loss of Fumarylacetoacetate Hydrolase Is Responsible for the Neonatal Hepatic-Dysfunction Phenotype of Lethal Albino Mice. Genes & development. 7 (12), 2298-2307 (1993).
- Paulk, N. K., et al. Adeno-associated virus gene repair corrects a mouse model of hereditary tyrosinemia in vivo. Hepatology. 51 (4), 1200-1208 (2010).
Tags
Genetik sag 133 CRISPR/Cas9 målrettet Integration homologi-medieret ende tiltræder In Vivo Embryo genmodificerede mus hydrodynamiske injektionErratum
Formal Correction: Erratum: CRISPR/Cas9-mediated Targeted Integration In Vivo Using a Homology-mediated End Joining-based Strategy
Posted by JoVE Editors on 03/10/2021.
Citeable Link.
An erratum was issued for: Studying TGF-β Signaling and TGF-β-induced Epithelial-to-mesenchymal Transition in Breast Cancer and Normal Cells. The phrases "surveyor assay" and "Surveyor Nuclease" have been updated to "T7E1 assay" to " T7 endonuclease I" respectively.
Step 1.2 in the Protocol has been updated from:
- Surveyor nuclease assay of sgRNA
NOTE: The targeting efficiency of the sgRNA used for the knock-in experiment is evaluated by surveyor nuclease assay (also known as T7 endonuclease I (T7EI) assay)17. Select the sgRNA with high DNA cleavage efficiency and a low distance between the sgRNA cutting site and the stop codon.
to:
- T7 endonuclease assay of sgRNA
NOTE: The targeting efficiency of the sgRNA used for the knock-in experiment is evaluated by T7 endonuclease (T7EI) assay17. Select the sgRNA with high DNA cleavage efficiency and a low distance between the sgRNA cutting site and the stop codon.
Figure 1 in the Representative Results has been updated from:
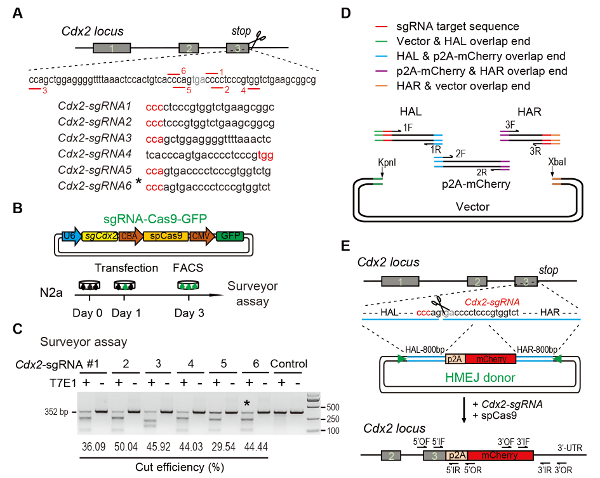
Figure 1: HMEJ-mediated targeted integration in vitro.
(A) Experimental scheme for selection of sgRNAs: Six different sgRNAs (Cdx2-sgRNA1~Cdx2-sgRNA6) around the stop codon of the Cdx2 locus with a higher rank and off-target potential were chosen based on online CRISPR design tool. The protospacer adjacent motif (PAM) sequence is in red. (B) Experimental design: The Cas9-CMV-GFP expression plasmids expressing sgRNA, Cas9, and GFP were introduced into N2a cells. GFP+ cells were sorted at day 3 for surveyor assay. (C) Surveyor assay for Cdx2 targeting: 6 different sgRNAs were designed for surveyor assay. Normal N2a cell genomic DNA serves as control. *, the sgRNA used for Cdx2-2A-mCherry knock-in experiment. (D) Schematic overview of construction of HMEJ donors using Gibson assembly. (E) Schematic overview of HMEJ-mediated gene targeting strategy at Cdx2 locus. HAL/HAR, left/right homology arm; triangles, sgRNA target sites; OF/OR, outer forward/reverse primer; IF/IR, inner forward/reverse primer. Figure modified from previous report10. Please click here to view a larger version of this figure.
to:
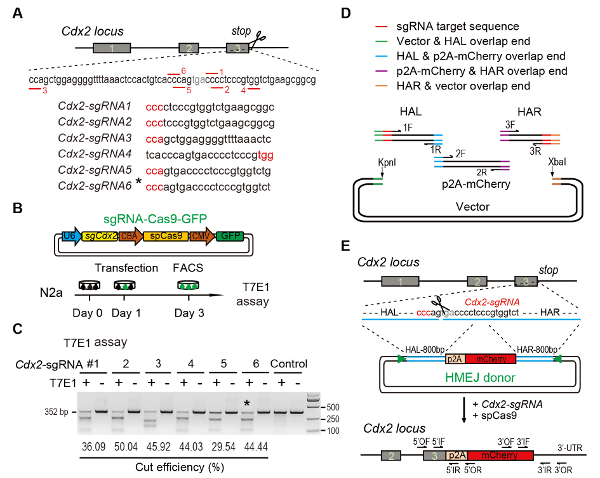
Figure 1: HMEJ-mediated targeted integration in vitro.
(A) Experimental scheme for selection of sgRNAs: Six different sgRNAs (Cdx2-sgRNA1~Cdx2-sgRNA6) around the stop codon of the Cdx2 locus with a higher rank and off-target potential were chosen based on online CRISPR design tool. The protospacer adjacent motif (PAM) sequence is in red. (B) Experimental design: The Cas9-CMV-GFP expression plasmids expressing sgRNA, Cas9, and GFP were introduced into N2a cells. GFP+ cells were sorted at day 3 for T7EI assay. (C) T7EI assay for Cdx2 targeting: 6 different sgRNAs were designed for T7EI assay. Normal N2a cell genomic DNA serves as control. *, the sgRNA used for Cdx2-2A-mCherry knock-in experiment. (D) Schematic overview of construction of HMEJ donors using Gibson assembly. (E) Schematic overview of HMEJ-mediated gene targeting strategy at Cdx2 locus. HAL/HAR, left/right homology arm; triangles, sgRNA target sites; OF/OR, outer forward/reverse primer; IF/IR, inner forward/reverse primer. Figure modified from previous report10. Please click here to view a larger version of this figure.
