

Summary
Unter Verwendung einer vormontierten Cas9 ist Ribonucleoprotein Komplex (RNP) eine leistungsfähige Methode für präzise und effiziente Genom-Bearbeitung. Hier heben wir ihre Nützlichkeit in einem breiten Spektrum von Zellen und Organismen, einschließlich der primären humanen Zellen und sowohl klassische und neue Modellorganismen.
Abstract
Standortspezifische eukaryotische Genom Bearbeitung mit CRISPR (gruppierten regelmäßig dazwischen kurze palindromische Wiederholungen)-Cas (CRISPR-assoziierten) Systeme geworden schnell ein Gemeinplatz unter den Wissenschaftlern, die eine Vielzahl von biologischen Fragen zu verfolgen. Benutzer beschäftigen in den meisten Fällen das Cas9 Protein von Streptococcus Pyogenes in einem Komplex mit einem leicht umprogrammierten Guide RNA (gRNA) abgeleitet. Diese Komponenten werden in Zellen eingebracht und durch eine Basis, die Kopplung mit einer ergänzenden Region des Genoms doppelsträngige DNA (DsDNA), spaltet das Enzym beide Stränge um eine Doppel-Strang-Pause (DSB) zu generieren. Nachbesserung führt zufällige einfügen oder löschen-Ereignisse (Indels) oder die Einbeziehung der Experimentator bereitgestellten DNA auf dem Gelände des Bruchs.
Die Verwendung von einem gereinigten Single-Guide RNA und Cas9 Protein, zu einem RNP vormontiert und direkt an Zellen, ist ein potenter Ansatz zur Erreichung hocheffiziente gen zu bearbeiten. RNP Bearbeitung verbessert vor allem die Rate der Gen Insertion, ein Ergebnis, das oft schwierig zu erreichen ist. Im Vergleich zu der Lieferung über ein Plasmid, führt das kürzere Fortbestehen der Cas9 RNP innerhalb der Zelle zu weniger Ziel-Ereignisse.
Trotz der Vorteile sind viele Gelegenheitsnutzer CRISPR gen Bearbeitung weniger mit dieser Technik vertraut. Um die Einstiegsschwelle zu senken, beschreiben wir ausführliche Protokolle zur Umsetzung der RNP-Strategie in einer Reihe von zusammenhängen, Hervorhebung seiner klare Vorteile und vielfältigen Einsatzmöglichkeiten. Wir decken in zwei Arten von primären humanen Zellen, T-Zellen und Hämatopoetischen Stammzellen/Vorläuferzellen (HSPCs) bearbeiten. Wir zeigen auch, wie Cas9 RNP Bearbeitung ermöglicht die einfache genetische Manipulation des gesamte Organismen, einschließlich der klassischen Modell Fadenwurm Caenorhabditis Elegans und mehr vor kurzem Modell Krustentier, Parhyale Hawaiensiseingeführt.
Introduction
Fder CRISPR-Cas9-System ermöglicht es Wissenschaftlern, gezielte Regionen des Genoms1zu ändern. Diese schnelle und kostengünstige Technologie revolutioniert Grundlagenforschung und verspricht eine profunde Auswirkung auf die Entwicklung von personalisierten Krankheit Therapien, Präzisionslandwirtschaft, und darüber hinaus2zu machen. CRISPR Bearbeitung ist ein Werkzeug, demokratisieren und Implementierung des Systems in ein neues Labor erfordert keine besonderen Expertise im Genom engineering, nur grundlegende Molekularbiologie Fähigkeiten. Forscher können nun bisher unlösbare Organismen mit ein paar alternative Mittel zur Genmanipulation3,4untersuchen. In den vergangenen fünf Jahren hat CRISPR Genom-Bearbeitung verwendet worden, mehr als 200 verschiedenen Wirbeltieren, Wirbellosen, Pflanze und mikrobielle Spezies zu entwickeln.
Adaptiert von der CRISPR prokaryotische Verteidigung Weg, die Kernelemente für standortspezifische Genom-Bearbeitung erforderlich sind das Cas9-Protein, in der Regel von S. Pyogenes und mit einer zusätzlichen nuklearen Lokalisierung Signal (NLS) und seinen spezialisierten Codon optimiert RNA Leitfaden5,6. Obwohl hier nicht behandelt, können andere Cas9 Orthologe oder CRISPR jedoch auch verwendet werden. Die natürlich vorkommenden gRNA besteht aus zwei separat transkribierten Stücke, die CRISPR-RNA (CrRNA) und der Trans-Aktivierung CrRNA (TracrRNA)7. Diese RNAs können in einer einzigen Abschrift, bekannt als die Single-Guide RNA (SgRNA)8fixiert werden. Die meisten Genom-Redakteure wählen den stromlinienförmigen SgRNA9, obwohl der Dual-Guide auch regelmäßig10,11verwendet. Experimentatoren 20-Nukleotid (nt) genomische DNA Ziel bestimmen, um sicherzustellen, dass es neben einer kurzen Lizenzierung Unterschrift erforderlich für Cas9 Anerkennung, genannt ein Protospacer angrenzenden Motiv (PAM), liegt und entwerfen eine gRNA, der komplementäre Sequenz12 enthält .
Einmal im Inneren der Zelle die RNP komplexe genomische Ziel lokalisiert, die gRNA Basenpaare mit dem komplementären DNA-Strang und dann das Enzym zerspaltet brechen beide DNA-Stränge zu einen Doppel-Strang zu generieren2. Handy-Reparatur-Mechanismus behebt die DSB durch eine von mindestens zwei Routen: über die fehleranfällige nicht-homologe Ende verbinden (NHEJ) Weg oder die Reparatur unter der Regie von Homologie (HDR), die nahtlos integriert DNA mit "Waffen" der Homologie zu beiden Seiten des Bruchs. Der ehemalige Reparatur Weg führt in der Regel zur Bildung von Indel und konsequente gen Störung, während Letzteres ermöglicht die Experimentatoren einfügen oder Ändern von DNA-Sequenzen1.
Die Bearbeitung Effizienz und Genauigkeit hängt die Mittel durch die Cas9 und gRNA in die Zelle eingeben. Diese Komponenten können an kultivierten Zellen, Embryonen oder Organismen in Form von Nukleinsäuren oder als eine vormontierte RNP komplexe13,14,15geliefert. Gemeinsamen Nukleinsäure-basierte Lieferung Methoden gehören die virale Transduktion, Transfektion oder Elektroporation von mRNA oder Plasmid DNA. Cas9 Protein und Guide RNA entstehen dann innerhalb der Zelle und sie verbinden, um einen Komplex bilden.
Die direkte Zustellung von RNP erfordert die separate Reinigung des Cas9 Proteins und Guide RNA. Dies kann intern oder das Protein und die SgRNA können aus einer von mehreren kommerziellen Anbietern erworben werden. Einmal erworben, werden die Cas9 und gRNA gemischt, um die enzymatisch zuständigen RNP-Komplex bilden und in Zellen durch direkte Injektion in befruchteten Eier/Embryonen, Lipid-basierte Transfektion16oder Elektroporation eingeführt. Der erste Bericht von RNP Bearbeitung beteiligten Injektion in C. Elegans Gonaden17. Mikroinjektion ist immer noch das bevorzugte Mittel der Embryonen und ganze Organismen RNP einzuführen, aber effektive Electroporation in18,19 und Ratte20 mausembryonen nachgewiesen wurde. Wir beschreiben Protokolle für die direkte Injektion RNP in C. Elegans Gonaden und p. Hawaiensis Embryonen und eine spezielle Art von Electroporation RNP liefern bei der Bearbeitung von primären humanen Zellen empfehlen. Diese Methode, Nucleofection, beinhaltet optimierte Elektroporation Programme und Zelle-spezifischen Lösungen und ermöglicht die RNP, Zytoplasma und Zellkern21einzugeben.
Genom-Bearbeitung mit RNP bietet einige deutliche Vorteile. Da die Proteine und RNA Komponenten vormontiert sind und Qualität vor der Auslieferung sichergestellt werden kann, vermeidet die RNP Bearbeitung viele Fallstricke, die Nukleinsäure-basierte Lieferung zugeordnet. Nämlich, besteht keine Gefahr der Cas9-kodierende DNA-Integration in das wirtsgenom mRNA ist nie für den Abbau ausgesetzt und es umgeht Probleme mit in Vivo gRNA oder Protein Ausdruck, Faltung und Assoziation22,23. Weiter, mit RNP führt um zu geringeren Toxizität und weit weniger Ziel Ereignisse als der Plasmid-basierten Ausdruck, ein Ergebnis der RNP kürzere Halbwertszeit im Inneren der Zelle24,25,26,27.
Zu guter Letzt führt RNP Bearbeitung nachweislich zu hohen Bearbeitung in einer Vielzahl von humanen Zelllinien, primäre Zellen wie Fibroblasten, embryonale Stammzellen (WSR), pluripotente Stammzellen (iSPCs), HSPCs induzierte, und T-16,24Zellen, 25,26,27,28,29; bei Wirbellosen wie C. Elegans, p. Hawaiensisund Fruchtfliegen3,17,30; in Wirbeltieren wie Zebrafisch, Mäuse und Ratten31,32; in Pflanzenarten Sie wie Arabidopsis, Tabak, Salat, Reis, Weinrebe, Apple, Mais und Weizen33,34,35,36; und in Chlamydomonas, Penicilliumund Candida Spezies37,38,39. Die Häufigkeit der Indel Bildung kann höher sein, bei der Verwendung von RNP im Vergleich zu den Plasmid-Lieferung und HDR-vermittelte DNA Einfügung kann einfacher sein,25,27,29zu erreichen.
Das hier beschriebene Protokoll nutzt die Cas9 RNP und ist eine effektive, leicht anpassbare Technik, die einfach anzuwenden, eine Vielzahl von biologischen Systemen40,41, vor allem in den Zellen, die sonst schwer zu arbeiten mit und in Organismen ohne etablierte Systeme für präzise Genmanipulation. Wir beginnen mit der Beschreibung wie zu entwerfen, zu erhalten und montieren die Cas9 RNP vor mit seiner Verwendung in ganz unterschiedlichen Modelltypen Zellen und Organismen. Hämatopoetischen Stammzellen/Progenitor Zellen (HSPCs) und T-Zellen bearbeitet werden, mit der gleichen Methode, Nucleofection, so dass sie zusammen in den Schritten 2 und 3 dieses Protokolls abgedeckt sind. Bearbeitung von Verfahren für C. Elegans werden beschrieben in den Schritten 4 und 5, und P. Bearbeiten von Hawaiensis fällt in den Schritten 6 und 7. Da der Erfolg eines Gens Bearbeitung Experiments in jedem Organismus durch Genotyp-Sequenzierung beurteilt werden kann, werden schließlich beschreibt mögliche Analysemethoden für alle Zellen und Organismen, die im Protokoll beschriebenen Teilschritte in Schritt 8 beschrieben.
Protocol
(1) RNP Assembly
-
Entwerfen Sie das Experiment im voraus, die RNA, DNA und Protein Komponenten vor der Zeit zu erwerben. Als einen ersten Durchlauf versuchen Sie eine der in Tabelle 1 aufgeführten positiven Kontrollen und verwenden Sie kommerziellen Reagenzien, die in der Tabelle Materialien beschrieben, um eine zuverlässige Versuchsanordnung und die Integrität des Materials zu gewährleisten. Zusätzliche Tipps zur Planung eines neuen Genom-Bearbeitung Experiments siehe Papiere auf diesem Thema12,42,43.
Hinweis: Nach der Montage wie in den nachfolgenden Schritten beschrieben, können RNPs im Voraus vorbereitet bei-80 ° c gelagert werden- Verwenden Sie nach der Wahl welche gen zum Ziel eine der kostenlosen Online-Tools, um eine optimale gRNA44,45,46,47,48zu entwerfen. Achten Sie darauf, ein Exon ausrichten, wenn in der Hoffnung, einen Knockout zu generieren.
Hinweis: Diese Werkzeuge helfen, ein Ziel-Site mit einer angrenzenden S. Pyogenes PAM identifizieren Sequenz, qualitativ hochwertige Score und geringe Ziel-Punktzahl. - S. Pyogenes Cas9 Proteins durch veröffentlichten Methoden8zu reinigen, oder von einem kommerziellen Anbieter zu kaufen.
- Bereiten Sie einen typischen Cas9-Puffer für die RNA-Verdünnung, RNP Vorbereitung und Protein-Lagerung, die 20 mM HEPES pH 7.5, 150 mM KCl, 10 % Glycerin und 1 mM DÄMMUND enthält. Verwenden Sie immer Nuklease-freies Wasser im Puffer, die verwendet werden, um Aufschwemmen oder verdünnen RNA um Abbau zu verhindern.
- Den Guide RNA (TracrRNA und CrRNA oder SgRNA) durch eine in-vitro- Transkription mit veröffentlichten Methoden zu produzieren, oder von einer Nukleinsäure-Synthese Unternehmen17,21,49, kaufen 50 , 51.
- Wenn ein Gen einfügen, synthetisieren oder kaufen ein Spender DNA-Vorlage.
- Die Proteine und RNA Aliquote bei-80 ° C und Tauwetter auf Eis unmittelbar vor der Anwendung zu speichern.
Hinweis: Jeder Frost-Tau-wechseln leicht senkt die Effizienz. Detaillierte, frei zugänglichen Protokolle für Cas9 Reinigung52 und die in-vitro- Transkription von SgRNAs53 sind an anderer Stelle zur Verfügung.
- Verwenden Sie nach der Wahl welche gen zum Ziel eine der kostenlosen Online-Tools, um eine optimale gRNA44,45,46,47,48zu entwerfen. Achten Sie darauf, ein Exon ausrichten, wenn in der Hoffnung, einen Knockout zu generieren.
- Wenn mit C. Elegansarbeiten, überspringen Sie Schritt 1.5. Überspringen Sie für das Protokoll p. Hawaiensis Schritt 1.6. Wenn Sie SgRNA verwenden, überspringen Sie Schritt 1.4. Fahren Sie mit Schritt 1.3 um eine gRNA für die Bearbeitung von Primärzelle zu montieren.
-
Montieren Sie eine gRNA durch Mischen äquimolaren Mengen von TracrRNA und CrRNA. 100 µL 80 µM gRNA Lager für etwa 50 Genom Bearbeitung Experimente zu machen.
- Die gRNA bei 37 ° C für 30 min inkubieren und dann langsam auf Raumtemperatur abkühlen lassen.
-
RNP Prep für HSPC und T Zelle bearbeiten: montieren eine komplexe RNP durch Mischen eine 1-2 X molaren Menge an gRNA zu 200 Pmol des Cas9-Proteins in einem Gesamtvolumen von 10 µL. sehr langsam hinzufügen konzentrierten Cas9 der gRNA (Pre-im Cas9-Puffer verdünnt) für ca. 30 s , macht schnell Kreise mit der Pipette, bringen die Endkonzentration Cas9 bis 20 µM.
- Bereiten Sie die Elektroporation Küvetten.
Hinweis: Dieses Protokoll ist speziell für das kommerzielle System gemäß der Tabelle der Materialien, aber RNP Bearbeitung kann auch mit anderen Geräten Elektroporation erzielt werden. - Jede Küvette 5 µL (100 Pmols, T-Zellen) oder 10 µL (200 Pmol, HSPCs) von RNP hinzufügen.
- Wenn hinzufügen einfügen neuen DNA, anstatt einen Knockout 1 µL von 100 µM (100 Pmol) einsträngige Oligonukleotid Spender DNA (SsODN)25,54,55 , Küvetten oder Vertiefungen der Platte.
- Überspringen Sie Schritt 2 für die nächsten Anweisungen in der Primärzelle Bearbeitung Protokoll.
- Bereiten Sie die Elektroporation Küvetten.
-
RNP Prep für die Bearbeitung von C. Elegans : montieren komplexe RNP durch Hinzufügen der folgenden Reagenzien um schaffen ein Finales Volumen von 20 µL (die Endkonzentrationen sind in Klammern angegeben): Cas9 (2 µM), HEPES pH 7,5 (10 µM), KCl (115 µM), CrRNA (12 µM) , TracrRNA (40 µM), und die Reparatur Vorlagen bei Bedarf (0,5 µM SsDNA oder bis zu 350 ng/µL DsDNA).
Hinweis: Die Effizienz einer Cas9-vermittelten DSB-templated Reparatur ist proportional zur Konzentration des Konstrukts DsDNA Reparatur; Also, je höher die Konzentration der Reparatur Vorlage, desto effizienter die vorgefertigten Reparatur. Eine Injektion von Mischungen mit mehr als 350 ng/µL DsDNA hat jedoch gezeigt worden, um die Lebensfähigkeit der injizierten Würmer zu reduzieren. Daher empfiehlt es sich, aber nicht mehr als 350 ng/µL DsDNA in die Mischung, um die Reparatur-Leistungsfähigkeit zu maximieren bei gleichzeitiger Minimierung der Letalität bis zu verwenden.- Fügen Sie mehrere CrRNAs gleichzeitig mehrere Loci abzielen, Bedarf für den co-CRISPR/co-Umwandlung Screening-Ansatz beschrieben in Schritt 5.4. Wenn Sie mehr als eine CrRNA hinzufügen, fügen Sie jeweils nacheinander auf die master-Mix.
Hinweis: Die Höhe der einzelnen CrRNA muss nicht identisch sein, und sogar verdoppelt die Gesamtkonzentration an CrRNAs in den master-Mix ohne Änderung der Konzentration der Cas9 scheint nicht zu stören, die Häufigkeit der Mutagenese an einen bestimmten Ort. Beispiele sind ausführlich in Paix Et Al. beschrieben. 56. - Mischen von pipettieren und drehen Sie die RNP-Lösung bei 16.000 x g für 5 s, um sicherzustellen, dass die Lösung an der Unterseite des Rohres gesammelt werden.
- Inkubieren Sie die Lösung bei 37 ° C für 15 m.
- Zentrifugieren Sie die Probe bei 16.000 x g für 1 min zu jeder Partikel Pellets, die die dünne gelangweilt Mikroinjektion Nadel verstopfen könnten. Verwenden Sie den überstand in den nachfolgenden Schritten.
- Überspringen Sie Schritt 4 für den Rest des Protokolls C. Elegans .
- Fügen Sie mehrere CrRNAs gleichzeitig mehrere Loci abzielen, Bedarf für den co-CRISPR/co-Umwandlung Screening-Ansatz beschrieben in Schritt 5.4. Wenn Sie mehr als eine CrRNA hinzufügen, fügen Sie jeweils nacheinander auf die master-Mix.
-
RNP Prep für die Bearbeitung von p. Hawaiensis : Einweg-Cas9 Aliquote durch das Verdünnen sie mit Nuklease-freies Wasser und Phenol-rot (zur Visualisierung von Injektionen), eine Endkonzentration von 6,25 µM Cas9 und 0,15 % Phenol rot vorzubereiten.
- Montieren Sie die RNP komplexe durch Mischen von 2-5 x molaren Überschuss an gRNA mit dem Cas9 Protein in einem Gesamtvolumen von 6 µl. hinzufügen 12 Pmol von Cas9, gRNA, 2 µM, gRNA Konzentration auf 4-8 µM und Phenol rot Konzentration 0,05 % die Endkonzentration Cas9 bringen.
- Inkubieren Sie der Mischung bei Raumtemperatur für 10 min bis hin zu komplexen die RNP.
- Fahren Sie mit Schritt 6 für die nächsten Anweisungen in der Bearbeitung von p. Hawaiensis Protokoll.
2. Handy-Kultur und Vorbereitung
Hinweis: Führen Sie Schritte 2.1.1 zu 3.3.3 in einem biologischen Sicherheitsschrank.
-
Kauf kryokonservierten Menschen mobilisiert peripherem Blut CD34+ HSPCs von einem Anbieter.
- Tauen Sie ~ 1 X106 HSPCs in einem 37 ° C Wasser Bad für 3 min und übertragen Sie sie auf eine 15 mL konische Rohr. Fügen Sie 10 mL Erweiterung serumfreien Medium aus eine kommerzielle Quelle hinzu und drehen Sie die Mischung bei 100 X g für 10 min. überstand zu entfernen und die Zellen in 2 mL ergänzt SFEM Aufschwemmen. Zellen im 6-Well Platten Teller und sie in einem 37 ° C Inkubator für 24-48 h vor der RNP Elektroporation Kultur.
- Zählen Sie die Zellen mit einem Hemocytometer und übertragen Sie die Gesamtzahl der HSPCs benötigt (150.000-200.000 HSPCs pro Küvette elektroporiert sein) auf einem Zentrifugenröhrchen.
- Drehen Sie das Rohr 100 X g für 10 min in die Zellen zu Pellets.
-
Menschlichen primären CD4 zu kaufen+ T-Zellen von einem Anbieter oder sie von menschlichen Vollblut durch die Dichte Gradienten Zentrifugation29isolieren.
- Vor der T-Zell-Aktivierung, Pre-Mantel Kultur 48-Well-Platten mit αCD3 (UCHT1) und αCD28 (CD28.2). Beschichten der Platten mit 500 µL 10 µg/mL αCD3 und 10 µg/mL αCD28 mit PBS-Puffer für mindestens 2 h bei 37 ° C.
Hinweis: Für einige Loci NHEJ erreicht werden ohne Pre-Stimulation, aber mit diesem Schritt seine Effizienz maximiert. - Kultur der T-Zellen für 48 h bei 37 ° C auf αCD3/αCD28 Antikörper gebundene Platten in einem RPMI komplette Medium [RPMI-1640 ergänzt mit 5 mM HEPES, kommerzielle Alternative zu L-Glutamin, 2 mM 50 µg/mL von Penicillin/Streptomycin, 50 µM 2-Mercaptoethanol, 5 mM nicht-essentiellen Aminosäuren, 5 mM Natrium Pyruvat und 10 % (Vol/Vol) FBS]. Kultur der T-Zellen bei einer Dichte von 2.000.000 T-Zellen in 500 µL Medien pro Bohrloch eine 48-Well-Platte.
- Anzahl der T-Zellen unter Verwendung eines Hemocytometer und Transfer experimentieren die Gesamtzahl der T-Zellen für die Elektroporation (100.000-1.000.000 T Zellen pro Küvette elektroporiert sein) zu einem Zentrifugenröhrchen.
- Drehen Sie das Rohr 90 X g für 8 min zu die Zellen zu Pellets. Wenn die Zellen Dichte Gradienten innerhalb von 2 Tagen getrennt wurden, drehen sie mit 200 X g für 8 min.
- Vor der T-Zell-Aktivierung, Pre-Mantel Kultur 48-Well-Platten mit αCD3 (UCHT1) und αCD28 (CD28.2). Beschichten der Platten mit 500 µL 10 µg/mL αCD3 und 10 µg/mL αCD28 mit PBS-Puffer für mindestens 2 h bei 37 ° C.
-
Für beide Zelltypen Aspirieren des Überstands mit einer Pipette/Vakuum, eventuell Luftblasen zu entfernen.
- Sanft Aufschwemmen der Zellen mit 20 µL der Elektroporation Puffer pro Küvette.
- Jede Küvette, die bereits 10 µL der RNP und Mix enthält auch durch pipettieren rauf und runter ohne Luftblasen fügen Sie 20 µL der Zellen (150.000-200.000 HSPCs oder 100.000-1.000.000 T-Zellen hinzu).
(3) RNP Electroporation
- Electroporate die Küvetten nach in eine Nucleofector ablegen. Verwenden Sie für die HSPCs den Puls Code ER100. Verwenden Sie für die T-Zellen den Puls Code EH-115.
-
HSPCs nur: Fügen Sie 100 µL ergänzt SFEM Medium (37 ° c erwärmt) für jede Küvette sofort nach Electroporation und lassen Sie die Zellen, für 10-15 min. erholen
- Übertragen Sie die Zellen auf Kultur in einer 96-Well Rundboden Platte und fügen Sie eine zusätzliche 100 µL des Mediums ergänzt SFEM für 24 h.
- Ändern sie in ein frisches ergänzt SFEM Medium und inkubieren sie für weitere 24-72 h.
- Entfernen Sie die Zellen für die Genotypisierung sie 48-96 h Post-Elektroporation. Drehen Sie die Zellen bei 300 X g für 5 min und entfernen Sie den überstand vor Beginn der DNA-Extraktion (Schritt 8.2).
-
T-Zellen nur: Fügen Sie 80 µL RPMI vervollständigen Nährmedien aus dem Reservoir für jede Küvette oder Brunnen, mit einer Multi-Kanal-Pipette (falls erforderlich) auf 37 ° C vorgewärmt.
- Inkubieren sie bei 37 ° C für 15 Minuten.
- Das Ziel Chicoréeblätter geeigneten Medien, Antikörper, Zytokine etc. hinzu und wärmen sie in einem Inkubator 37 ° C vor.
- Übertragen Sie 107 µL der elektroporiert Zellen aus den Brunnen auf einem Rundboden-96-Well-Platte mit einer Multi-Kanal-Pipette (falls erforderlich).
- Informationen zur Bewertung der Bearbeitung Ergebnisse fahren Sie mit Schritt 8.
4. C. Elegans Vorbereitung
-
1 Tag vor der Mikroinjektion: bereiten Sie die Agarose-Pads für die Mikroinjektion.
- Machen Sie eine 3 % ige (w/V) Agarose-Lösung in Wasser durch Zugabe von Agarose in Wasser und die Lösung zum Kochen auf einer heißen Platte oder in der Mikrowelle zu bringen.
- Arrangieren Sie 24 mm x 50 mm x 1,5 mm Deckglas Folien auf einem Tisch zu und verwenden Sie ein Glas Pasteurpipette, um einen kleinen (~ 15 µL) Tropfen Agarose-Lösung auf der Folie platzieren. Schnell abflachen der Agarose-Drop indem man ein anderes Deckglas an der Spitze. Lassen Sie die Agarose zu festigen und dann Entfernen eines der Deckgläsern.
- Lassen Sie die Agarose-beschichtete Deckglas bildoben auf einer Tischplatte über Nacht zu trocknen. Nach 24 h speichern Sie die Agarose-Pads in einem sauberen, trockenen Behälter.
Hinweis: Diese können unbegrenzt verwendet werden.
- Ziehen Sie die Mikroinjektion Nadeln: Filamente Borosilikatglas Kapillaren mit (Außendurchmesser 1,0 mm und innerer Durchmesser 0,58 mm), ziehen Sie die Nadeln basierend auf Mello und Feuer57 und andere Ressourcen-58. Die Nadeln können sofort verwendet werden oder können in einem sauberen, trockenen Behälter, verspannt durch Ton stützen gelagert werden.
- Für die Wartung der Würmer, bereiten eine Nematode Wachstum Medien (NGM) Agar in Petri Platten gegossen und mit OP50 Bakterien entdeckt (für Protokolle auf standard C. Elegans Wartung und Rezepte für Wachstummedia, siehe Stiernagle59).
- Die Würmer für die Mikroinjektion zu inszenieren: 12-24 h vor der Mikroinjektion wählen L4 inszeniert Hermaphroditen auf eine neue NG-Agar-Platte mit OP50 Bakterien und inkubieren sie über Nacht bei 20 ° C. Wählen Sie für jede Cas9 Ziel/Injektion Mischung ~ 30 Würmer auf den Teller.
-
Tag der Mikroinjektion: Laden die gezogenen Mikroinjektion Nadel mit der RNP überstand vorbereitete Lösung im Schritt 1.5.
- Pipette des Überstands von 1.5.4 Schritt in einer gezogenen Kapillare Pipette und Hinterfüllung der Lösung aus der Kapillare Pipette in die vorbereiteten Mikroinjektion Nadel (in der Regel weniger als 0,1 µL laden).
- Montieren Sie die geladene Nadel auf der Mikroinjektion Apparat mit einem Mikromanipulator verbunden. Legen Sie den Einspritzdruck Apparat auf 250 kPa und dem Gleichgewicht Druck bis 25 kPa.
-
Zurück zu brechen die geladene Nadelspitze zu eine Spitzen Nadel Kante erzeugen. Legen Sie eine 15 x 15 mm x 1,5 mm quadratisch Deckglas auf der Oberseite ein 24 mm x 50 mm x 1,5 mm Deckglas.
- Überlagern Sie ein Rand des quadratischen Deckglas mit Öl Halocarbon 700.
- Positionieren Sie die Nadel in das Öl am Rande der 15 mm quadratisch Deckglas.
- Mit der einen Hand, Führer der Mikroskoptisch und Deckglas, Bürsten Sie die Folie oben und entlang der Kante der Nadel während der Injektion Pedal/Taste. Brechen Sie die Nadelspitze zurück, Erhöhung des Durchflusses der Flüssigkeit aus der Nadel. Erzielen Sie eine optimale Durchflussmenge durch eine Injektion, die Strömung entlang der Kante der Nadel, bilden ~ 1 Blase/s verwechseln.
- Bestätigen Sie, dass die L4-Würmer nahm 12-24 h vor der Mikroinjektion Entwicklungsgeschichtlich inszenierten junge Erwachsene am Tag der Injektion. Wählen Sie die jungen Erwachsenen Würmer an eine NG-Agar-Platte, die OP50 Bakterien fehlt und es ermöglichen Sie ihnen, um für 5 min zu kriechen. Dies reduziert die Menge der Bakterien auf die Injektion Pad, Minimierung der Nadel Clogs übertragen.
- Legen Sie eine Agarose Injektion Pad/deckgläschen auf eine Dissektion Umfang. Mit einem Wurm-Pick, lag eine kleine Spur von Halocarbon Öl an einem Rand des Pads.
-
Heben Sie mit dem Wurm Pick in Öl beschichtet, mehrere Würmer aus der NG-Agar-Platte und in die Spur des Öls. Positionieren Sie mit einem feinem Haar befestigt, eine Pipette, wie z. B. eine Wimper oder Katze Whisker die Würmer in parallel, sanft schob die Würmer in der Agarose-Pad. Bis komfortabel mit der Mikroinjektion Verfahren, nur montieren und einem Wurm zu einem Zeitpunkt zu injizieren.
Hinweis: Die trockene Agarose wird die Feuchtigkeit von den Würmern, wodurch sie zur Einhaltung der Pad Docht. Folglich muss man schnell arbeiten wie die Würmer auszutrocknen können.- Einmal in position und angeschlossen an das Pad, überlagern die Würmer mit einem anderen paar Tropfen Öl Halocarbon (~ 20 µL) von der Spitze des Wurms zu wählen.
5. C. Elegans Gonade Mikroinjektion mit RNPs und nach der Injektion zu kümmern
Hinweis: Die Mikroinjektion Protokoll ist adaptiert von Mello und Feuer57und60,61an anderer Stelle ausführlich beschrieben.
-
Legen Sie das Deckglas mit den montierten Würmern auf die Injektion-Mikroskop. Positionieren Sie unter einer niedrigen Vergrößerung (5 X Objektiv, 10 X Okularen) die Würmer, die senkrecht auf der Injektionsnadel.
- Umstellung auf eine hohe Vergrößerung (40 X Objektiv, 10 X Okularen), positionieren Sie die Nadel direkt neben der Gonade-Arm, der Region in der Nähe von den Kernen in Mitte - zu spät-Pachytene entspricht.
- Mit dem Mikromanipulator, bewegen Sie die Nadel vor dem Wurm, die Nagelhaut etwas deprimierend. Mit einer Hand Tippen Sie auf der Seite der Mikroskoptisch, die Nadel durch die Kutikula Ruck. Injektion Pedal/Taste drücken und langsam die Gonade Arm mit der Injektion-Mischung füllen und entfernen Sie die Nadel.
- Wiederholen Sie diesen Schritt mit dem anderen Arm der Gonade.
-
Sobald die Würmer injiziert werden, entfernen Sie das Deckglas/Agarose-Pad und legen Sie es unter dem sezierenden Mikroskop.
- Mit einer gezogenen Kapillare Pipette, verdrängen Sie das Öl aus der Würmer durch pipettieren einen M9-Puffer über sie. Führen Sie diese Behandlung um die Würmer aus dem Agar freizugeben.
- Nach 10 min wenn die Würmer im Puffer um Prügel sind, verschieben Sie sie in eine NG-Agar-Platte mit OP50 Bakterien mit der gezogenen Kapillare Pipette. Legen Sie die Platte bei 20 ° C für ca. 2-3 h, bis die Würmer erholt haben und bewegen.
- Nach der Wiederherstellung einzeln die Würmer auf NG-Agarplatten mit OP50 und übertragen Sie die Platten in einem Inkubator 25 ° C.
-
Ermöglichen die P-0-Würmer wachsen und legen Nachkommen für 3 Tage injiziert. Bildschirm der F-1 -Nachwuchs.
- Wenn co-CRISPR oder CO-Konvertierung62,mit63,64,65, wählen Sie dann die Würmer Kandidat für das Screening basierend auf, ob sie die mutierten Phänotyp des Referenz-Gens. Individuell auf neue NG-Agarplatten mit OP50 übertragen Sie diese markierten Würmer und gestatten Sie ihnen, F2 Nachkommen lag bei 20 ° C.
Hinweis: Der Phänotyp für ein co-CRISPR-Screening oder Auswahl verwendet soll eine frühe Schätzung für den Erfolg des Cas9 zu bearbeiten. - Wenn der co-CRISPR-Phänotyp nicht vorhanden ist, microinject eine Positivkontrolle Plasmid, bei der Verbesserung der Effizienz der Mikroinjektion.
Hinweis: zum Beispiel unter anderem ein Plasmid in die Injektion-Mischung, die mCherry-Tags MYO-2 kodiert hilft die Injektion Effizienz bewerten. Würmer, die erfolgreich mit pCFJ90 injiziert haben einige Nachkommen mit fluoreszierenden Rachen.
- Wenn co-CRISPR oder CO-Konvertierung62,mit63,64,65, wählen Sie dann die Würmer Kandidat für das Screening basierend auf, ob sie die mutierten Phänotyp des Referenz-Gens. Individuell auf neue NG-Agarplatten mit OP50 übertragen Sie diese markierten Würmer und gestatten Sie ihnen, F2 Nachkommen lag bei 20 ° C.
- Untersuchen Sie die F-1 -Würmer auf das Vorhandensein der gewünschten Bearbeitungen. Wählen Sie die F-1 -Mutter zu einem einzelnen Brunnen einer 96-Well-Platte, lösen sie und prüfen Sie ihre DNA durch Einfügen-spezifische PCR Verstärkung, DNA-Sequenzanalyse oder Vermesser Nuklease Assay (CEL-1)66.
Hinweis: Diese Tests können durchgeführt werden, bei Verwendung eines co-CRISPR/co-Umwandlung oder anderen Screening oder Auswahl Regime65,66,67,68. - Informationen zur Bewertung der Bearbeitung Ergebnisse fahren Sie mit Schritt 8.
6. p. Hawaiensis Vorbereitung
- bereichern Sie 1 Tag vor der Mikroinjektion für die frühen Embryonen durch die Einrichtung von ein paar-Tank am Vorabend; neu getrennte Weibchen werden frisch befruchteten Embryonen enthalten. Vgl. Rehm Et al. 69 für Details.
- Am Tag der Mikroinjektion, sammeln die einzelligen Parhyale Embryonen (0-4 h nach Befruchtung) von trächtigen Weibchen mit 0,02 % Nelkenöl im Meerwasser zu betäuben und sanft kratzen die Embryonen aus ihrem ventralen Brut Beutel mit einer Flamme gezogen und Abgerundete Glaspipette und ein stumpf paar #3 Zangen.
7. p. Hawaiensis Embryo Mikroinjektion mit RNPs und nach der Injektion zu kümmern
- Oben beschriebenen Hinterfüllung zog Kapillarrohr mit ca. 1 µL des RNP-Injektion-Mix.
-
Verwenden Sie komprimiertem Stickstoff, um jedes Embryo microinject wie in Rehm Et Al. beschrieben 69.
- Die Parhyale Embryonen unter dem sezierenden Mikroskop mit einem Microinjector und einem Mikromanipulator zu injizieren. Laden Sie 1,5 µL der Injektion-Mix in den hinteren Teil eine gezogene Kapillarrohr (4 Zoll - 1,0 mm mit Fäden gezogen mit einer Mikropipette ziehen Apparat) mit einem Microloader-PIPETTENSPITZE.
- Richten Sie die Nadel auf der injektionsapparat und brechen Sie die Spitze der Nadel (eine sehr kleine Menge) mit einer Pinzette in den sezierenden Anwendungsbereich. Kalibrieren Sie das Volumen durch Einspritzen in Halocarbon Öl 700 und Messung des Durchmessers der Blase geliefert.
- Geschnitten Sie eine "Mulde" aus dem Härtemittel mit einer Rasierklinge. Mit Filter sterilisiert Meerwasser füllen Sie auf halbem Weg zu, und richten Sie die Parhyale Embryonen in den Trog zu stabilisieren.
- Spritzen Sie die Embryonen mit der Mikroinjektion Setup, jedes Embryo während der Injektion mit einer Zange zu stabilisieren. Verwenden Sie nach der Injektion eine Glaspipette Transfer, um die Embryonen übertragen, eine frische 60 mm Kulturschale auf halbem Weg mit Filter sterilisiert Meerwasser gefüllt.
-
Wenn die first Division in Form einer 2-Zell-Embryo (4-6 h nach Befruchtung) bereits aufgetreten ist, generieren Sie vollständig mutierte Tiere durch die Injektion von beiden Blastomeren. Um eine totale Spaltung von 2-Zellstadium sicherzustellen, Co injizieren Sie die Blastomeren mit FITC oder TRITC Dextran und beachten Sie, dass das Signal auf eine einzelne Blastomere unter einer Leuchtstofflampe sezieren Umfang nach der Injektion beschränkt.
- Alternativ zu generieren "Hälfte-Mutant" Tiere durch die Injektion nur eines der beiden Blastomeren im 2-Zell-Stadium (etwa geteilten links-rechts je nach Gewebe und Position entlang der Achse A-P).
- Eine Zelle in einem 8-Zellen-Embryo (7,5-9 h nach Befruchtung) zu beschränken, die Bearbeitung auf einem einlagigen Keim zu injizieren. Siehe Gerberding Et al. 70 für eine Karte der frühen Blastomere Linien.
-
Inkubieren Sie die Embryonen in 60 mm Kultur Gerichte (nicht mehr als 25 pro Schale), auf halbem Weg mit Filter sterilisiert Meerwasser, "Pre-Sauerstoff" mit einem Aquarium-Wäscher oder durch Schütteln kräftig gefüllt.
- Legen Sie die Gerichte von Embryonen in eine lose verschlossen Plasticware ausgekleidet mit feuchten Papiertüchern, Luftfeuchtigkeit und legen Sie sie in einem Inkubator 26 ° C mit einer 12 h Hell-Dunkel-Zyklus.
- Übertragen Sie die Überlebenden Embryonen sauberem Meerwasser Gerichte alle paar Tage.
Hinweis: Embryonen können bei Zimmertemperatur kultiviert werden, obwohl sie sehr viel langsamer entwickeln werden.
-
Zerlegen Sie und reparieren Sie die Embryonen in verschiedenen Stadien für eine Expressionsanalyse von in Situ Hybridisierung oder Antikörper Färbung (siehe Browne Et al. 71 für ein staging Führer und zusätzliche Referenzen für Dissektion und Fixierung72, in Situ Hybridisierung73und74Färbung Antikörper).
- Machen Dissektion Nadeln Einfädeln einem gebogenen Stück wolframdraht ca. 0,5 in Länge in das Ende eines Insulin-Nadel. Schärfen Sie die Nadel in Natronlauge unter Strom. Verwenden Sie eine 1 mL Spritze, als der Griff der Dissektion Nadel.
- Füllen Sie einen Brunnen von einer 3-Brunnen Glasschale auf halbem Weg mit einer frisch zubereiteten Lösung 9 Teile PEM Puffer (0,1 M Rohre pH 6,95, 2 mM der EGTA, 1 mM MgSO4), 1-Teil 10 X PBS und 1 Teil 32 % PFA. Legen Sie 3-5 Embryonen in die Schüssel und stecken Sie ein kleines Loch in jedes Embryo mit einer scharfen Wolfram-Nadel Poke und ein leicht abgestumpft um zu stabilisieren, so dass das Eigelb heraus fließen und das Fixiermittel in laufen.
- Mit einer geschärften Wolfram Nadeln, sanft zu necken entfernt die äußeren zwei Membranen umgebenden Parhyale Embryos. Zerlegen sie in Fixiermittel, die Embryonen robuster zu machen aber die Arbeit schnell zu verhindern, dass die Membran immer fest mit dem Embryo, die Membran Entfernung schwieriger macht. Lassen Sie die Embryonen für eine Gesamtmenge von 15-20 min. für Antikörper Färbung oder 40-50 min für in Situ Hybridisierung zu beheben.
- Bild live Jungtiere und für morphologische und Verhaltensstörungen Phänotypen zu analysieren oder zu beheben und für genauere Analysen zu beflecken. Erhöhen Sie die Jungtiere zur sexuellen Reife in 2-3 Monaten, Knockout und transgenen Linien (siehe Kontarakis und Pavlopoulos75 für Jungtier Pflege und andere nützliche Details) zu etablieren.
8. Bewertung bearbeiten Ergebnisse
- Falls zutreffend, suchen Sie eine optische oder funktionale Phänotyp in den bearbeiteten Zellen oder Organismen.
Hinweis: Wird dieser Prozess durch Anwendung unterschiedlich, und einige Beispiele sind am Ende ihrer entsprechenden Protokoll Schritte oben beschrieben. Analysieren Sie nach der Korrektur der Sichelzellen-Mutation in HSPCs die Hämoglobin-Herstellung von differenzierten Erythroblasts mittels HPLC (Abb. 1A). Ein Knockout von der IL-2-Rezeptor-Gens in T-Zellen kann durch Oberfläche Färbung bestätigt werden und flow Cytometry (Abbildung 1B). C. Elegans und p. Hawaiensis Phänotypen zu bewerten, beobachten Sie die tierischer Morphologie und Verhalten unter dem Licht oder fluoreszierende Mikroskop (Zahlen 1 und 1 D). - Um festzustellen, die Effizienz und die Art der genomischen Edits generiert, die Pools der bearbeiteten Zellen lysiert und ihre genomischen DNA mit einem kommerziellen Extraktion Kit21extrahieren.
-
Für eine schnelle Einschätzung der Indel Bildung, PCR-Verstärkung mindestens 200 Basenpaaren rund um den Schnitt vor Ort und führen eine T7-endonuclease1 (T7E1)76 oder Vermesser (CEL-1 Nuklease) assay77.
- Wenn eine Indel Formation auf Cas9 geschnittene Standort- oder erfolgreiche HDR erstellen oder eine bekannte Einschränkung-Website zu entfernen, erwägen Sie eine Beschränkung Enzym Verdauung, um die Bearbeitung Effizienz6zu schätzen. Die Beschränkung Fragment Länge Polymorphismus (RFLP) Assay kann eine komfortable Möglichkeit, die Effizienz zu prüfen, wenn es zur Verfügung stehen passiert.
- Für eine genaue Quantifizierung der Bearbeitung Effizienz und Bestimmung der vorherrschende Bearbeitung Ergebnisse, senden die PCR-Amplifikate für ein standard Sanger Sequenzierung mit vorwärts und rückwärts Primern.
Hinweis: Wenn einer einzigen Klon oder eines Organismus zu analysieren, ist die Analyse der Sanger Ergebnisse einfach, wie in Abbildung 2Agezeigt. Wenn einen Pool von Zellen zu analysieren, dann analysieren Sie die Chromatogramme mit dem Onlinetool78, wie in Abbildung 2Bdargestellt. - Führen Sie für eine vollständige Quantifizierung und Sequenzen der Bearbeitung der Ergebnisse Tiefe Sequenzierung27,54, wie in Abbildung 2Cdargestellt.
- Um einen bestimmten Satz von Ziel-Änderungen, PCR-verstärken die vorhergesagten aus Ziel-Sites zu beurteilen und zur NGS senden. Um den Nachweis von chromosomalen Translokationen ermöglichen, führen Sie GUIDE-Seq79 oder hohem Durchsatz, Translokation genomweite Sequenzierung (HTGTS)80. Führen Sie für ein vollständiges Bild der Ziel-Bearbeitungen in einer klonalen Population Sequenzierung des gesamten Genoms (WGS)81,82,83.
Hinweis: Gibt es eine Vielzahl von Methoden für die Quantifizierung - und off-Target Genom Bearbeitungen, erklärt weiter in verschiedenen Beitrag84,85,86Artikel.
Representative Results
Diese Experimente zeigen wie vormontierte Cas9 RNP verwendet werden, kann um die Genome der primäre Zellen und ganze Organismen zu verändern. Forscher reinigen oder kaufen Cas9 Protein und SgRNA, die zwei Komponenten des Komplexes Praegen und führen die RNP in ihren Zellen oder Organismus von Interesse. Wenn man genug Zeit für die Bearbeitung um entstehen und für die Nachkommen der nächsten Generation (falls zutreffend) geboren werden, zu überprüfen, für Phänotypen und/oder Zellen für die Genotypisierung zu sammeln. Phänotypen können über funktionelle Assays, Ausdruck-Assays, Visualisierung (nach Augenmaß oder mit Mikroskopie) oder andere Methoden, je nachdem das Experiment beobachtet werden.
Zum Beispiel können HSPCs, die bearbeitet wurden, um die β-Globin-Mutation zu korrigieren, die Sichelzellenanämie verursacht in Erythrozyten differenzieren und untersucht für die Produktion von gesunden oder Sichel Hämoglobin27,87 (Abbildung 1 A). T-Zellen bearbeitet, knock out High-Affinity IL-2 Rezeptor-Gens, CD25 (IL2RA), können von Oberfläche beflecken und Flow Cytometry88analysiert, und funktionell analysiert, um zu erkennen, eine Signalisierung Reaktion auf IL-2 Stimulation (Abbildung 1B ). T-Zellen können auch vielfach klinisch wichtig, die Bewertung der verschiedenen Phänotypen, einschließlich die Wirksamkeit der HIV-Infektion89 erfordern umprogrammiert werden und die in Vivo Antitumor-Wirksamkeit von CAR-T-Zellen11.
Mit einem co-CRISPR/co-Umwandlung Screening-Ansatz, sind C. Elegans Würmer gleichzeitig an zwei Loci62bearbeitet. HDR in das Dpy-10 Referenz-gen mit einem SsODN Reparatur vorlagenergebnisse in einer leicht erzielte dominante Dpy-10 Gain-of-Function-Mutation. Heterozygot F1dpy-10(gof) Tiere sind Roller (Rol) und homozygot dpy-10(gof) Tiere sind plump (Dpy). Das Vorhandensein des Phänotyps traten bei diesen Tieren zeigt, dass Cas9 die Bearbeitung und verbessert die Chancen eine Bearbeitung Veranstaltung an den zweiten Locus in Rol oder Dpy F1 Tiere zu identifizieren. 33-50 % der injizierten P0 Würmer mindestens 20 F1 -nachkommen, die Rol oder Dpy90nachgeben sollte ein erfolgreiche Bearbeitung Experiment führen. Es ist dann möglich, nicht-Rol Tiere, Dpy-10 in Wildtyp zurückzukehren, und wählen für die homozygot Bearbeiten von Interesse zu wählen. Als Faustregel gilt sollte die Hälfte die Konzentration der CrRNA für das co-CRISPR-Referenz-gen, dass von den CrRNA das Gen des Interesses gezielt. Wenn eine Bearbeitung im gen des Interesses nicht wiederhergestellt wird, können das Verhältnis der beiden CRISPR-RNAs erhöht die Wahrscheinlichkeit der Wiederherstellung der gewünschten Mutation angepasst werden. Zum Beispiel wird erhöhen des CrRNA für das Gen des Interesses bezogen auf die Referenz-gen CrRNA Erhöhung des Anteils der Würmer besitzen Bearbeitungen im gen des Interesses in der Bevölkerung von Würmern, die auch Änderungen bei den Referenz-gen-Locus besitzen. CO-Konvertierung Frequenzen variieren, aber die Preise sind in der Regel 20-60 %, oft nachgeben homozygote Bearbeitungen in der F-1 Generation (Abbildung 1C).
P. Hawaiensis Jungtiere, die bearbeitet wurden, um knock out des Abdominal-B -Gens (Abd-B) zeigen klare morphologischen Anomalien3 (Abbildung 1D). Dieses Gen ist erforderlich für richtige Bauch Musterung und seine Störung Ergebnisse beim thorakalen Typ springen und laufen Beine ersetzen die Schwimmen und Anker Beine, die in der Regel zu präsentieren, auf dem Bauch.
Ermittlung Genom Bearbeitung Ergebnisse der genotypischen Ebene erfordert Sequenzierung oder eine in-vitro- Assay, der Sequenz Änderungen erkennt. Hier zeigen wir repräsentative Sequenzierungsdaten von unserem Modell Zelltypen und Organismen, Hervorhebung der verschiedenen Ansätze zur Quantifizierung bearbeiten. Beachten Sie, dass die Figur Etiketten generalisiert sind, weil alle hier gezeigte Methoden für jedes biologische System angewendet werden können.
Sequenzierung-basierte Ansätze unterscheiden sich in technische Komplexität und Tiefe der Ergebnisse. Für klonale bearbeiteten Populationen oder leicht trennbar einzelner Organismen können bearbeitete Einzelpersonen nach genomische DNA-Extraktion sequenziert werden. Standardergebnisse Sanger-Sequenzierung werden die Sequenz-Änderung an der Cas9-Schnitt Stelle in einem bestimmten Individuum mit hypothetischen Frameshifts zeigen, die seine Funktion (Abb. 2A) stören würde. Das online-Tool für die Sequenzierung verwendet ist eine andere Sanger-Sequenzierung basierenden Ansatz, der die gemischte Bevölkerung, anstatt einzelne Mutanten78angewendet werden können. Sequenzen sind mit einem online-Tool analysiert, die Bearbeitung Gesamtwirkungsgrad sowie vorherrschende Sequenz Ergebnisse annähern können. Die repräsentativen Daten sind in Abbildung 2Bdargestellt.
Die gründlichste Sequenzierung hier beschriebene Methode ist Tiefe Sequenzierung (manchmal auch als Hochdurchsatz- oder Next-Generation Sequenzierung). Diese Methode bietet DNA-Sequenzen von individueller Genome in eine gemischte Bevölkerung. Diese Daten können in eine Vielzahl von Möglichkeiten veranschaulicht werden. Hier haben wir einzelne Sequenzierung liest aus bearbeiteten Zellen abhängig vom Bearbeitungs Ergebnis (Abbildung 2C) klassifiziert. Die meisten Zellen bearbeitet werden über den NHEJ Weg, was gewöhnlich in gen Störung führt. In anderen Fällen hat das Zielgen, für eine Alternative Version über HDR27getauscht.
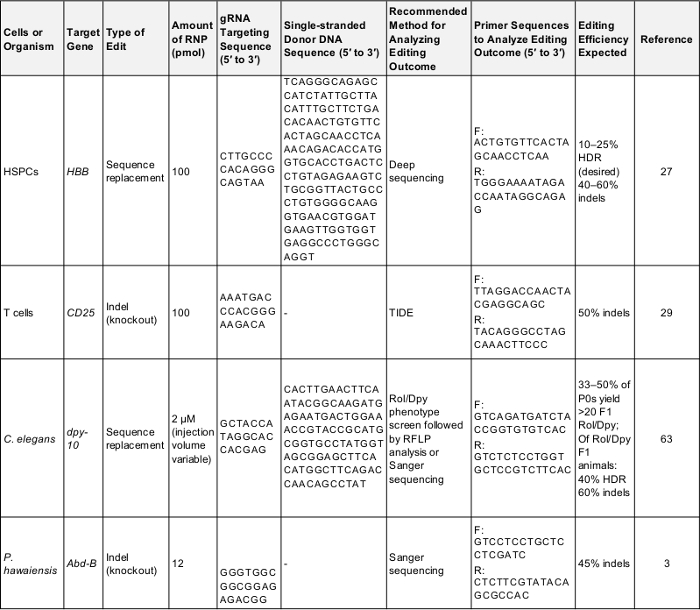
Tabelle 1: Positive steuert für vorläufige Genom Bearbeitung Experimente. Diese Tabelle zeigt die wichtige Informationen benötigt, um eine erstmalige Genom Bearbeitung Experiment in einzelnen Zellen und Organismen, die in diesem Protokoll beschrieben durchführen. Nach diesen Parametern ist wahrscheinlich um ein erfolgreiches Ergebnis zu erzielen, das verwendet werden können, um das Protokoll zu testen oder als Grundlage zum Vergleich einmal zielt des Experimentators ein Gen von ihrem eigenen Interesse. F: nach vorn, R: rückwärts, HDR: unter der Regie von Homologie Reparatur. Klicken Sie hier, um diese Tabelle herunterzuladen.

Abbildung 1 : Vertreter phänotypischen ergibt sich aus Cas9 RNP Bearbeitung von primären humanen Zellen und Organismen. (A) Dies ist eine HPLC-Ablaufverfolgung zeigt, dass nach erfolgreicher Genom Bearbeitung, HSPCs, die in fortgeschrittenen Erythroblasts unterschieden werden funktioneller Hämoglobin als Hämoglobin Sichel produzieren wird. Mutierte Erythrozyten erzeugen Sichel Hämoglobin (HbS), während erfolgreich bearbeitet Zellen gesund Hämoglobin (HbA und HbA2) sowie fetalen Hämoglobins (HbF) erzeugen werden. Die Extinktion ist in beliebigen Einheiten (au) grafisch dargestellt. Dieses Panel wurde erstmals veröffentlicht in DeWitt et al. 27. es ist Nachdruck mit freundlicher Genehmigung von der American Association for Advancement of Science. (B) auf der linken Seite, für jede Bedingung zeigt dieses Panel Flow Cytometry Daten zeigen, dass die Oberfläche gebeizt T-Zellen nicht ausdrücken CD25 nach CD25 gen mit RNP ausgeschlagen wurde. Die CD25 Fülle ist auf der x-Achse mit der Zellengröße auf der y-Achse aufgetragen. Auf der rechten Seite, für jede Bedingung zeigt dieses Fenster die Phospho-Stat5 (pStat5) Quantifizierung nach Induktion mit IL-2. Die Signalisierung wird reduziert, wenn der IL-2-Rezeptor fehlt (CD25 KO) ist. Die pStat5 Fülle ist auf der x-Achse aufgetragen und die Daten, die aus drei verschiedenen Ebenen der IL-2 Eingang vertikal verglichen. (C) dieses Panel zeigt einen Caenorhabditis Elegans co CRISPR/co Umwandlung Bildschirm gezielt Dpy-10 als Co-Konvertierung-Marker. Zwei guide RNAs Ziel zwei Loci, Dpy-10 und Ihre Lieblings-gen (Yfg), in der gleichen P-0-eingespritzte Tier. HDR bei Dpy-10 ergibt sich einen Rol oder Dpy Phänotyp. Die Auswahl der Rol-Dpy F1 Tiere erhöht die Chancen der Identifizierung von Änderungen an den zweiten Locus. (D) dieses Panel zeigt, dass Wildtyp Parhyale Hawaiensis Jungtiere normalen Bäuche mit Schwimmen und Anker Beine haben. Die Abd-B Knock-out-Jungtiere (F0 Personen) entwickeln ein Bauch in Richtung Brustkorb verwandelt. So sind Schwimmen und Ankern Beine verschwunden und ersetzt durch die springen und zu Fuß Beine mit einem normalen Thorax verbunden. Bitte klicken Sie hier für eine größere Version dieser Figur.

Abbildung 2 : Typische Ergebnisse vom Ergebnis-Analyse-Methoden zu bearbeiten. (A) dieses Panel zeigt Beispiele für die Sanger Sequenzierung Ergebnisse aus einzelnen F1 p. Hawaiensis Organismen, einschließlich der Wildtyp-Sequenz und drei verschiedenen Indels, die die Genfunktion stören durch die Verlagerung der offenen Leserahmen. (B) diese Flut Ergebnisse zeigen die Bandbreite der Einfügungen und löschen Ereignisse, die an einer Cas9-Ziel-Site in einem Pool von sequenzierten T-Zellen. Die x-Achse zeigt die Länge eines gegebenen Insertion oder Deletion im Nukleotide. (C) diese Tiefe Sequenzierung Ergebnisse zeigen keine Genom-Bearbeitung ohne Nucleofection oder gRNA und erfolgreiche Bearbeitung mit intakten Cas9 RNP, gruppiert nach DNA-Reparatur-Ergebnis in HSPCs. Bitte klicken Sie hier für eine größere Version dieser Figur.
Discussion
Aufbau einer robusten Genoms Bearbeitung Protokoll in einer Zelle Linie oder Organismus des Interesses erfordert die Optimierung und empirische Prüfung von mehreren wichtigen Parameter, in diesem Abschnitt behandelten. Versucht ein paar Variationen der hier vorgestellten allgemeine Ansätze ist ausdrücklich erwünscht. Die wesentliche Einschränkung dieses Protokolls ist, die Anwendung dieser Methoden auf andere Zellen oder Organismen führen zu einem anderen Ergebnis abhängig von der untersuchten Spezies und ein experimentelles Design, das führt zu einem hocheffizienten gen Knockout kann DNA Einfügung nicht fördern. Daher empfehlen wir, beginnend mit den Methoden, die hier vorgestellten und Fehlerbehebung, wie unten beschrieben.
Problembehandlung bei Genom Reagenz Qualität bearbeiten:
Generieren oder den Kauf hochwertiger Reagenzien ist ein entscheidender Schritt in jedem Genom Protokoll bearbeiten. Cas9 Protein kann gereinigt im Labor oder im Handel erworben werden. Viele Protokolle beachten Sie eine Endkonzentration für Cas9 im RNP Rezepte, aber das optimale gen Bearbeitung Aktivität hängt die spezifische Aktivität des einzelnen Cas9 Protein Vorbereitung, die je nach Quelle unterschiedlich ist. Sobald das Protokoll hier vorgestellten funktioniert, prüfen, optimieren die Höhe der RNP von Titration Cas9 Ebenen verwendet, um eine optimale Konzentration zu etablieren: eine hochspezifische Ziel DNA Dekolleté ohne unnötige Ziel Spaltung verursacht durch übermäßige Cas940.
Auch kann Guide RNA Reinheit und Homogenität Determinanten des Genoms Bearbeitung Erfolg22. Erworbene SgRNAs oder Einzelkomponenten für CrRNA und TracrRNA sind in der Regel qualitativ hochwertige Reagenzien und eine Vielzahl von chemischen Modifikationen stehen zur Verfügung, um Probleme mit der RNA-Abbau zu bekämpfen oder zusätzliche Funktionen, um die RNP91verleihen. Einige Gruppen haben viel höhere Effizienz mit solchen Reagenzien, Bearbeitung, so dass sie möglicherweise einen Versuch nach dem masteringprozess Wert und/oder wenn beobachtet, während chemisch modifiziert gRNAs möglicherweise nicht notwendig für standard Genom Bearbeitung Experimente, gRNA Abbau erscheint eine Ausgabe22,91. In-vitro- Transkription und anschließende Gel Reinigung ist eine kostengünstige Alternative, die für routinemäßige Genom Bearbeitung Experimente17,21,49,50ausreichend sein kann. Darüber hinaus mehrere Ansätze, die werden häufig angewendet, um homogene gRNA Populationen in Vivo, einschließlich Ribozym und tRNA-basierten Exzision der einzelnen Führer zu produzieren, kann verlängert werden in Vitro RNA Vorbereitung Reiniger generieren Produkte-92.
Leitfaden-RNA und DNA Spender design-Tipps:
Guide RNA Auswahl ist ein kritischer Faktor bei der Erreichung hocheffiziente zielgerichtet bearbeiten und gleichzeitig die Chancen der Ziel-Spaltung. Zur Reiseführer-Auswahl zu erleichtern, haben mehrere Studien Hochdurchsatz-Bildschirme gepaart mit Next Generation Sequencing Sequenz Merkmale der erfolgreichen Führer47,79,93,94Kompilieren verwendet, 95,96. Diese Features wurden zur prädiktive Algorithmen und online-Tools, die im Leitfaden Auswahl44,45,46,47,48zu entwickeln. Solche Algorithmen werden auf Bildschirmen mit DNA-basierten Systemen zur Anleitung RNA Ausdruck geerdet. Führungen sind mit einem Pol III-Promotor ausgedrückt, und ihr Ausdruck ist daher anfällig für die Grenzen verbunden mit Pol III Transkription, wie vorzeitige Beendigung, wenn Spuren von Uracil97,98auftreten, 99. Jedoch RNPs genutzt mit in-Vitro-synthetisierten Guide RNAs umgeht diese Bedenken und vereinfacht die Einschränkungen auf Reiseführer Design. Ein gemeinsames Merkmal, das von diesen Algorithmen entstanden und wurde in zahlreichen Studien mit hochwirksamen Genom-Bearbeitung, bestätigt ist das Vorhandensein von Purin, besonders ein Guanin, 3 ' Ende der Führer gezielt Sequenz. Dieses Feature "Leitfaden" hat zwischen den Organismen von Säugetieren bis hin zu C. Elegans, Fruchtfliegen und Zebrafisch65,100,101sehr erfolgreich. Darüber hinaus ist die Gestaltung Führer mit einem GG-Dinucleotide am 3 ' Ende der Guide targeting Region für C. Elegans, eine wirksame Strategie für die Vorhersage von hochwirksamen Guide RNAs65. Testen Sie im Idealfall mehrere Hilfslinien parallel zu bestimmen, welche für eine bestimmte Anwendung am erfolgreichsten ist.
Beim Versuch, eine DNA-Sequenz des Genoms einzuführen, ist das Design des Spenders oder Schablone DNA auch entscheidend. Einsträngige Oligonukleotid-Geber (SsODNs) sind zuverlässiger als andere typische Reparatur Vorlagen, linearen Doppelstrang und Plasmid DNA54,55,102eingefügt. An einigen Loci kann HDR Effizienz mit SsODNs verbessert werden, die komplementär zu den nicht-Zielorganismen oder Vertriebenen DNA-Strang und besitzen Homologie-Arme, die asymmetrische Länge27,55. Da die Reparatur Vorlage an der geschliffenen Stelle eingefügt werden ist und die gezielte Abfolge beinhaltet, müssen Schritte unternommen werden, um zu verhindern, dass Cas9 Spaltung des Spenders DNA vor oder nach dem genomischen einsetzen. Dies wird erreicht durch stille Mutationen an der PAM-Sequenz oder Samen Region, die Anerkennung von Cas9 unter Beibehaltung der Funktion des eingefügten Gene21,103zu vermeiden. Obwohl auch Einzel-Nukleotid-Änderungen an der PAM Bindung104abzuschaffen dürften, versuchen Sie, mindestens vier Nukleotide sicherheitshalber zu ändern.
Bedeutung und zukünftige Anwendungen:
Genom-Bearbeitung mit CRISPR-Cas9 ist eine leistungsfähige Methode ermöglicht einfache Genmanipulation von jedem Organismus entstanden. Bearbeitung mit Cas9 RNP braucht ein bisschen mehr Mühe am Anfang aber ist einfach zu bedienen, sobald Reagenzien und Protokolle in einem Labor hergestellt werden. Bearbeiten von Zellen mit vormontierten RNP anstelle von Plasmid DNA führt zu höhere allgemeine Bearbeitung Wirkungsgrade, einschließlich der schwer zu erreichen-gen Aufnahme über HDR, mit weniger Ziel Effekte24,25,26 , 27 , 29. Darüber hinaus Experimentatoren vermeiden Probleme mit Genexpression, RNA Abbau, Proteinfaltung und die Zuordnung zwischen gRNA und Cas9 Moleküle synthetisiert separat innerhalb der Zelle22,23. RNP Bearbeitung auch Sicherheitsbedenken über insertional Mutagenese umgeht und nachhaltig Ausdruck, die entstehen können, wenn virale Liefermethoden sind klinisch verwendet14. Aufgrund dieser Vorteile bevorzugen viele Wissenschaftler, die Durchführung von präklinischen, Proof-of-Concept-Experimente RNP Bearbeitung für menschliche therapeutische Anwendungen. In Vivo und Ex Vivo RNP-basierte Genom Bearbeitung Ansätze sind in der Entwicklung zu behandeln oder sogar heilen eine Vielzahl von Bedingungen von Erbkrankheiten wie Duchenne Muskeldystrophie105 und Sichelzellenanämie27 bis HIV-29 und Krebs11. Interessanterweise ist Cas9 RNP zunehmend als eine Lieferung Methode für Landtechnik, weil dadurch der Pflanzen33,34,36bearbeiten "DNA-frei" beschäftigt.
Disclosures
Die Autoren Alexander Marson und Jacob E. Corn sind Mitbegründer von Spotlight Therapeutika. Jacob E. Corn ist ein Berater Mission Therapeutika und seinem Labor hat gesponserte Forschungsunterstützung von AstraZeneca und Pfizer erhalten. Alexander Marson ist Berater für Juno Therapeutics und Pakt Therapeutika und seinem Labor erhielt gesponserten Forschungsförderung von Juno Therapeutics, Epinomics und Sanofi. Sein Labor hat auch Patente im Zusammenhang mit Cas9 RNP Technologie beantragt.
Acknowledgments
Wir danken vielen bisherigen Mitglieder unseres Labors und der Bay Area Genom Bearbeitung Gemeinschaft für ihre Beiträge zur Entwicklung dieser Methoden. Wir danken Ross Wilson für kritisch Lesen dieses Manuskript.
Alexander Marsons Forschung stützt sich auf ein Geschenk von Jake Aronov und eine National Multiple Sclerosis Society gewähren (CA 1074-A-21). Alexander Marson hält den Career Award für Mediziner aus dem Burroughs Wellcome Fonds und ist ein Chan Zuckerberg Biohub Ermittler. Jacob E. Corn Forschung unterstützt Li Ka Shing Foundation, das Erbe medizinische medizinische Forschungsinstitut und California Institute für die Regenerationsmedizin. Behnom Farboud und Barbara J. Meyer der Forschung ist Teil von NIGMS Grant R01 GM030702, Barbara J. Meyer, finanziert, die ein Ermittler des Howard Hughes Medical Institute ist. Erin Jarvis und Nipam H. Patel Forschung wird teilweise durch die NSF Bewilligung IOS-1257379 finanziert und Erin Jarvis bestätigt Unterstützung durch eine NSF GRFP und ein Philomathia Graduate Fellowship.
Materials
| Name | Company | Catalog Number | Comments |
| Reagents/Materials | |||
| DNA oligonucleotides | Integrated DNA Technologies | - | IDT will provide custom DNA sequences, including those in Table 1 |
| Guide RNAs | Synthego | - | Synthego will provide high-quality sgRNAs for S. pyogenes Cas9, including custom sgRNAs containing the targeting sequences included in Table 1 |
| Purified Cas9 protein (EnGen Cas9 NLS, S. pyogenes) | New England Biosciences | M0646T | If possible, purifying Cas9 in-house or purchasing from local core facilities is a less expensive option |
| Normal peripheral blood CD34+ stem/progenitor cells | AllCells | PB032-2 | |
| StemSpan SFEM | StemCell Technologies | 09650 | |
| StemSpan CC110 | StemCell Technologies | 02697 | |
| P3 Primary Cell 4D-Nucleofector X Kit | Lonza | V4XP-3032 | |
| RPMI-1640 Medium, With sodium bicarbonate, without L-glutamine, liquid | Sigma | R0883-6X500ML | |
| EasySep™ Human T Cell Isolation Kit | Stemcell | 17951 | |
| cell culture plate, 96 wells, round | Fisher Scientific | 3799 | |
| CTS (Cell Therapy Systems) Dynabeads CD3/CD28 | Life Tech | 40203D | |
| Reombinant Human IL-2 | UCSF Pharmacy | NA | |
| SepMate-50 500-pack IVD | Stemcell Technologies | 85460 | |
| OP50 Escherichia coli | Caenorhabditis Genetics Center | OP-50 | https://cgc.umn.edu/ |
| Nematode Growth Media agar in petri dishes | - | - | See Stiernagle, T (ref. 59) |
| Standard borosilicate glass capillaries with filament: 4 in (100 mm), 1/0.58 OD/ID | World Precision Instruments | 1B100F-4 | |
| Single-barrel standard borosilicate glass capillaries: 6 in (152 mm), 2/1.12 OD/ID | World Precision Instruments | 1B200-6 | |
| Cover glass; 24 × 50 mm | Thermo Fisher Scientific | 12-544E | |
| Cover glass; 22 × 22 mm | Thermo Fisher Scientific | 12-518-105K | |
| Apex LE agarose | Genesee Scientific | 20-102 | |
| Halocarbon oil 700 | Sigma-Aldrich | H8898-100ML | |
| pCFJ90 plasmid | Addgene | 19327 | |
| Compressed nitrogen | - | ||
| 60 mM culture dishes | BD | ||
| Capillary tubes with filament: 4 in (1.0 mm) | World Precision Instruments | T2100F-4 | |
| Sylgard 184 | Dow Corning | ||
| Petri dishes (100 × 15 mm) | - | ||
| Tungsten wire (0.005 in. diameter) | Ted Pella | ||
| Perfluoroalkoxy alkane (PFA) | - | ||
| Marine salt | - | ||
| 9" pasteur pipettes | - | ||
| Phenol red | - | ||
| Nuclease-free water | - | ||
| Equipment | |||
| 4D Nucleofector | Lonza | AAF-1002X | |
| MZ75 Stereomicroscope | Leica | Out-of-production. Current model is the M80 Stereomicroscope | |
| Axio Vert35 inverted phase contrast fluorescent microscope | Zeiss | Out-of-production. Current model is the Axio VertA.1 | |
| Laser-based micropipette puller (for C. elegans protocol) | Sutter Instrument | FG-P2000 | |
| Picoliter Microinjector (for C. elegans protocol) | Warner Instruments | PLI-100A | |
| Three-axis Joystick oil hydraulic micromanipulator | Narishige International | MO-202U | |
| Coarse manipulator | Narishige International | MMN-1 | |
| Micropipette puller (for P. hawaiensis protocol) | Sutter Instrument | P-80/PC | |
| Microinjector (for P. hawaiensis protocol) | Narishige | IM300 | |
| Microloader pipette tips | Eppendorf | 5242956003 | |
| NG-agar |
References
- Komor, A. C., Badran, A. H., Liu, D. R. CRISPR-based technologies for the manipulation of eukaryotic genomes. Cell. , 1-17 (2016).
- Barrangou, R., Horvath, P. A decade of discovery: CRISPR functions and applications. Nature Microbiology. 2, 1-9 (2017).
- Martin, A., Serano, J. M., et al. CRISPR/Cas9 mutagenesis reveals versatile roles of Hox genes in crustacean limb specification and evolution. Current Biology. 26 (1), 14-26 (2016).
- Goldstein, B., King, N. The future of cell biology: emerging model organisms. Trends in Cell Biology. 26 (11), 818-824 (2016).
- Mali, P., Yang, L., et al. RNA-guided human genome engineering via Cas9. Science. 339 (6121), 823-826 (2013).
- Cong, L., Ran, F. A., et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 339 (6121), 819-823 (2013).
- Deltcheva, E., Chylinski, K., et al. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature. 471 (7340), 602-607 (2011).
- Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J. A., Charpentier, E. A Programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 337 (6096), 816-821 (2012).
- Nowak, C. M., Lawson, S., Zerez, M., Bleris, L. Guide RNA engineering for versatile Cas9 functionality. Nucleic Acids Research. 44 (20), 9555-9564 (2016).
- Jiang, W., Cox, D., Zhang, F., Bikard, D., Marraffini, L. A. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nature Biotechnology. , 1-9 (2013).
- Rupp, L. J., Schumann, K., et al. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Scientific Reports. 7 (1), 737 (2017).
- Graham, D. B., Root, D. E. Resources for the design of CRISPR gene editing experiments. Genome Biology. 16, 260 (2015).
- Wang, H., La Russa, M., Qi, L. S. CRISPR/Cas9 in genome editing and beyond. Annual Review of Biochemistry. 85, 2270 (2016).
- Nelson, C. E., Gersbach, C. A. Engineering delivery vehicles for genome editing. Annual Review of Chemical and Biomolecular Engineering. 7, 637-662 (2016).
- Yin, H., Kauffman, K. J., Anderson, D. G. Delivery technologies for genome editing. Nature Reviews Drug Discovery. 16 (6), 387-399 (2017).
- Zuris, J. A., Thompson, D. B., et al. Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nature Biotechnology. 33 (1), 73-80 (2015).
- Cho, S. W., Lee, J., Carroll, D., Kim, J. -S., Lee, J. Heritable gene knockout in Caenorhabditis elegans by direct injection of Cas9-sgRNA ribonucleoproteins. Genetics. 195 (3), 1177-1180 (2013).
- Wang, W., Kutny, P. M., et al. Delivery of Cas9 protein into mouse zygotes through a series of electroporation dramatically increases the efficiency of model creation. Journal of Genetics and Genomics. 43 (5), 319-327 (2016).
- Chen, S., Lee, B., Lee, A. Y. -F., Modzelewski, A. J., He, L. Highly efficient mouse genome editing by CRISPR ribonucleoprotein electroporation of zygotes. Journal of Biological Chemistry. 291 (28), 14457-14467 (2016).
- Remy, S., Chenouard, V., et al. Generation of gene-edited rats by delivery of CRISPR/Cas9 protein and donor DNA into intact zygotes using electroporation. Scientific Reports. 7 (1), 16554 (2017).
- DeWitt, M. A., Corn, J. E., Carroll, D. Genome editing via delivery of Cas9 ribonucleoprotein. Methods. , 1-7 (2017).
- Hendel, A., Bak, R. O., et al. Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nature Biotechnology. 33 (9), 985-989 (2015).
- Thyme, S. B., Akhmetova, L., Montague, T. G., Valen, E., Schier, A. F. Internal guide RNA interactions interfere with Cas9-mediated cleavage. Nature Communications. 7, 11750 (2016).
- Kim, S., Kim, D., Cho, S. W., Kim, J., Kim, J. -S. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Research. 24 (6), 1012-1019 (2014).
- Lin, S., Staahl, B. T., Alla, R. K., Doudna, J. A. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. eLife. 3, 04766 (2014).
- Liang, X., Potter, J., et al. Rapid and highly efficient mammalian cell engineering via Cas9 protein transfection. Journal of Biotechnology. 208, 44-53 (2015).
- DeWitt, M. A., Magis, W., Bray, N. L., Wang, T. Selection-free genome editing of the sickle mutation in human adult hematopoietic stem/progenitor cells. Science Translational Medicine. 8 (360), (2016).
- Ramakrishna, S., Kwaku Dad, A. -B., Beloor, J., Gopalappa, R., Lee, S. -K., Kim, H. Gene disruption by cell-penetrating peptide-mediated delivery of Cas9 protein and guide RNA. Genome Research. 24 (6), 1020-1027 (2014).
- Schumann, K., Lin, S., et al. Generation of knock-in primary human T cells using Cas9 ribonucleoproteins. Proceedings of the National Academy of Sciences of the United States of America. 112 (33), 10437-10442 (2015).
- Lee, J. -S., Kwak, S. -J., et al. RNA-guided genome editing in Drosophila with the purified Cas9 protein. G3: Genes, Genomes, Genetics (Bethesda, MD). 4 (7), 1291-1295 (2014).
- Sung, Y. H., Kim, J. M., et al. Highly efficient gene knockout in mice and zebrafish with RNA-guided endonucleases. Genome Research. 24 (1), 125-131 (2014).
- Menoret, S., De Cian, A., et al. Homology-directed repair in rodent zygotes using Cas9 and TALEN engineered proteins. Scientific Reports. 5, 14410 (2015).
- Woo, J. W., Kim, J., et al. DNA-free genome editing in plants with preassembled CRISPR-Cas9 ribonucleoproteins. Nature Biotechnology. 33 (11), 1162-1164 (2015).
- Malnoy, M., Viola, R., et al. DNA-free genetically edited grapevine and apple protoplast using CRISPR/Cas9 ribonucleoproteins. Frontiers in Plant Science. 7, 1904 (2016).
- Svitashev, S., Schwartz, C., Lenderts, B., Young, J. K., Mark Cigan, A. Genome editing in maize directed by CRISPR-Cas9 ribonucleoprotein complexes. Nature Communications. 7, 13274 (2016).
- Liang, Z., Chen, K., et al. Efficient DNA-free genome editing of bread wheat using CRISPR/Cas9 ribonucleoprotein complexes. Nature Communications. 8, 14261 (2017).
- Shin, S. -E., Lim, J. -M., et al. CRISPR/Cas9-induced knockout and knock-in mutations in Chlamydomonas reinhardtii. Scientific Reports. 6, 27810 (2016).
- Pohl, C., Kiel, J. A. K. W., Driessen, A. J. M., Bovenberg, R. A. L., Nygård, Y. CRISPR/Cas9 based genome editing of Penicillium chrysogenum. ACS Synthetic Biology. 5 (7), 754-764 (2016).
- Grahl, N., Demers, E. G., Crocker, A. W., Hogan, D. A. Use of RNA-protein complexes for genome editing in non-albicans Candida species. mSphere. 2 (3), (2017).
- Rivera-Torres, N., Kmiec, E. B. A standard methodology to examine on-site mutagenicity as a function of point mutation repair catalyzed by CRISPR/Cas9 and ssODN in human cells. Journal of Visualized Experiments. (126), (2017).
- Nandal, A., Mallon, B., Telugu, B. P. Efficient generation and editing of feeder-free IPSCs from human pancreatic cells using the CRISPR-Cas9 system. Journal of Visualized Experiments. (129), (2017).
- Mohr, S. E., Hu, Y., Ewen-Campen, B., Housden, B. E., Viswanatha, R., Perrimon, N. CRISPR guide RNA design for research applications. The FEBS Journal. 283 (17), 3232-3238 (2016).
- Bauer, D. E., Canver, M. C., Orkin, S. H. Generation of genomic deletions in mammalian cell lines via CRISPR/Cas9. Journal of Visualized Experiments. (95), e52118 (2015).
- Hsu, P. D., Scott, D. A., et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nature Biotechnology. 31 (9), 827-832 (2013).
- Heigwer, F., Kerr, G., Boutros, M. E-CRISP: fast CRISPR target site identification. Nature Methods. 11 (2), 122-123 (2014).
- Moreno-Mateos, M. A., Vejnar, C. E., et al. CRISPRscan: designing highly efficient sgRNAs for CRISPR-Cas9 targeting in vivo. Nature Methods. 12 (10), 982-988 (2015).
- Labun, K., Montague, T. G., Gagnon, J. A., Thyme, S. B., Valen, E. CHOPCHOP v2: a web tool for the next generation of CRISPR genome engineering. Nucleic Acids Research. 44, 272-276 (2016).
- Haeussler, M., Schönig, K., et al. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biology. 17 (1), 148 (2016).
- Lo, T. -W., Pickle, C. S., et al. Precise and heritable genome editing in evolutionarily diverse nematodes using TALENs and CRISPR/Cas9 to engineer insertions and deletions. Genetics. 195 (2), 331-348 (2013).
- Bassett, A., Liu, J. -L. CRISPR/Cas9 mediated genome engineering in Drosophila. Methods. 69 (2), 128-136 (2014).
- Prior, H., Jawad, A. K., MacConnachie, L., Beg, A. A. Highly efficient, rapid and co-CRISPR independent genome editing in Caenorhabditis elegans. G3: Genes, Genomes, Genetics. , Bethesda, MD. (2017).
- Hirsh, A. Cas9 expression and purification protocol. protocols.io. , (2017).
- DeWitt, M. A., Wong, J. In vitro transcription of guide RNAs. protocols.io. , (2017).
- Yang, L., Guell, M., et al. Optimization of scarless human stem cell genome editing. Nucleic Acids Research. 41 (19), 9049-9061 (2013).
- Richardson, C. D., Ray, G. J., DeWitt, M. A., Curie, G. L., Corn, J. E. Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nature Biotechnology. 34 (3), 339-344 (2016).
- Paix, A., Folkmann, A., Seydoux, G. Precision genome editing using CRISPR-Cas9 and linear repair templates in C. elegans. Methods. 121-122, 86-93 (2017).
- Mello, C., Fire, A. DNA transformation. Methods in Cell Biology. 48, 451-482 (1995).
- Sutter Pipette Cookbook. , Available from: https://www.sutter.com/PDFs/pipette_cookbook.pdf (2017).
- Stiernagle, T. Maintenance of C. elegans. WormBook: the online review of C. elegans biology. , (2006).
- Evans, T. C. Transformation and microinjection. WormBook: the online review of C. elegans biology. , (2006).
- Berkowitz, L. A., Knight, A. L., Caldwell, G. A., Caldwell, K. A. Generation of stable transgenic C. elegans using microinjection. Journal of Visualized Experiments. (18), (2008).
- Kim, H., Ishidate, T., et al. A co-CRISPR strategy for efficient genome editing in Caenorhabditis elegans. Genetics. 197 (4), 1069-1080 (2014).
- Arribere, J. A., Bell, R. T., Fu, B. X. H., Artiles, K. L., Hartman, P. S., Fire, A. Z. Efficient marker-free recovery of custom genetic modifications with CRISPR/Cas9 in Caenorhabditis elegans. Genetics. 198 (3), 837-846 (2014).
- Ward, J. D. Rapid and precise engineering of the Caenorhabditis elegans genome with lethal mutation co-conversion and inactivation of NHEJ repair. Genetics. 199 (2), 363-377 (2015).
- Farboud, B., Meyer, B. J. Dramatic enhancement of genome editing by CRISPR/Cas9 through improved guide RNA design. Genetics. 199 (4), 959-971 (2015).
- Wood, A. J., Lo, T. -W., et al. Targeted genome editing across species using ZFNs and TALENs. Science. 333 (6040), 307 (2011).
- Friedland, A. E., Tzur, Y. B., Esvelt, K. M., Colaiácovo, M. P., Church, G. M., Calarco, J. A. Heritable genome editing in C. elegans via a CRISPR-Cas9 system. Nature Methods. 10 (8), 741-743 (2013).
- Dickinson, D. J., Ward, J. D., Reiner, D. J., Goldstein, B. Engineering the Caenorhabditis elegans genome using Cas9-triggered homologous recombination. Nature Methods. 10 (10), 1028-1034 (2013).
- Rehm, E. J., Hannibal, R. L., Chaw, R. C., Vargas-Vila, M. A., Patel, N. H. Injection of Parhyale hawaiensis blastomeres with fluorescently labeled tracers. Cold Spring Harbor Protocols. 2009 (1), (2009).
- Gerberding, M., Browne, W. E., Patel, N. H. Cell lineage analysis of the amphipod crustacean Parhyale hawaiensis reveals an early restriction of cell fates. Development (Cambridge, England). 129 (24), 5789-5801 (2002).
- Browne, W. E., Price, A. L., Gerberding, M., Patel, N. H. Stages of embryonic development in the amphipod crustacean, Parhyale hawaiensis. Genesis. 42 (3), New York, NY. 124-149 (2005).
- Rehm, E. J., Hannibal, R. L., Chaw, R. C., Vargas-Vila, M. A., Patel, N. H. Fixation and dissection of Parhyale hawaiensis embryos. Cold Spring Harbor Protocols. 2009 (1), (2009).
- Rehm, E. J., Hannibal, R. L., Chaw, R. C., Vargas-Vila, M. A., Patel, N. H. In situ hybridization of labeled RNA probes to fixed Parhyale hawaiensis embryos. Cold Spring Harbor Protocols. 2009 (1), (2009).
- Rehm, E. J., Hannibal, R. L., Chaw, R. C., Vargas-Vila, M. A., Patel, N. H. Antibody staining of Parhyale hawaiensis embryos. Cold Spring Harbor Protocols. 2009 (1), (2009).
- Kontarakis, Z., Pavlopoulos, A. Transgenesis in non-model organisms: the case of Parhyale. Methods in Molecular Biology. 1196, Clifton, NJ. 145-181 (2014).
- Kim, H. J., Lee, H. J., Kim, H., Cho, S. W., Kim, J. -S. Targeted genome editing in human cells with zinc finger nucleases constructed via modular assembly. Genome Research. 19 (7), 1279-1288 (2009).
- Qiu, P., Shandilya, H., D'Alessio, J. M., O'Connor, K., Durocher, J., Gerard, G. F. Mutation detection using Surveyor nuclease. BioTechniques. 36 (4), 702-707 (2004).
- Brinkman, E. K., Chen, T., Amendola, M., van Steensel, B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Research. 42 (22), 168 (2014).
- Tsai, S. Q., Zheng, Z., et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nature Biotechnology. 33 (2), 187-197 (2015).
- Frock, R. L., Hu, J., Meyers, R. M., Ho, Y. -J., Kii, E., Alt, F. W. Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nature Biotechnology. 33 (2), 179-186 (2015).
- Smith, C., Gore, A., et al. Whole-genome sequencing analysis reveals high specificity of CRISPR/Cas9 and TALEN-based genome editing in human iPSCs. Cell Stem Cell. 15 (1), 12-13 (2014).
- Veres, A., Gosis, B. S., et al. Low incidence of off-target mutations in individual CRISPR-Cas9 and TALEN targeted human stem cell clones detected by whole-genome sequencing. Cell Stem Cell. 15 (1), 27-30 (2014).
- Kim, D., Bae, S., et al. Digenome-seq: genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nature Methods. 12 (3), 237-243 (2015).
- Hendel, A., Fine, E. J., Bao, G., Porteus, M. H. Quantifying on- and off-target genome editing. Trends in Biotechnology. 33 (2), 132-140 (2015).
- O'Geen, H., Yu, A. S., Segal, D. J. How specific is CRISPR/Cas9 really. Current Opinion in Chemical Biology. 29, 72-78 (2015).
- Tsai, S. Q., Joung, J. K. Defining and improving the genome-wide specificities of CRISPR-Cas9 nucleases. Nature Reviews Genetics. 17 (5), 300-312 (2016).
- Hoban, M. D., Cost, G. J., et al. Correction of the sickle cell disease mutation in human hematopoietic stem/progenitor cells. Blood. 125 (17), 2597-2604 (2015).
- Simeonov, D. R., Gowen, B. G., et al. Discovery of stimulation-responsive immune enhancers with CRISPR activation. Nature. , (2017).
- Hultquist, J. F., Schumann, K., et al. A Cas9 ribonucleoprotein platform for functional genetic studies of HIV-host interactions in primary human T cells. Cell Reports. 17 (5), 1438-1452 (2016).
- Paix, A., Wang, Y., et al. Scalable and versatile genome editing using linear DNAs with microhomology to Cas9 sites in Caenorhabditis elegans. Genetics. 198 (4), 1347-1356 (2014).
- Lee, K., Mackley, V. A., et al. Synthetically modified guide RNA and donor DNA are a versatile platform for CRISPR-Cas9 engineering. eLife. 6, (2017).
- Minkenberg, B., Wheatley, M., Yang, Y. CRISPR/Cas9-enabled multiplex genome editing and its application. Progress in Molecular Biology and Translational Science. 149, 111-132 (2017).
- Doench, J. G., Fusi, N., et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nature Biotechnology. 34 (2), 184-191 (2016).
- Doench, J. G., Hartenian, E., et al. Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nature Biotechnology. , 1-8 (2014).
- Liu, H., Wei, Z., Dominguez, A., Li, Y., Wang, X., Qi, L. S. CRISPR-ERA: a comprehensive design tool for CRISPR-mediated gene editing, repression and activation. Bioinformatics (Oxford, England). 31 (22), 3676-3678 (2015).
- Wu, X., Scott, D. A., et al. Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nature Biotechnology. 32 (7), 670-676 (2014).
- Bogenhagen, D. F., Brown, D. D. Nucleotide sequences in Xenopus 5S DNA required for transcription termination. Cell. 24 (1), 261-270 (1981).
- Cozzarelli, N. R., Gerrard, S. P., Schlissel, M., Brown, D. D., Bogenhagen, D. F. Purified RNA polymerase III accurately and efficiently terminates transcription of 5S RNA genes. Cell. 34 (3), 829-835 (1983).
- Chen, B., Gilbert, L. A., et al. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell. 155 (7), 1479-1491 (2013).
- Gagnon, J. A., Valen, E., et al. Efficient mutagenesis by Cas9 protein-mediated oligonucleotide insertion and large-scale assessment of single-guide RNAs. PLoS ONE. 9 (5), 98186 (2014).
- Ren, X., Yang, Z., et al. Enhanced specificity and efficiency of the CRISPR/Cas9 system with optimized sgRNA parameters in Drosophila. Cell Reports. 9 (3), 1151-1162 (2014).
- Ran, F. A., Hsu, P. D., Wright, J., Agarwala, V., Scott, D. A., Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nature Protocols. 8 (11), 2281-2308 (2013).
- Serano, J. M., Martin, A., et al. Comprehensive analysis of Hox gene expression in the amphipod crustacean Parhyale hawaiensis. Developmental Biology. 409 (1), 297-309 (2016).
- Sternberg, S. H., Redding, S., Jinek, M., Greene, E. C., Doudna, J. A. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature. , 1-17 (2014).
- Lee, K., Conboy, M., et al. Nanoparticle delivery of Cas9 ribonucleoprotein and donor DNA in vivo induces homology-directed DNA repair. Nature Biomedical Engineering. 1 (11), 889-901 (2017).
