

ERRATUM NOTICE
Important: There has been an erratum issued for this article. Read more …
Summary
Il cluster regolarmente intervallate proteine brevi ripetizioni/CRISPR palindromi associato 9 (CRISPR/Cas9) sistema fornisce uno strumento promettente per l'ingegneria genetica e apre la possibilità di integrazione mirata dei transgeni. Descriviamo un fine omologia-mediata entrare (HMEJ)-base di strategia per DNA efficiente mirata integrazione in vivo e mirate terapie geniche utilizzando CRISPR/Cas9.
Abstract
Come una promettente piattaforma di editing del genoma, il sistema CRISPR/Cas9 ha un grande potenziale per la manipolazione genetica efficiente, soprattutto per l'integrazione mirata dei transgeni. Tuttavia, a causa della scarsa efficienza di ricombinazione omologa (HR) e le varie mutazioni indel di non-omologo fine unirsi (NHEJ)-basato su strategie in cellule di divisione, in vivo genome pubblica rimane una grande sfida. Qui, descriviamo una fine omologia-mediata entrare (HMEJ)-CRISPR/Cas9 sistema efficiente in vivo precisa mirata integrazione basato. In questo sistema, il genoma mirato e il donatore vector contenente armi di omologia (~ 800 bp) fiancheggiato da destinazione di RNA (sgRNA) guida singola sequenze sono spaccate da CRISPR/Cas9. Questa strategia basata su HMEJ realizza integrazione efficiente transgene in zigoti del mouse, così come in epatociti in vivo. Inoltre, una strategia basata su HMEJ offre un approccio efficace per la correzione di fumarylacetoacetate idrolasi (Fah) mutazione negli epatociti e Salva Fah-deficit indotto da topi di insufficienza epatica. Presi insieme, concentrandosi su mirata integrazione, questa strategia basata su HMEJ fornisce uno strumento promettente per una varietà di applicazioni, inclusa la generazione di modelli animali geneticamente modificati e terapie geniche mirate.
Introduction
L'editing genomico precisa, mirata è spesso necessaria per la produzione di modelli animali geneticamente modificati e terapie cliniche. Molto sforzo è stato fatto per sviluppare varie strategie per efficiente genoma mirata modifiche, ad esempio l'apparato nucleasi a dita zinco (ZFN), nucleasi di trascrizione attivatore-come effettore (TALENs) e sistemi di CRISPR/Cas9. Queste strategie creare rotture a doppio filamento del DNA mirate (DSB) nel genoma e trarre vantaggio intrinseci sistemi di riparazione del DNA, come ricombinazione omologa (HR)1,2, basi-mediata fine unirsi (MMEJ)3 , 4 , 5e alla fine non-omologo unendo (NHEJ)6,7,8 per indurre l'integrazione mirata di transgeni1,9. La strategia basata su HR è attualmente il più comunemente usato genoma modificando l'approccio, che è molto efficiente in linee cellulari, ma non prontamente accessibile alle cellule di divisione a causa del relativo avvenimento limitata verso la fine della fase S/G2. Così, la strategia basata sulla non è applicabile per in vivo l'editing genomico. Recentemente, la strategia basata su NHEJ è stata sviluppata per efficiente gene knock-in mouse tessuti8. Tuttavia, il metodo basato su NHEJ introduce solitamente indels alle giunzioni, rendendo difficile generare l'editing genomico preciso, soprattutto quando si tenta di costruire in-frame fusione geni8. Integrazione mirata basata su MMEJ è capace di editing preciso genomico. Tuttavia, solo modestamente aumenta l'efficienza di integrazione mirata in precedenti relazioni5. Di conseguenza, migliorare l'efficienza della precisa integrazione mirata in vivo è urgente per applicazioni terapeutiche ampio3.
In un lavoro pubblicato recentemente, abbiamo dimostrato un fine omologia-mediata entrare (HMEJ)-base di strategia, che ha mostrato la massima efficienza di integrazione mirata a tutti segnalati strategie sia in vitro che in vivo10. Qui, descriviamo un protocollo per l'istituzione del sistema HMEJ, e anche la costruzione dei vettori di RNA (sgRNA) singolo-guida il gene di interesse e il donatore di targeting vettori harboring siti di destinazione sgRNA e ~ 800 bp di braccia di omologia (Figura 1) . In questo protocollo, descriviamo la procedura dettagliata per la generazione di topi knock-in DNA e brevi passi per l'integrazione mirata in tessuti in vivo. Inoltre, uno studio di proof-of-concept della strategia basata su HMEJ dimostrato la sua capacità di correggere Fah mutazione e salvare Fah- / - fegato guasto topi, che ulteriore ha rivelato il suo potenziale terapeutico.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
Tutte le procedure tra cui animali soggetti sono state approvate dal comitato etico di ricerca biomedica presso gli istituti di Shanghai per scienze biologiche (CAS).
1. progettazione dei plasmidi di donatore
-
Selezione di sgRNA
- Utilizzare strumenti di progettazione online CRISPR per prevedere sgRNAs la destinazione regione11,12,13,14,15. Per il locus di Cdx2 , progettare sei diversi sgRNAs (Cdx2-sgRNA1 ~Cdx2-sgRNA6) intorno alla fermata codone con maggiore potenziale di rango e inferiore off-target (Figura 1A)16.
- Linearizzare 2 µ g di vettori di espressione Cas9-CMV-EGFP e sgRNA di BbsI digestione (1 µ l di BbsI per 2 ore a 37 ° C ad una concentrazione finale di 1 U / µ l in un volume di 20 µ l). Quindi purificare il prodotto con kit di purificazione di gel con un gel di agarosio 1% in tampone TAE × 1.
- Mix una coppia di oligonucleotidi sgRNA in 10 µ l di 1 × buffer di ligasi del DNA T4 per una concentrazione finale di 50 µM. Incubare la soluzione di oligo utilizzando un gradiente di temperatura da 95 ° C a 25 ° C con un tasso di cambiamento di temperatura di 5 ° C/5 min (95 ° C per 5 min quindi 90 ° C per 5 min, 85 ° C per 5 min, ecc.), che sarà temprare i oligos.
- Mix 4 µ l di ricotto prodotto, 2 µ l di vettore linearizzato con 1 µ l di T4 DNA ligasi in 10 µ l di tampone di 1 × T4 DNA ligasi e quindi legare a 22 ° C per 1-2 h (Figura 1B).
-
Surveyor nuclease dosaggio della sgRNA
Nota: L'efficienza di targeting di sgRNA utilizzato per l'esperimento di knock-in viene valutata dall'analisi di surveyor nuclease (noto anche come T7 endonucleasi ho (T7EI) analisi)17. Selezionare il sgRNA con alta efficienza di clivaggio del DNA e una bassa distanza tra il sito di taglio sgRNA e il codone di stop.- Transfect vettori di espressione di Cas9-sgRNA-EGFP in linee di cellule N2a coltivate in DMEM completati con 10% siero bovino fetale, 1% PSG e 1% aminoacidi non essenziali per la transfezione kit (Vedi Tabella materiali). Incubare le cellule transfected a 37 ° C in 5% CO2.
- Dopo 48 h di incubazione, raccogliere 5.000 cellule trasfettate (GFP+) di fluorescenza-attivato delle cellule ordinano (FACS) utilizzando cellule trasfettate con non come un controllo.
- Digerire le cellule raccolte in 2-5 µ l oflysis tampone (0,1% Triton X-100, 0.1% Tween 20 e 100 µ g/mL proteinasi K) a 56 ° C per 30 min e poi calore inattivare proteinasi K a 95 ° C per 10 min.
- Amplificare il campione dalla PCR annidata (tabella 1) utilizzando il protocollo del produttore. La dimensione dei prodotti di PCR è impostata a 300-500 bp.
- Mescolare 1 µ l di prodotto di lisi con DNA polimerasi e una coppia di primer esterno riconoscendo la sequenza attorno al sito di destinazione sgRNA (0,1 µM, concentrazione finale) (tabella 1) ed eseguire la PCR primaria in un volume di 20 µ l.
- Attivazione della polimerasi di DNA a 95 ° C per 5 min ed eseguire la PCR primaria per 30 cicli a 95 ° C per 30 s, 60 ° C per 30 s e 72 ° C per 24 s (1 min/1 kb), con un'estensione finale a 72 ° C per 5 min.
- Eseguire la PCR secondaria utilizzando 1 µ l di prodotto PCR primario e una coppia di primers interna nidificati.
- Denaturare e ri-tempri 300-600 ng del prodotto PCR purificato in 20 µ l di tampone di reazione di 1 × T7EI (50 mM NaCl, 10 mM Tris-HCl, 10mm MgCl2, pH di 1 mM DTT 7,9) utilizzando un gradiente di temperatura da 95 ° C a 25 ° C con un tasso di 5 ° C/5 min.
- Aggiungere 1 µ l di enzima T7EI per i prodotti di PCR Ricotti e digest a 37 ° C per 2 h. Quindi eseguire il prodotto di digestione su gel di agarosio 2% in tampone TAE 1x 120 V per 40 min, fino a quando i frammenti sono separati (Vedi Tabella materiali).
- Utilizzare ImageJ per determinare le intensità di banda del taglio e del DNA uncut. Calcolare la frequenza di indel utilizzando i metodi come precedentemente segnalato9 (Figura 1C).
-
Costruzione del vettore di donatore
Nota: Per generare dei vettori-donatore HMEJ per gene Cdx2 , costruire un donatore del DNA (800 bp HAL-p2A-mCherry-800 bp HAR) fiancheggiato con 23 nt Cdx2-sgRNAs targeting sequenza ad entrambe le estremità (Figura 1D e 1 di figuraE). Il PAM di sequenza di destinazione era adiacente all'estremità del braccio omologo. Assemblea di Gibson è raccomandato per donatore HMEJ clonazione.- Amplificare il braccio di sinistra omologia di bp (HAL) 800 con forward primer-1F (contenente 15-20 nt sovrapposizione sequenza dal vettore, 23 nt sequenza targeting Cdx2-sgRNA e circa 20 nt sequenza da HAL) e reverse primer-1R (contenente 15-20 bp sovrapposizione sequenza da p2A-mCherry e circa 20 nt sequenza da HAL) alla concentrazione finale di 0,1 µM usando il DNA genomic del mouse a 200 ng / µ l (Figura 1D, tabella 1).
- Amplificare il frammento di inserimento p2A-mCherry con forward primer-2F (contenente la sequenza di sovrapposizione di nt 15-20 da HAL e circa 20 nt dal frammento di inserimento) e reverse primer-2R (contenente la sequenza di sovrapposizione di nt 15-20 da HAR e circa 20 nt da il frammento di inserimento) alla concentrazione finale di 0,1 µM utilizzando DNA genomico o plasmide con sequenze di reporter a 100 ng / µ l o 30 ng / µ l (Figura 1D, tabella 1).
- Amplificare il braccio di destra omologia di bp (HAR) 800 con forward primer-3F (contenente 15-20 nt sovrapposizione sequenza dal vettore, 23 nt sequenza targeting Cdx2-sgRNA e circa 20 nt sequenza da HAR) e reverse primer-3R (contenente 15-20 nt sovrapposizione sequenza da p2A-mCherry e circa 20 nt sequenza da HAR) alla concentrazione finale di 0,1 µM usando il DNA genomic del mouse a 200 ng / µ l (Figura 1D, tabella 1).
- Eseguire tutti i prodotti PCR su gel di agarosio all'1% in 1 × TAE tampone e purificare i prodotti di PCR delle dimensioni previste dal kit di estrazione del gel, secondo le istruzioni del produttore (tabella 1).
- Digerire 50-100 ng di un vettore di costrutto con KpnI e XbaI. Mix 2 µ l del vettore linearizzato a 30-40 ng / µ l con tre PCR amplificati frammenti (1 µ l per ogni, 100-200 ng / µ l) 2 x Gibson mix. Aggiungere H2O per regolare il volume finale di 10 µL.Incubate il mix a 50 ° C per 60 min.
- Trasformare competente Escherichia coli cellule con tutti il prodotto assemblato ed estrarre il plasmide costruisce da kit di estrazione del DNA secondo le istruzioni del produttore. Verificare il donatore HMEJ di sequenziamento del DNA.
2. genome Editing in embrioni di topo utilizzando il metodo basato su HMEJ
-
Produzione di mRNA Cas9
- Per la preparazione di Cas9 mRNA, aggiungere la sequenza del promotore T7 per la Cas9 regione di codificazione dall'amplificazione di PCR utilizzando la coppia di primer appropriato elencata nella tabella 1. Aggiungere il primer Cas9 F/R a una concentrazione finale di 0,1 µM e 20 ng di Cas9 esprimendo vettore da 1 × alta fedeltà mix di polimerasi del DNA. Regolare il volume finale di 50 µ l con H2O.
- Attivazione della polimerasi di DNA a 95 ° C per 5 min ed eseguire la PCR per 36 cicli a 95 ° C per 30 s, 60 ° C per 30 s e 68 ° C per 4 min (min 1/1 kb), con un'estensione finale a 68 ° C per 10 min.
- Purificare il prodotto di PCR di T7-Cas9 per trascrizione in vitro (IVT) e poi trascrivere 0,5-1 µ g di DNA da kit di trascrizione di mRNA a 37 ° C per 8 h in un volume totale di 20 µ l, secondo le istruzioni del produttore (Vedi Tabella materiali).
- Aggiungere 1 µ l di dnasi alla miscela per rimuovere il modello di DNA a 37 ° C per 15 min. Aggiungi una coda poli-A per 45 min a 37 ° C e recuperare il mRNA Cas9, kit di purificazione di RNA, secondo le istruzioni del produttore (Vedi Tabella materiali).
-
Produzione di sgRNA
- Generare il modello di sgRNA guidato da un promotore T7 con la polimerasi del DNA di alta fedeltà come sopra. Scegliere un'impalcatura di sgRNA contenente vettoriale come il modello. I primers utilizzati sono elencati nella tabella 1.
- Purificare il prodotto PCR di T7-sgRNA ed usare 0,5-1 µ g di DNA come il modello per la trascrizione in vitro di sgRNA utilizzando un kit di trascrizione breve RNA a 37 ° C per 6 h in un volume totale di 20 µ l, secondo le istruzioni del produttore (vedere tabella materiali < / c11 >).
- Aggiungere 1 µ l di dnasi alla miscela e continuare l'incubazione a 37 ° C per 15 minuti togliere la mascherina del DNA. Purificare il sgRNAs con kit di purificazione di RNA, come sopra (Vedi Tabella materiali).
- Diluire il sgRNA a 500 ng / µ l in acqua RNAsi-libera e conservare i campioni a − 80 ° C fino a 3 mesi.
Nota: Ribonucleoproteine CRISPR (RNP) sono un'alternativa sostituzione con un migliore taglio efficienza18,19,20.
-
Coltura di collezione, microiniezione e in vitro dell'embrione
- Superovulate B6D2F1 (C57BL/6 × DBA2J) topi femminili (7-8 settimane) di gonadotropina del siero di cavalla gravida (PMSG), seguiti da gonadotropina corionica umana (hCG) 48 ore più tardi. Dopo l'iniezione di hCG, le femmine di casa con i maschi B6D2F1 durante la notte.
- Sacrificare le femmine di CO2 anestesia, 24 h dopo l'iniezione di hCG. Raccogliere gli embrioni fecondati dal loro ovidotti (con 30-50 embrioni per ogni femmina) nel terreno di M2.
- Posto gli embrioni fecondati (circa 300 uova un giorno iniettabile) nel mezzo di KSOM (5,55 g/L NaCl, KCl, 0,05 g/L KH2PO4, 0,05 g/L MgSO4•7H2O, 0,04 g/L glucosio, 1,12 g/L sodio 0,19 g/L lattato, 2,1 g/L NaHCO3 , piruvato di sodio 0,02 g/L, 0,25 g/L CaCl2•2H2O, 0,004 g/L EDTA, 0,146 g/L L-glutamina e 1 g/L albumina di siero bovino) a 37 ° C in un incubatore con 5% CO2.
- Mix Cas9 mRNA (100 ng / µ l), sgRNA (50 ng / µ l) e donatore HMEJ (100 ng / µ l) di vettore e aggiungere H2O per regolare il volume finale di 10 µ l. mettere il composto sul ghiaccio.
- Tirare aghi capillari (diametro esterno 1,0 mm, diametro interno mm 0,78 con filamento) utilizzando un estrattore micropipetta (parametri: calore, pressione, 300; velocità, 80; ritardo, 200; tirare, 60, 74. Vedi tabella materiali). Aghi delle imprese sarebbe una sostituzione alternativa per la microiniezione.
- Iniettare un probabile volume della miscela nel citoplasma degli zigoti con pronuclei ben definite in una goccia di mezzo CZB HEPES contenente 5 µ g/mL citocalasina B utilizzando un microinjector con costante flusso impostazioni (Figura 2A) (Vedi Tabella materiali)21.
Nota: Deve essere iniettato ogni gruppo degli zigoti entro 20-30 min Cytochalasin B potrebbe aumentare la redditività dello zigote del mouse dopo l'iniezione. In alternativa, microinjection può essere azionato con il sistema piezo, come descritto in precedenza22. - Cultura gli zigoti iniettati nel mezzo KSOM a 37 ° C, inferiore al 5% di CO2 fino a quando la blastocisti fase dopo 3,5 giorni per l'osservazione della fluorescenza (figure 2B e 2C).
-
Trasferimento dell'embrione e la generazione di topi
- Compagno di estro topi femminili ICR con vasectomia topi maschi ICR lo stesso giorno come iniezione.
- Cultura gli zigoti iniettati nella fase 2 celle a 37 ° C in 5% CO2e trasferire 25-30 2 celle gli embrioni in ovidotti di femmine pseudopregnant ICR a 0,5 giorno coitum dell'alberino (dpc). Destinatario madri offrono cuccioli a 19,5 dpc.
-
Genotipizzazione del mouse
- Estrarre il DNA genomico del mouse da campioni di punta o coda utilizzando un kit di estrazione del DNA, secondo le istruzioni del produttore (Vedi Tabella materiali).
- Identificare svincolo 5' e 3' di knock-in eventi utilizzando 200-400 ng di DNA genomic misurato tramite spettrometria UV/vis come un modello per eseguire il amplification PCR.
- Attivazione della polimerasi di DNA a 95 ° C per 5 min ed eseguire la PCR per 38 cicli a 95 ° C per 30 s, 60 ° C per 30 s e 72 ° C per 1 min (min 1/1 kb), con un'estensione finale a 72 ° C per 10 min. Per il 5' junction, utilizzare il primer forward a monte dell'HAL, con quella inversa sul frammento knock-in (p2A-mCherry). Per quanto riguarda 3' incrocio, è necessario utilizzare il primer avanti sul frammento knock-in (p2A-mCherry), con quello inverso sulla valle di HAR (tabella 1).
- Eseguire 6 µ l del prodotto della PCR su gel di agarosio 1% in tampone TAE × 1 e controllo per la dimensione del frammento previsto. Quindi verificare loro DNA sequencing (Figura 2D).
3. HMEJ-base In Vivo Genome Editing negli epatociti
- Inserire destinatario mouse C57BL/6J (8 settimane) in un dispositivo trattenente e mettere la coda attraverso la fessura.
- Mescolare HMEJ donatore vettori (30 µ g) e vettori di espressione di spCas9 (30 µ g) in 2 mL di soluzione salina. Per l'esperimento di controllo, è necessario sospendere vettori di HMEJ donatore (30 µ g) in 2 mL di soluzione salina (Figura 3A).
- Pulire la coda di topo con etanolo al 70%. Inserire l'ago nella coda della vena e iniettare la soluzione di DNA plasmidico entro 5-7 s. rimuovere l'ago e rilasciare il mouse dal dispositivo trattenente.
- Sacrificare i topi di CO2 anestesia dopo 5-9 giorni dopo l'iniezione. Irrorare la via di topi con soluzione fisiologica allo 0,9%, seguita da paraformaldeide 4% utilizzando una pompa peristaltica e difficoltà del fegato durante la notte a 4 ° C.
- Disidratare il tessuto con 30% di saccarosio durante la notte, fino a quando scende fino al fondo del tubo.
- Sezione del tessuto congelato con uno spessore di 10 µm per i campioni del fegato.
- Risciacquare le sezioni tre volte in tampone fosfato 0,1 M (PB) e li incubare con l'anticorpo primario: Rabbit anti-mCherry (diluito in 5% NGS) overnight a 4 ° C.
- Lavare sezioni tre volte in PB e quindi li incubare con l'anticorpo secondario: Cy3-AffiniPure Goat Anti-Rabbit IgG per 2 h a temperatura ambiente su agitatore orbitale.
- Colorante di contrasto le sezioni con DAPI per 20 min e montaggio con glicerina su vetrini per l'osservazione della fluorescenza (Figura 3B) ulteriormente.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Editing basato su HMEJ genomico in embrioni di topo: Per definire l'efficienza knock-del metodo basato su HMEJ in zigoti mouse, abbiamo consegnato Cas9 mRNA, sgRNA targeting del gene Cdx2 e il donatore HMEJ in zigoti del mouse, che è stato progettato per fondere un gene reporter p2A-mCherry al codone ultimo di Cdx2 gene (Figura 2A). Gli zigoti iniettati sviluppato in blastocisti nella cultura. Per valutare l'efficienza di knock-in, abbiamo analizzato la fluorescenza mCherry con un microscopio a fluorescenza, e abbiamo trovato che il 12,9% delle blastocisti ricezione donatori HMEJ erano positive per mCherry, che è stato rigorosamente espresso nella delle cellule trofoblastiche (figure 2B 2 C). Ordinando i topi positivi di PCR, abbiamo anche trovato che tutti esaminati eventi di integrazione sono stati precisi nella cornice integrazioni alle giunzioni di 5' e 3' (Figura 2D).
Editing genomico basati su HMEJ in tessuti adulti e terapia genica mediata da HMEJ: Per studiare se l'editing genomico basati su HMEJ può essere applicata nei tessuti adulti, abbiamo inserito la cassetta mCherry destra prima il codone di arresto del gene Actb di trasdurre costrutti - HMEJ Actbdi fegati del topo C57/B6J dalla coda-vena iniezione idrodinamica (Figura 3A). Dopo 7 giorni di iniezioni, abbiamo trovato che quasi la metà degli epatociti trasfettate espresso mCherry come macchiato sulle sezioni del fegato (Figura 3B).
Per esplorare la possibilità di utilizzare una strategia basata su HMEJ per la terapia genica, abbiamo impiegato le idrolasi di fumarylacetoacetate (Fah)-topi deficienti. Il Fah- / - mouse è una consolidata tirosinemia ereditaria di tipo I modello di topo (HTI), che ospita un frammento di inserimento nell'esone 5 del gene di Fah , causando mutazioni frameshift nel seguente sequenza23. Per mantenere Fah- / - topi, abbiamo trattato i topi di Fah- / - con un inibitore a Monte della via catabolica di tirosina, 2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione (NTBC)24. Qui abbiamo deciso di vedere se la correzione genica MMEJ - e HMEJ-mediata potrebbe salvare Fah mutazione nel topo Fah- / . Abbiamo iniettato idrodinamicamente Cas9 costrutto insieme Fah- MMEJ o costrutti di Fah- HMEJ, progettati per inserire Fah cDNA dell'essone 5 di 14 in introne 4 del gene di Fah , Fah - / - mouse (fegatini Figura 3 C). una settimana dopo l'iniezione, il NTBC è stato ritirato per indurre danni al fegato (Figura 3C). Dopo il ritiro di NTBC, Fah-corretti epatociti di Fah- / - topi che ricevono Fah- HMEJ e costrutti di Cas9 ha mostrato la proliferazione più efficace rispetto a metodo basato su MMEJ (Figura 3D ).

Figura 1 : HMEJ-mediata mirata integrazione in vitro.
Il regime sperimentale (A) per la selezione di sgRNAs: sei diverse sgRNAs (Cdx2-sgRNA1 ~Cdx2-sgRNA6) intorno al codone di arresto del locus Cdx2 con un potenziale più alto rango e fuori bersaglio sono stati scelti sulla base progettazione CRISPR on-line strumento. La sequenza di motivi adiacenti (PAM) di protospacer è in rosso. Disegno sperimentale (B): plasmidi di espressione The Cas9-CMV-GFP che esprimono GFP, Cas9 e sgRNA sono stati introdotti nelle cellule N2a. Le cellule GFP+ sono state ordinate al giorno 3 per l'analisi di surveyor. Geometra (C) analisi per il targeting di Cdx2 : 6 diversi sgRNAs sono stati progettati per l'analisi di surveyor. N2a normale delle cellule DNA di genomic è funge da controllo. *, il sgRNA utilizzato per Cdx2-2A-mCherry knock-in esperimento. (D) lo schema di costruzione dei donatori HMEJ utilizza l'assembly di Gibson. (E) descrizione schematica di HMEJ-mediata del gene targeting strategia Cdx2 locus. Braccio di omologia HAL/HAR, sinistra/destra; triangoli, sgRNA siti di destinazione; DI / OR, primer esterno di avanti/indietro; Se / IR, interna avanti/indietro primer. Figura modificata da precedente relazione10. Clicca qui per visualizzare una versione più grande di questa figura.

Figura 2 : L'editing genomico in embrioni di topo via HMEJ-mediata mirata integrazione
(A) schema sperimentale di microiniezione: una miscela di Cas9 mRNA (100 ng / µ l), sgRNA (50 ng / µ l) e donatore plasmidi (100 ng / µ l) sono state iniettate in zigoti del mouse. (B) immagini di fluorescenza rappresentativo degli embrioni del mouse modificati dalla strategia di HMEJ. Bar, 20 µm. (C) Knock-in efficienza indicata dalla percentuale di mCherry+ blastocisti. Numero sopra ogni bar, totale blastocisti conteggiati. (D) analisi dei topi gene-modificato Cdx2 locus di sequenza. Prodotti della PCR amplificati dai siti di giunzione 5' e 3' sono stati sequenziati. Braccio superiore, omologia; viola, p2A; rosso, mCherry; HAR o HAL, braccio omologo destro o sinistro. Linee tratteggiate segnano la regione omessa per maggiore chiarezza. Figura modificata da precedente relazione10. Clicca qui per visualizzare una versione più grande di questa figura.

Figura 3 : HMEJ-mediata mirata integrazione in vivo.
(A) descrizione schematica dell'iniezione della vena della coda idrodinamica. Una miscela di plasmidi che esprimono sgRNA e sequenza di donatore e plasmidi che esprimono spCas9 sono stati trasportati al fegato tramite iniezione della vena della coda idrodinamica. (B) immagini rappresentative di immunofluorescenza degli epatociti. Le sezioni del fegato sono stati raccolti 7 giorni dopo l'iniezione. Barra della scala, 50 µm. GFP, transfected le cellule. (C) plasmidi di entrambi MMEJ - o HMEJ-mediata strategia di sostituzione genica il progettato per inserire introne 4 del gene di Fah Fah cDNA dell'essone 5 e 14 sono stati consegnati in fegati del topo Fah- / - da iniezione idrodinamica. NTBC su: topi Fah- / - sono stati mantenuti su acqua NTBC; NTBC fuori: prelievo di acqua NTBC (il primo giorno del ritiro NTBC è stato definito come giorno 0, che è il 7 ° giorno dopo l'iniezione). (D) Fah colorazione immunoistochimica di sezioni del fegato da Fah- / - topi iniettati con plasmidi MMEJ o HMEJ. Barra di scala, 100 µm. figura modificata da precedenti rapporti5,10. Clicca qui per visualizzare una versione più grande di questa figura.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
I passaggi più critici nella costruzione di plasmidi di donatore HMEJ sono: (1) selezione di sgRNA con alta efficienza di clivaggio del DNA e bassa distanza tra sito di taglio sgRNA e codone di arresto e costruzione (2) corretta del donatore HMEJ. CRISPR/Cas9-mediata cleavage su entrambi vettoriale di donatore del transgene (contenente siti di destinazione sgRNA e ~ 800 bp omologia braccia) e genoma mirato è necessario per l'integrazione mirata efficiente e precisa in vivo. I passaggi più critici della generazione di topi knock-in utilizzando il metodo basato su HMEJ sono: (1) la preparazione di alta qualità di Cas9 mRNA e sgRNA (nessuna degenerazione esiste in Cas9 mRNA e sgRNA) e (2) la preparazione del plasmide di donatore HMEJ di alta qualità. Il plasmide viene illustrato senza effetti tossici sullo sviluppo embrionale.
Recentemente, un metodo basato su NHEJ era stato segnalato anche per editing8genomico per un efficiente in vivo . Tuttavia, vari tipi di mutazioni indel solitamente sono state indotte alle giunzioni, come descritto in precedenti relazioni8, rendendo difficile ottenere una precisa integrazione. Qui, la strategia basata su HMEJ che abbiamo descritto in precedenza ha mostrato precisa integrazione mirata con quasi nessun mutazioni indel. Così, una strategia basata su HMEJ potrebbe essere una piattaforma ideale per la sostituzione di una sequenza mutata (ad esempio, una mutazione di punto) con quello corretto, che non è applicabile per il metodo basato su NHEJ.
Il mosaicism è un grave problema per l'editing di gene in embrioni. Iniezione di proteine Cas9 invece di mRNA in una prima fase embrionale può raggiungere transgene knock-in uno stadio di cellule senza mosaicismo. Per applicazioni cliniche, consegna dei sistemi CRISPR/Cas9 in tessuti adulti è comunque impegnativo.
Ci sono molti usi potenziali futuri di editing genomico basati su HMEJ. Può essere utilizzato per generare modelli animali geneticamente modificati. Considerando la sua alta knock-in efficienza negli embrioni, questo metodo potrebbe ridurre significativamente il numero di animali necessario per la generazione di modelli animali geneticamente modificati e soprattutto apre la possibilità di generare modelli genetici primate non umano. L'editing genomico basati su HMEJ può tipi di singola cella lignaggio traccia nei tessuti dell'adulto, che è particolarmente utile per modelli animali, poiché non vi è una mancanza di modelli animali disponibili, come ad esempio i primati non umani. Può essere utilizzato per terapie geniche mirate: l'applicazione più attraente di una strategia basata su HMEJ è la terapia genica per clinica USA. In questo studio, abbiamo corretto la mutazione di Fah di tirosinemia ereditaria di tipo I topi da iniezione idrodinamica dei vettori indicati. Tuttavia, la consegna del sistema CRISPR/Cas9 in tessuti adulti è ancora l'importante sfida tecnica per uso clinico, come iniezione idrodinamica è improbabile da essere eseguita in pazienti. Attualmente, ulteriore miglioramento della strategia di consegna è urgentemente necessaria prima di tradurre questo metodo basato su HMEJ in clinica.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Gli autori non hanno nulla a rivelare.
Acknowledgments
Questo lavoro è stato supportato da CAS strategico priorità Research Program (XDB02050007, XDA01010409), la nazionale Hightech di R & D programma (programma 863; 2015AA020307), National Natural Science Foundation of China (NSFC concede 31522037, 31500825, 31571509, 31522038), China Youth mille talenti programma (HY), rottura attraverso il progetto dell'Accademia delle scienze cinese, Shanghai città comitato della scienza e della tecnologia del progetto (16JC1420202 a HY), il Ministero della scienza e della tecnologia della Cina (la maggior parte; 2016YFA0100500).
Materials
| Name | Company | Catalog Number | Comments |
| pX330 | Addgene | 42230 | |
| pAAV vector | Addgene | 37083 | |
| pX260 | Addgene | 42229 | |
| AAV_Efs_hSpCas9_NLS_FLAG-SV40 | Addgene | 97307 | AAV vector for encoding a human codon-optimized SpCas9 driven by EFs promoter |
| AAV_Actb HMEJ donor_U6_sgRNA_EF1a_GFP_polyA | Addgene | 97308 | HMEJ donor for fusing a p2A-mCherry reporter to mouse Actb. EGFP driven by EF1a promoter and U6-driven sgRNAs targeting Actb. AAV backbone. |
| AAV_Cdx2 HMEJ donor | Addgene | 97319 | HMEJ donor for fusing a p2A-mCherry reporter to mouse Cdx2. |
| Lipofectamine 3000 Transfection Reagent | Life Technology | L3000015 | |
| Nuclease-Free Water | Life Technologies | AM9930 | |
| Bbs I | New England Biolabs | R0539S | |
| NEB Buffer 2 | New England Biolabs | B7002S | |
| T7 endonuclease I | New England Biolabs | M0302L | |
| NEBuilder HiFi DNA Assembly Master Mix | New England Biolabs | E2621L | |
| Plasmid EndoFree-Midi Kit | Qiagen | 12143 | |
| MMESSAGE MMACHINE T7 ULTRA | Life Technologies | AM1345 | |
| MEGACLEAR KIT 20 RXNS | Life Technologies | AM1908 | |
| MEGASHORTSCRIPT T7 KIT 25 RXNS | Life Technologies | AM1354 | |
| Flaming/Brown Micropipette Puller | Sutter Instrument | P-97 | Micropipette Puller (parameters: heat, 74; pull, 60; velocity, 80; time/delay, 200; pressure, 300) |
| Borosilicate glass | Sutter Instrument | B100-78-10 | type of capillaries (outer diameter 1.0 mm, inner diameter 0.78 mm with filament) |
| FemtoJet microinjector | Eppendorf | ||
| Freezing microtome | Leica | CM1950-Cryostat | thickness of 40 μm for brain, 10 μm for liver |
| Rabbit anti-mCherry | GeneTex | ||
| Cy3-AffiniPure Goat Anti-Rabbit IgG | Jackson Immunoresearch | ||
| DMEM | Gibco | 11965092 | |
| FBS | Gibco | 10099141 | |
| NEAA | Gibco | 11140050 | |
| Pen,Strep,Glutamine | Gibco | 10378016 | |
| Gel Extraction Kit | Omega | D2500-02 | |
| FACS | BD AriaII | ||
| PMSG | Ningbo Sansheng Medicine | S141004 | |
| HCG | Ningbo Sansheng Medicine | B141002 | |
| Cytochalasin B | Sigma | CAT#C6762 | |
| KSOM+AA with D-Glucose and Phenol Red | Millipore | CAT#MR-106-D | |
| M2 Medium with Phenol Red | Millipore | CAT#MR-015-D | |
| Mineral oil | Sigma |
References
- Yang, H., et al. Generation of Mice Carrying Reporter and Conditional Alleles by CRISPR/Cas-Mediated Genome Engineering. Cell. 154 (6), 1370-1379 (2013).
- Hockemeyer, D., et al. Genetic engineering of human pluripotent cells using TALE nucleases. Nature Biotechnology. 29 (8), 731-734 (2011).
- Nakade, S., et al. Microhomology-mediated end-joining-dependent integration of donor DNA in cells and animals using TALENs and CRISPR/Cas9. Nature Communications. 5, 5560 (2014).
- Hisano, Y., et al. Precise in-frame integration of exogenous DNA mediated by CRISPR/Cas9 system in zebrafish. Scientific reports. 5, 8841 (2015).
- Yao, X., et al. Cas9 - Mediated Precise Targeted Integration In Vivo Using a Double Cut Donor with Short Homology Arms. EBioMedicine. , (2017).
- Auer, T. O., Duroure, K., De Cian, A., Concordet, J. P., Del Bene, F. Highly efficient CRISPR/Cas9-mediated knock-in in zebrafish by homology-independent DNA repair. Genome research. 24 (1), 142-153 (2014).
- Maresca, M., Lin, V. G., Guo, N., Yang, Y. Obligate ligation-gated recombination (ObLiGaRe): custom-designed nuclease-mediated targeted integration through nonhomologous end joining. Genome Research. 23 (3), 539-546 (2013).
- Suzuki, K., et al. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature. 540 (7631), 144-149 (2016).
- Cong, L., et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 339 (6121), 819-823 (2013).
- Yao, X., et al. Homology-mediated end joining-based targeted integration using CRISPR/Cas9. Cell Research. 27 (6), 801-814 (2017).
- Han, D. W., et al. Direct reprogramming of fibroblasts into epiblast stem cells. Nature Cell Biology. 13 (1), 66-71 (2011).
- Han, D. W., et al. Direct Reprogramming of Fibroblasts into Neural Stem Cells by Defined Factors. Cell Stem Cell. , (2012).
- Ambasudhan, R., et al. Direct reprogramming of adult human fibroblasts to functional neurons under defined conditions. Cell Stem Cell. 9 (2), 113-118 (2011).
- Sparman, M., et al. Epigenetic reprogramming by somatic cell nuclear transfer in primates. Stem Cells. 27 (6), 1255-1264 (2009).
- Schatten, G., Mitalipov, S. Developmental biology: Transgenic primate offspring. Nature. 459 (7246), 515-516 (2009).
- Hsu, P. D., et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nature Biotechnology. 31 (9), 827-832 (2013).
- Cong, L., et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science. 339 (6121), 819-823 (2013).
- Quadros, R. M., et al. Easi-CRISPR: a robust method for one-step generation of mice carrying conditional and insertion alleles using long ssDNA donors and CRISPR ribonucleoproteins. Genome Biology. 18 (1), 92 (2017).
- Park, K. E., et al. Targeted Gene Knockin in Porcine Somatic Cells Using CRISPR/Cas Ribonucleoproteins. International journal of molecular sciences. 217 (6), (2016).
- Woo, J. W., et al. DNA-free genome editing in plants with preassembled CRISPR-Cas9 ribonucleoproteins. Nature biotechnology. 33 (11), 1162-1164 (2015).
- Harms, D. W., et al. Mouse Genome Editing Using the CRISPR/Cas System. Current protocols in human genetics. 83, 11-27 (2014).
- Yang, H., Wang, H., Jaenisch, R. Generating genetically modified mice using CRISPR/Cas-mediated genome engineering. Nature protocols. 9 (8), 1956-1968 (2014).
- Grompe, M., et al. Loss of Fumarylacetoacetate Hydrolase Is Responsible for the Neonatal Hepatic-Dysfunction Phenotype of Lethal Albino Mice. Genes & development. 7 (12), 2298-2307 (1993).
- Paulk, N. K., et al. Adeno-associated virus gene repair corrects a mouse model of hereditary tyrosinemia in vivo. Hepatology. 51 (4), 1200-1208 (2010).
Tags
Genetica problema 133 CRISPR/Cas9 integrazione mirata fine omologia-mediata entrare In Vivo embrione geneticamente topi iniezione idrodinamicaErratum
Formal Correction: Erratum: CRISPR/Cas9-mediated Targeted Integration In Vivo Using a Homology-mediated End Joining-based Strategy
Posted by JoVE Editors on 03/10/2021.
Citeable Link.
An erratum was issued for: Studying TGF-β Signaling and TGF-β-induced Epithelial-to-mesenchymal Transition in Breast Cancer and Normal Cells. The phrases "surveyor assay" and "Surveyor Nuclease" have been updated to "T7E1 assay" to " T7 endonuclease I" respectively.
Step 1.2 in the Protocol has been updated from:
- Surveyor nuclease assay of sgRNA
NOTE: The targeting efficiency of the sgRNA used for the knock-in experiment is evaluated by surveyor nuclease assay (also known as T7 endonuclease I (T7EI) assay)17. Select the sgRNA with high DNA cleavage efficiency and a low distance between the sgRNA cutting site and the stop codon.
to:
- T7 endonuclease assay of sgRNA
NOTE: The targeting efficiency of the sgRNA used for the knock-in experiment is evaluated by T7 endonuclease (T7EI) assay17. Select the sgRNA with high DNA cleavage efficiency and a low distance between the sgRNA cutting site and the stop codon.
Figure 1 in the Representative Results has been updated from:
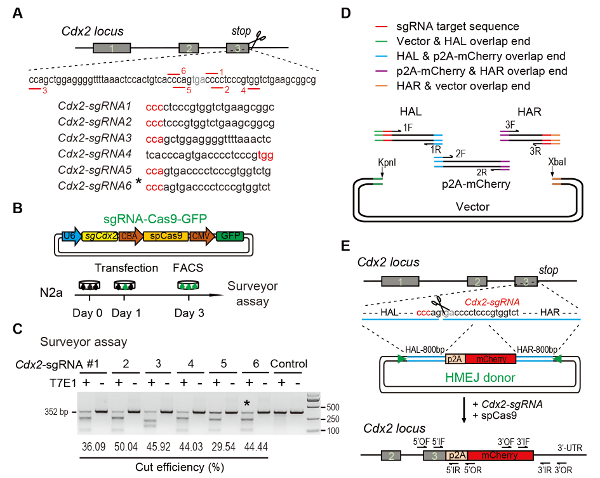
Figure 1: HMEJ-mediated targeted integration in vitro.
(A) Experimental scheme for selection of sgRNAs: Six different sgRNAs (Cdx2-sgRNA1~Cdx2-sgRNA6) around the stop codon of the Cdx2 locus with a higher rank and off-target potential were chosen based on online CRISPR design tool. The protospacer adjacent motif (PAM) sequence is in red. (B) Experimental design: The Cas9-CMV-GFP expression plasmids expressing sgRNA, Cas9, and GFP were introduced into N2a cells. GFP+ cells were sorted at day 3 for surveyor assay. (C) Surveyor assay for Cdx2 targeting: 6 different sgRNAs were designed for surveyor assay. Normal N2a cell genomic DNA serves as control. *, the sgRNA used for Cdx2-2A-mCherry knock-in experiment. (D) Schematic overview of construction of HMEJ donors using Gibson assembly. (E) Schematic overview of HMEJ-mediated gene targeting strategy at Cdx2 locus. HAL/HAR, left/right homology arm; triangles, sgRNA target sites; OF/OR, outer forward/reverse primer; IF/IR, inner forward/reverse primer. Figure modified from previous report10. Please click here to view a larger version of this figure.
to:
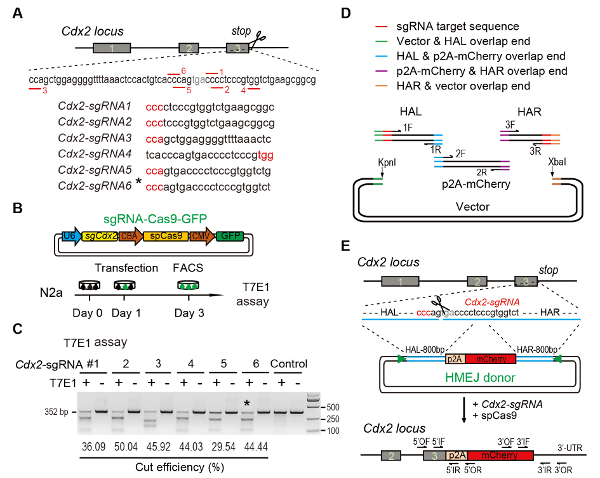
Figure 1: HMEJ-mediated targeted integration in vitro.
(A) Experimental scheme for selection of sgRNAs: Six different sgRNAs (Cdx2-sgRNA1~Cdx2-sgRNA6) around the stop codon of the Cdx2 locus with a higher rank and off-target potential were chosen based on online CRISPR design tool. The protospacer adjacent motif (PAM) sequence is in red. (B) Experimental design: The Cas9-CMV-GFP expression plasmids expressing sgRNA, Cas9, and GFP were introduced into N2a cells. GFP+ cells were sorted at day 3 for T7EI assay. (C) T7EI assay for Cdx2 targeting: 6 different sgRNAs were designed for T7EI assay. Normal N2a cell genomic DNA serves as control. *, the sgRNA used for Cdx2-2A-mCherry knock-in experiment. (D) Schematic overview of construction of HMEJ donors using Gibson assembly. (E) Schematic overview of HMEJ-mediated gene targeting strategy at Cdx2 locus. HAL/HAR, left/right homology arm; triangles, sgRNA target sites; OF/OR, outer forward/reverse primer; IF/IR, inner forward/reverse primer. Figure modified from previous report10. Please click here to view a larger version of this figure.
