1. Sample preparation
- Preparation of a target sample
- Seed 2.5 x 105 HeLa cells on a 35-mm glass-bottom dish and grow them in 2 mL of growth medium (Dulbecco's modified eagle medium with L-glutamine and sodium pyruvate supplemented with 10% fetal bovine serum [FBS]) at 37 °C and a CO2 concentration of 5%. Alternatively, seed 6 x 104 HeLa cells on an 8-well chambered coverglass and grow them in 0.5 mL of growth medium at 37 °C and a CO2 concentration of 5%. Use #1.5 coverglass (with thickness of 0.170 mm) for high-resolution microscopy.
NOTE: Use 1/4 volumes for the 8-well chambered coverglass for further steps except steps 1.1.5, 1.1.6, 1.1.8, 1.1.11 and 1.2.5 where the volume is 100 µl irrespective of the type of container. - After about 24 h of incubation, replace the solution with 2 mL of 3.7% formaldehyde in phosphate-buffered saline (PBS). After gentle mixing, continue to fix cells for 15 min at room temperature (RT) on a rotation platform.
CAUTION: Work in a fume hood. - Wash cells with 2 mL of PBS, 3x each for 5−10 min on a rotation platform.
NOTE: Follow local regulations to dispose formaldehyde waste. - Permeabilize cells with 2 mL of 0.1% Triton X-100 in PBS for 5 min on a rotation platform, followed by three washes with 2 mL of PBS, each for 5−10 min on a rotation platform.
- Incubate cells in 100 µL of 1% bovine serum albumin (BSA) in PBS for 1 h at RT on a rotation platform.
- Replace the solution with 100 µL of a mixture of primary antibodies (anti-emerin polyclonal antibody14 and anti-tubulin monoclonal antibody15) in 1% BSA in PBS at appropriate dilutions (1/500 for anti-emerin and 1/100 for anti-tubulin), and incubate overnight at 4 °C.
- Wash cells with 2 mL of PBS, 3−5x each for 10 min on a rotation platform.
- Replace the solution with 100 µL of a mixture of secondary antibodies (anti-rabbit IgG with Alexa Fluor 488 and anti-mouse IgG with Alexa Fluor 555) in 1% BSA in PBS at 1/500 dilution, and incubate for 3−4 h at RT.
- Wash cells with 2 mL of PBS, 3−4x each for 5 min on a rotation platform.
- Replace the solution with 2 mL of 0.5 µg/mL DAPI in PBS to stain DNA for 30 min at RT on a rotation platform.
NOTE: Instead of DAPI, Hoechst 33342 at the same concentration can also be used. - Replace the solution with ~100 µL of mounting medium (Table of Materials).
- Seed 2.5 x 105 HeLa cells on a 35-mm glass-bottom dish and grow them in 2 mL of growth medium (Dulbecco's modified eagle medium with L-glutamine and sodium pyruvate supplemented with 10% fetal bovine serum [FBS]) at 37 °C and a CO2 concentration of 5%. Alternatively, seed 6 x 104 HeLa cells on an 8-well chambered coverglass and grow them in 0.5 mL of growth medium at 37 °C and a CO2 concentration of 5%. Use #1.5 coverglass (with thickness of 0.170 mm) for high-resolution microscopy.
- Preparation of a biological calibration sample
- Prepare fixed cells as described in steps 1.1.1−1.1.4.
NOTE: Preferably, samples are prepared on a chambered coverglass to place both the target and reference samples in separate chambers on the same coverslip (Table 1, right-most). This preparation is preferable when the experiment requires the highest correction accuracy. When the sample needs to be prepared on a regular coverslip, use precision coverslips with low thickness variation ("No. 1.5H") to insure reproducibility. - Prepare the stock solution of fluorescent dye-conjugated phalloidin in 1.5 mL of methanol and store at -20 °C. Mix phalloidin stock solution in PBS at the following dilution: 1/100 for phalloidin with Alexa Fluor 405, 1/1,000 for phalloidin with Alexa Fluor 488, and 1/200 for phalloidin with Alexa Fluor 594.
- Replace the solution with 1 mL of the phalloidin mixture prepared in step 1.2.2 and incubate for 30 min at RT on a rotation platform.
- Wash cells with 2 mL of PBS, 3−5x each for 10 min on a rotation platform.
- Replace the solution with ~100 µL of mounting medium.
NOTE: The calibration samples can be stored at 4 °C or -20 °C for repeated use.
- Prepare fixed cells as described in steps 1.1.1−1.1.4.
2. Acquisition of reference images
- Crosstalk reference images
- Place the target sample prepared in step 1.1 on a wide-field microscope.
- Acquire a fluorescence image of the target in blue, green, and red channels.
NOTE: For 3D images, the Z step size in the reference image can be different from that in the target image as long as the image file contains information for the step size. The Z step size for reference and target images is preferably less than half of the optical resolution of the Z axis, which is calculated by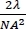 for a diffraction-limited microscopy, where λ is the wavelength in nanometers and NA is the numerical aperture of the objective lens. For example, if the axial resolution is 550 nm, then use a Z step smaller than 275 nm. No time series is required for the reference image.
for a diffraction-limited microscopy, where λ is the wavelength in nanometers and NA is the numerical aperture of the objective lens. For example, if the axial resolution is 550 nm, then use a Z step smaller than 275 nm. No time series is required for the reference image. - Subsequently, to acquire a fluorescence image of the reference, select the excitation light only for DAPI, and choose to acquire in blue, green, and red channels. Acquire the reference image exactly at the same stage position and Z height, in case of a 3D stack.
NOTE: For longer emission wavelengths, increased illumination intensity and exposure time will be required.
- Bright-field reference images
- Place the target sample prepared in step 1.1 on a wide-field microscope.
- Acquire a fluorescence image of the target in blue, green, and red channels.
NOTE: Z step size preferably fulfills the Nyquist criterion as described in step 2.1.2. - Acquire a bright field image of the target in blue, green, and red channels exactly at the same stage position as in 2.2.2 and the same Z height in case of a 3D stack.
- Biological calibration reference images
- Place the target sample prepared in step 1.1 on a 3D-SIM microscope.
- Acquire a fluorescence image of the target in blue, green, and red channels by 3D-SIM. Reconstruct the super-resolved image.
NOTE: The type of microscope can be of any type such as 3D-SIM, confocal, STED, etc. Z step size preferably fulfills the Nyquist criterion as described in step 2.1.2. - Acquire multiple fluorescence images of the reference prepared in step 1.2 at different stage positions similar to the target image (step 2.3.2) by 3D-SIM. Reconstruct the super-resolved images..
NOTE: The total number of pixels in XY must be the same in all reference images. The step size in Z must be the same in all reference images. The total number of Z sections is preferred to be the same as that in the target image, but this is not an absolute requirement. The XY position on the stage for these reference images does not matter because the position or coverslip is already different from the position of the target sample and furthermore the difference in chromatic shift on a single coverslip is less than 15 nm3. Imaging conditions including objective lens, observation temperature, immersion oil, pinhole size in confocal microscopy, and tilt angle in highly inclined illumination microscopy16, should all match the reference for the best performance. If the microscope equips multiple cameras to acquire multiple channels simultaneously, the reference images should be acquired as often as weekly to correct the instrumental drift.
3. Correction of chromatic shift using Chromagnon software
- Using a web browser, go to https://github.com/macronucleus/Chromagnon/releases, and download the newest binary release of Chromagnon.
NOTE: Binary releases are available for Windows, Mac, and some Linux versions. - Extract the program and put the executable file at a convenient location. On a Windows or Mac double-click the file to open it, or else execute the binary file from the command line on a Linux system.
NOTE: The graphical user interface as in Figure 1 will open. The program may be prevented from opening by double clicking for the first time because of a security reason. In this case, use the platform-dependent method to open the program downloaded from the internet.

Figure 1: A screenshot of Chromagnon graphical user interface. Please click here to view a larger version of this figure.
- Drag and drop the reference files in the "Reference box" (Figure 1) or click Reference files (Figure 1A) to open a file selector dialog. Depending on the file formats, if the program asks to install a Java Development Kit (JDK), press yes and the user will be navigated to the download page. Download and install the JDK for the operating system as instructed. Chromagnon should be able to read the image file format after restarting the program.
NOTE: Chromagnon can read most image file formats (multipage 'tif', 'czi', 'nd2', 'oib', 'lif', 'dv', etc.). The original microscope image format is preferred at this step because metadata can be lost when an image format is converted to a multipage tif file. If the channel name is shown as 0, 1, 2, etc. instead of wavelengths such as 528, 609 (Figure 2A, green boxes), then Chromagnon does not know the identity of the channels in the image. In this case, ensure that the order of the channels and the pixel size in the reference file match those in the target file. For example, if the order of channels in the reference is green and red, then the order of channels in the target must also be green and red, but not red and green.

Figure 2: Example screenshot for loading multiple files. (A) A case in which all reference images have the corresponding target images. Channel names are correctly identified by wavelengths (indicated by a green box) in the image files used in this example. (B) A case in which a single reference image is used to correct multiple target images. (C) A case in which multiple reference images (indicated by a red box) are averaged, and the resulting reference image after averaging is used to correct multiple target images. Please click here to view a larger version of this figure.
- Drag and drop the target files in the "Target box" (Figure 1) or click Target files (Figure 1B) to open a file selector dialog.
NOTE: If there are multiple target images and each of them have corresponding reference images, the corresponding reference image and target image must be on the same row in the respective reference and target boxes (Figure 2A). A single reference image can also be used to align multiple target images (Figure 2B). - Check the crop margins checkbox if unchecked (Figure 1C).
NOTE: If this checkbox is checked, margins resulting from the alignment are cropped. Check this option for general use but uncheck if a pixel-level comparison is required between images before and after alignment. - Choose an output image format from the choice list (Figure 1D).
NOTE: The available formats are 'tif' (ImageJ17 format), 'dv', and 'ome.tif'; the 'ome.tif' format is only available after installing JDK. - Specify the suffix for the output filename (Figure 1E, the default value is '_ALN'), which is added to the target filename.
- For crosstalk reference images with high signal-to-noise ratio, use local alignment from the choice list of Local align (Figure 1F), choose Projection to use local alignment and None to disable it. Use a minimum window size of 60 (Figure 1G).
NOTE: When using local alignment, all target images must have corresponding reference images (Figure 2A). Local alignment is not recommended for biological calibration reference images as local chromatic shifts vary from one sample to the other (Table 1). For the same reason, single local alignment cannot be applied to many target images (Figure 2B). As an exception, it can be used when the sample of interest is only at the surface of the coverslip, and the field of view is filled with bright objects, just like the multicolor fluorescent bead samples. - For multiple biological calibration reference images, check the average references option (Figure 1H) to measure a single alignment parameter from the averaged image, and the single alignment parameter is then applied to all target images (Figure 2C). Choose None for Local align (Figure 1F).
- Click Run all (Figure 1I) to start measurements; the alignment parameters are applied to the target images.
NOTE: After measuring chromatic shifts from the reference files, the program makes files with extension "chromagnon.csv" or "chromagnon.tif". The type of output file depends on whether local alignment is in effect (Figure 1F). If alignment is done without local alignment, the output is "chromagnon.csv", while if local alignment is being used, the output is "chromagnon.tif". The file names are the same as the reference file except for the specified suffix (Figure 1J, the default is without suffix) and the extension ("chromagnon.csv" or "chromagnon.tif"). A detailed description of the alignment process can be found in the "Chromagnon.log" file created in the same folder. - Wait until the corrected image shows up in the viewer (Figure 3).
NOTE: Dragging the image with the mouse moves the image and moving the mouse wheel changes the zoom. Move the slider (Figure 3A) to change Z (and/or T when applicable) section for display. If the viewer is too slow to refresh an image, click the Load whole data into memory button (Figure 3B) to stop it accessing the data on the hard disk. Dragging the left or right edge of color boxes (Figure 3C) can control the minimum or maximum values for display. Clicking the button for each channel (Figure 3D) toggles between showing or hiding the selected channel in the viewer. Right clicking on the color box (Figure 3D) enables users to choose the colors for display, or change the display options of the color bar, and it also allows users to specify the exact minimum and maximum values for display scaling by choosing "scale to …". Clicking the Orthogonal view button (Figure 3E) shows the images of ZY and XZ views. Moving the cross lines changes the position to show in the side views.

Figure 3: A screenshot of the image viewer. Please click here to view a larger version of this figure.
- To check if the measurement was performed correctly, drag and drop the reference images into the "Reference box" and "Target box". Run the program, and check if the images are perfectly overlapped.
NOTE: When available, "chromagnon.csv" or "chromagnon.tif" files can be used as references in order to skip measurements. If the chromatic shift in the image remains uncorrected, try solutions summarized in Table 2. - To access the alignment parameters in the sample, or when the alignment parameters need to be edited manually, open "chromagnon.csv" files directly with any text editor or spreadsheet software. When it is edited, save it as "csv".
NOTE: The values are in pixels for "tz", "ty", "tx", and in degrees for "r", and in magnification factors for "mz", "my", and "mx".- Alternatively, load a "chromagnon.csv" or "chromagnon.tif" file into Chromagnon's Reference or Target box (Figure 1), then double-click it to open the alignment editor (Figure 4).
- To see how much a channel is shifted when the parameters are edited, drag and drop the reference image file onto the bottom area (Figure 4A) to open the image in the editor. Upon editing the values in the table (Figure 4B) and hitting the enter/return key, observe how the image of the corresponding channel moves as the values changed. After editing, click save as… (Figure 4C) to save the changes.

Figure 4: A screenshot of an alignment parameter editor. Please click here to view a larger version of this figure.
- To check the local alignment map, load the "chromagnon.tif" via Chromagnon's Reference or Target box (Figure 1), and double-click it to open the alignment editor (Figure 4). Click view (Figure 4D) to view the local shift indicated by lines whose lengths are magnified by the factor specified in the magnification choice list (Figure 4E).
NOTE: By specifying the "original image file" (Figure 4F), the shifts are mapped along with the original image.
4. Generating a microscope-specific local alignment map
- Dilute 2 µL of multicolor fluorescent beads of 200 nm diameter in 18 µL of ethanol.
- Vortex the solution and place 10 µL of the bead solution at the center of a glass bottom dish. Make sure to use #1.5 coverglass (with thickness of 0.170 mm) for high-resolution microscopy.
- Leave the dish at RT for 1 h to dry completely.
- Place the dish on a microscope to be calibrated and focus on the fluorescent beads.
- Obtain 2D or 3D images with the microscopy to be calibrated. Obtain multiple images by changing the stage position.
- Open Chromagnon graphical user interface.
- Drag and drop the bead image files on the "Reference box" (Figure 1) or click Reference files (Figure 1A) to open a file selector dialog.
- Check the average references option (Figure 1H). From the choice list of Local align (Figure 1F), choose Projection. Use a minimum window size of 60 (Figure 1G). Click Run all (Figure 1I) to start measurements.
NOTE: A ".chromagnon.tif" file is created, in which the local alignment map of the microscope is stored. - After measurement, click Extra parameters (Figure 1K). From the Local distortion of your microscope instrument box, click the choice list and choose New… to open a dialog. Drag and drop the "chromagnon.tif" file generated in step 4.8 into the file name box or click choose file button to open a file selector dialog. Enter the name of the microscope and click OK.
- When measuring chromatic shifts from crosstalk or biological calibration reference images, click Extra parameters (Figure 1K). From the Local distortion of your microscope instrument box, choose the name of the microscope specified in step 4.9. Click OK.
NOTE: The choice of the "Local distortion of your microscope instrument" is not saved after the program is shut down. - Proceed to chromatic correction without local alignment as in section 3.
NOTE: It is also possible to use local alignment in addition to microscope calibration. In this case, local alignment starts with microscope calibration.
An example of chromatic shift correction using a crosstalk reference image is shown in Figure 5. The image was obtained with a wide-field microscope equipped with a single camera. Fluorescence emission from DAPI was used as a reference (Figure 5A,B) to correct the blue, green, and red channels. The image comprises 3 channels of 60 Z slices, each composed of 256 x 256 pixels. The images were deconvolved before measuring the chromatic shifts. Measuring the local chromatic shifts using Chromagnon took 51 s on a Mac with Intel Core i7 (quad core, 8-threads, 2.7 GHz), 16 GB RAM and 1 TB flash storage. The alignment parameter was applied to the target image (Figure 5C,D), which has exactly the same number of voxels as the reference image. Preparing the aligned file took 3 s. As a result of trimming the edge pixels during the alignment process (crop margins checkbox in Figure 1C), the number of voxels was reduced after alignment (Figure 5B, 51 Z slices, 252 x 251 pixels). DNA in the anaphase bridge (indicated by arrowheads) is seen incorrectly outside of the nuclear envelope before alignment (Figure 5C, obvious in the bottom panel showing the XZ view), but as expected inside the envelope after alignment (Figure 5D).
An example of chromatic shift correction using a biological calibration reference image is shown in Figure 6. The images were obtained with a SIM microscope equipped with three cameras. Three images of HeLa cells stained with phalloidins conjugated to blue, green, and red dyes were averaged (Figure 6A). The reference image comprises 3 channels of 76 Z slices, each composed of 1,024 x 1,024 pixels. Measuring chromatic shifts using Chromagnon without local alignment required 194 s on the Mac system described above. The parameter was applied to a target image consisting of 3 channels of 73 Z slices, each composed of 1,024 x 1,024 pixels. Generation of the aligned file took 25 s. The XZ view shows incorrect channel positions along the Z, and slightly along the X directions (Figure 6A,C) but this misregistration was corrected after alignment (Figure 6B,D).

Figure 5: An example of alignment with a crosstalk reference image. HeLa cells were stained with DAPI for DNA (shown in magenta), Alexa Fluor 488 (shown in yellow) for nuclear envelope, and Alexa Fluor 555 (shown in blue) for microtubules. Images were acquired by 3D wide-field microscopy with a single camera and deconvolved. (A,B) A representative crosstalk reference image using DAPI emission, before and after alignment by Chromagnon. Three color channels are shown as overlaid. (C,D) An optical section of the 3D stack in three channels before and after alignment. Axial chromatic aberration is obvious at the anaphase bridge shown by arrowheads. Scale bar in panel A indicates 5 µm for all panels. Please click here to view a larger version of this figure.

Figure 6: An example of alignment with a biological calibration reference image Images were acquired with 3D-SIM equipped with three cameras. (A,B) A reference image averaged from three images before (A) and after (B) alignment. HeLa cells were stained with phalloidin conjugated with Alexa Fluor 405, 488 or 594. (C,D) The target image before (C) and after (D) alignment. HeLa cells were stained with DAPI for DNA (shown in magenta), Alexa Fluor 488 (shown in yellow) for nuclear envelope, and Alexa Fluor 594 (shown in blue) for microtubules. Scale bar in panel A indicates 5 µm for all panels. Please click here to view a larger version of this figure.
The procedure for chromatic correction is a trade-off between accuracy and effort. To save needless efforts, it is better to know how much accuracy is required for your study. The highest accuracy may not be required for conventional wide-field (live) imaging, and thus, bright field reference images are often sufficient to correct the chromatic shift. Similarly, when the imaging condition and environment is constant, repeated use of a biological calibration will save time. On the other hand, if a highly accurate registration is desired, high-quality crosstalk or biological calibration reference images are necessary. For the best performance, reference images should be obtained with as similar conditions and timings as the target images as possible. As long as both reference and target images are obtained by the same microscopy, higher spatial resolution will improve the correction accuracy. If deconvolution is available for both reference and target images, then implementing this before correction may improve the correction accuracy. Also, for the best performance, the sampling theorem for the optical (Z) axis should be fulfilled in both the reference and target file for precise subpixel interpolation (protocol step 2.1.3).
Failure to correct chromatic shift leads to incorrect conclusions. Furthermore, using the wrong calibration may even worsen the chromatic shifts rather than correcting them, and this therefore needs to be avoided. We have summarized the possible causes of failures, and their common solutions, in Table 2. To examine the cause of a failure, in the first place, it is necessary to visually check if the chromatic shift in the reference image is precisely corrected (protocol step 3.12). Most failures are due to the quality of the reference images and are easily remedied as per the descriptions in Table 2. Regarding to the quality of reference images, it is important to note that the accuracy of global alignment decreases if the entire field of view is not filled with the sample (Figure 7, Table 2). Compared to the good example shown in Figure 7A, the bad example shown in Figure 7B contains only three nuclear envelopes in the upper-left region, and Chromagnon failed to align a part of this image. This is because the global alignment method of Chromagnon splits the field of view into four regions (Figure 7C) in order to measure the differences in rotation and magnification with high accuracy3. This method, if correctly operated, is one order more accurate than other linear methods such as the log polar transformation and simplex methods3. If any of the four regions are unavailable, then Chromagnon will switch to less effective linear methods. Therefore, for the best performance, the examples shown in Figure 7B and Figure 7C are undesirable, and the four regions should be filled with objects. Users can check if any quadratic region of the field of view is unavailable for measurement by looking at the log file ("Chromagnon.log"; see protocol step 3.10). Fortunately, this problem can be easily overcome by averaging multiple biological calibration images or using local alignment for crosstalk or bright-field reference images (Table 2). Contrary to the case of failure to correct reference images, failure to correct target images is more difficult to identify. Because such failures arise due to differences in file formats, imaging conditions, imaging timings, imaging/alignment methods between the reference and target images (Table 2), users should always be careful when using reference images that are obtained in different conditions/timings from the target images. Some example images are available for testing (https://github.com/macronucleus/Chromagnon) to obtain concrete idea of the good and bad example images.
| Problem | Cause | Solution |
| Failed to correct the reference image | Low contrast | Acquire a higher contrast image if possible. If a bright-field reference image is used, reacquire the image in a water-based solution to obtain higher contrast of the cell. Alternatively, try applying computational noise reduction (e.g. Gaussian filtering). Turn off local alignment, which is more sensitive to noise. |
| Contamination of unrelated images | Remove the source of the unrelated images in the sample if possible. For crosstalk reference images, check the excitation spectra of the dyes used for the target images. If the dyes are excited during acquisition of a crosstalk image (e.g. Alexa Fluor 568 or 594), consider other dyes (e.g. Alexa Fluor 555). If dusts on the camera chip creates an obvious channel difference, clean the camera chip or use a computational flat-fielding method. | |
| An extremely bright spot made by a cosmic ray | Acquire the image again if possible. Alternatively, try applying computational noise reduction (e.g. Median or Gaussian filtering). | |
| Deconvolution artifacts (artificial signals at the axial and lateral edges) | Trim the edge pixels or Z sections after deconvolution. If one side is trimmed, the other side should also be trimmed to maintain the image center. | |
| The Z step size too sparse | A Z stack should be acquired to fulfill the Nyquist criterion as written in protocol 2.1.3. | |
| Optical aberration | Spherical aberration is the major aberration caused by users. Choose the right objective lens for the sample and use a coverslip thickness of 170 µm. If the objective lens is equipped with a correction ring, adjust it to find the position where the highest fluorescence count is obtained from the focus. In the case of an oil-immersion objective without a correction ring, adjust the refractive index of the immersion oil that increases the fluorescence count at the focus. | |
| Field of view is unfilled (Fig. 7) | In the case of biological calibration reference images, average many images. In the case of crosstalk or bright-field reference images, use local alignment. | |
| An unidentified software bug | Report the issue through GitHub (https://github.com/macronucleus/Chromagnon/issues) | |
| Failed to correct the target image | Metadata of the image file is lost | Use the original microscope file format which contains complete metadata, and avoid converting to a multipage tiff file before processing. Use the same ordering of channels as written in protocol 3.3. |
| Wrong alignment methods for the given microscopy | Do not apply the local alignment method when measuring from biological calibration reference images to target images. Do not use crosstalk reference images other than wide-field microscopy. | |
| Differences in imaging conditions | Keep the imaging conditions constant between the reference and the target images as written in protocol 2.3.3. | |
| Differences in sample (including coverslip) | Always use the same mounting medium, coverslip (e.g. No. 1.5H) and a similar depth of focus. | |
| Microscope drift since the calibration was last made | Make a calibration as often as every two weeks. Keep the temperature constant, and use a floating table to avoid hardware drift of the microscope. |
Table 2: Troubleshooting for chromatic correction.

Figure 7: Examples of reference images. Nuclear envelope in fission yeast cells labeled with GFP and mCherry. Images were acquired with conventional wide-field microscopy. Chromatic shifts were corrected using Chromagnon without local alignment using the images themselves as reference images. Images were then deconvolved to show the details. (A) A good example with many objects in the field of view. (B) A bad example with objects only at the top-left corner. Misalignment is obvious at a certain region of the image. (C) An undesirable example where one of the quadrisection (separated by dotted cross lines) is empty. Scale bar in panel A indicates 5 µm for the full field view and 1.25 µm for the enlarged view and is applicable to all panels. Please click here to view a larger version of this figure.
In this protocol, we described three different reference types (Table 1). Among them, crosstalk reference images and biological calibration reference images need further careful discussion. For crosstalk reference images, samples stained with DAPI or Hoechst 33342, and mounted in glycerol or commercial mounting media can be efficiently used to align the blue, green, and red channels. Similarly, Alexa Fluor 488 can be used to align the green and red channels. However, obtaining crosstalk fluorescence is often difficult since many blue dyes except DAPI and Hoechst are dimmer and decay faster than most green and red dyes. Furthermore, the emission spectra of modern dyes are narrower, which makes the alignment of more than three channels by this method challenging. Attention should also be paid to some common red dyes (e.g., Alexa Flour 568 and 594, but not Alexa Fluor 555) that can be excited by violet light, which prevent obtaining high-contrast crosstalk images from blue dyes. Another drawback is that this method cannot measure the chromatic aberration of excitation light paths in multicolor excitation, because only a single excitation wavelength is used for excitation (Table 1). As most advanced microscopy uses altered illumination optics, the application of this method is limited. Still, its higher correction accuracy is sufficiently advantageous for it to be described in this protocol. In general, a crosstalk image should be taken after a target image to prevent bleaching or phototoxic effects. For SMLM observed with the wide-field mode, a reference image should be acquired before acquiring a target image as fluorescence dyes can be bleached while imaging.
Biological calibration reference images allow users to easily align any desired number of channels at the cost of additional sample preparation. Another advantage of biological calibration reference images is the availability of "averaging" multiple references that helps fill all fields of view. This method may suffer from differences in imaging conditions if the calibration sample is prepared on a different slide. Most of this problem can be solved if both targets and references are prepared on the same slide by using commercial chambered coverglasses (Table 1), and other imaging conditions are kept constant as in protocol step 2.3.3. In this case, a correction accuracy similar to that of crosstalk reference images can be expected3. The protocol to use phalloidin as shown here is one of the easiest ways to stain a single cellular structure with multiple colors. There are numerous possible scenarios to prepare biological calibration samples. For immunostaining, a sample can be labeled with a single primary antibody followed by staining with secondary antibodies of multiple colors. In this way, a single target structure can be labeled with multiple colors. Alternatively, 5-ethynyl-2'-deoxyuridine, detected by "click" chemistry labels newly synthesized DNA in multiple colors at high density, as described in detail previously8. For live cells, it is useful to prepare a transgenic strain harboring two copies of a gene that are fused to GFP or mCherry to label the same structure with two colors. If the copy number of the gene is critical as often observed for membrane proteins, a single copy of the gene can be tandemly fused to GFP and mCherry (Figure 7). Photoconvertible fluorescent proteins, such as mEOS218, can also be used by illuminating a moderate level of violet light to obtain both protein species with or without photoconversion. Under low oxygen conditions, GFP can also be used as a photoconvertible protein from green to red19,20. Choosing the right calibration sample will thus make the experiment more robust.
