Fuente: Michael S. Lee1 y Tonya J. Webb1
1 Departamento de Microbiología e Inmunología, Facultad de Medicina de la Universidad de Maryland y el Centro Integral del Cáncer Marlene y Stewart Greenebaum, Baltimore, Maryland 21201
La inmunohistoquímica (IHC) y la inmunocitoquímica (ICC) son técnicas utilizadas para visualizar la expresión y localización de antígenos específicos utilizando anticuerpos. El primer uso publicado de IHC fue en 1941 cuando Albert Coons utilizó la técnica para visualizar la presencia de antígeno neumocócico en secciones de tejido de ratones infectados con Pneumococcus (1). El nombre, inmunohistoquímica, se deriva de las raíces “inmuno-“, en referencia a los anticuerpos, y “histo-“, en referencia a las secciones de tejido utilizadas en IHC. La raíz “cito-” en inmunocitoquímica destaca la diferencia clave entre ICC e IHC. Mientras que IHC utiliza secciones de tejido entero, ICC utiliza células que han sido aisladas de tejido o cultivadas en cultivo. La diferencia en las muestras utilizadas significa que la preparación de muestras difiere técnicamente entre iHC y la CPI, pero de lo contrario los protocolos para ICC e IHC son idénticos y uno encontrará que los términos se utilizan con frecuencia indistintamente.
Tanto en IHC como en ICC, los anticuerpos con etiquetas químicas o fluorescentes, como la peroxidasa o la rodamina, respectivamente, se utilizan para visualizar la distribución de cualquier antígeno de interés a través de la unión específica del anticuerpo etiquetado al antígeno. En el caso de IHC, las rodajas finas de tejido se inmovilizan en una diapositiva para mantener la estructura del tejido antes de ser manchado, permitiendo la visualización de antígenos en el contexto de tejidos enteros (Figura 1). En el caso de la CPI, las células se distribuyen uniformemente en una diapositiva antes de ser teñidas, permitiendo la visualización de la distribución de antígenos dentro de las células individuales, pero no dentro de la estructura de cualquier tejido específico. Debido a las similitudes entre los dos protocolos, este protocolo se centrará en iHC para abordar las complejidades adicionales de la preparación de muestras involucradas en el IHC.
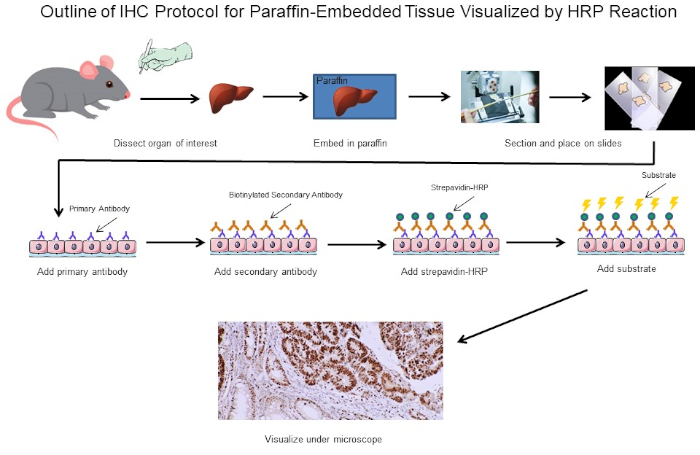
Figura 1: Esquema del Protocolo IHC. Esquema visual de un protocolo IHC para tejido incrustado en parafina diseccionado desde un ratón. Este protocolo utiliza un anticuerpo secundario biotinilado y estrepavidina-HRP para visualizar la ubicación de la unión de anticuerpos. Otras opciones, como los anticuerpos etiquetados fluorescentemente, también son posibles. Haga clic aquí para ver una versión más grande de esta figura.
La primera decisión importante al realizar IHC es cómo preparar las secciones de tejido con el fin de mantener la estructura del tejido durante todo el proceso de tinción. Las dos opciones principales son secciones fijas de formalina de tejido incrustado en parafina o secciones frescas de tejido congelado. No hay una respuesta simple sobre qué método utilizar, ya que depende de qué análisis posterior se llevará a cabo. La formalina-fijación de los tejidos incrustados de parafina generalmente se piensa para preservar mejor la morfología del tejido para obtener imágenes óptimas, mientras que la congelación de tejido fresco puede preservar la función proteica para ensayos posteriores fuera de IHC. Además, se ha demostrado que las secciones de tejido congelado fresco son más adecuadas para el análisis de la expresión génica (2). Una tercera consideración es si los anticuerpos para su antígeno de interés son adecuados para secciones de tejido fijo o congelado, ya que algunos anticuerpos solo se han optimizado para un tipo específico de sección y pueden no funcionar para otros. Por último, también hay que determinar cuánto tiempo necesitan para almacenar las secciones de tejido, ya que las muestras congeladas frescas deben mantenerse a -80 oC y no pueden durar más de un año, mientras que las secciones fijas se pueden almacenar durante mucho más tiempo a temperatura ambiente. Estas son algunas de las principales consideraciones para determinar si se deben utilizar secciones fijas de formalina de tejido incrustado en parafina o secciones frescas de tejido congelado. En última instancia, si uno tiene suficiente tejido, puede ser mejor sólo tener algunos de ambos.
En este experimento, nos propusimos determinar si la expresión de ciclina D1 se incrementó en el bazo agrandado a partir de un modelo espontáneo de ratón de desarrollo de linfoma. Las muestras de tejido esplénico se aislaron primero de ratones de tipo salvaje, ratones transgénicos que no tienen linfoma o ratones transgénicos que tienen linfoma desarrollado espontáneamente. Las muestras de tejido del bazo se fijaron en paraformaldehído, incrustadas en parafina, seccionadas, teñidas con un anticuerpo primario anticiclina D1 de ratón seguido de un anticuerpo secundario anti-ratón de caballo, y desarrolladas con 3,3-diaminobenzidina (DAB). Las secciones fueron contrarrestadas en Harris Hematoxylin Solution y luego las secciones fueron imágenes con aumento 20X.
Reactivos
Secciones integradas en Parafina
- 4% Paraformaldehyde (PFA)
- Etanol (anhidro desnaturalizado, grado histológico 100%, 95%, 80%, 75% y 50%). Se puede diluir a partir de 100% de stock utilizando agua destilada doble (ddH2O)
- Xileno
- Diapositiva de vidrio compatible con IHC para asegurar que la sección de tejido permanezca unida durante todo el procedimiento. Las guías de vidrio compatibles con IHC tienen un recubrimiento especializado y están disponibles en varios minoristas. Si realiza ICC, utilice una diapositiva con cámara. Las diapositivas acotadas permiten que las células se salpiquen en las cámaras y se coloquen en la incubadora hasta que las células se adhieran a la diapositiva y alcancen la confluencia adecuada, momento en el que las cámaras se pueden retirar y la tinción puede continuar de la misma manera que IHC.
- Parafina
- 0.3% Peróxido de hidrógeno (H2O2)/metanol: Para preparar, añadir 1 mL 30% H2O 2 O2 a 99 mL de metanol. Conservar a -20oC
- Tampón de recuperación de antígenos: tampón de citrato IHC pH 6.0
Secciones Frescas Congeladas
- Compuesto de incrustación de temperatura de corte óptima (OCT)
- Fijador óptimo: 4% PFA o acetona que se ha enfriado a -20 oC
Tinción
- Búfer de bloqueo: debe ser determinado por el usuario. Un ejemplo es el suero de caballo diluido en 1X PBS
- Anticuerpo primario diluido: ver las especificaciones del fabricante
- Anticuerpo secundario biotinylato diluido: ver las especificaciones del fabricante
- Peroxidasa de ávidina-caballo diluida (HRP): Sólo para la visualización de peroxidasa. Consulte las especificaciones del fabricante.
- DAB u otro sustrato compatible
- Contramancha (opcional)
- Etanol (anhidro desnaturalizado, grado histológico 100% y 95%)
- Xileno
- Montaje Organo/Limoneno
IHC and ICC have a vast range of applications. For example, one use of IHC is to examine the expression of oncogenes in spontaneous mouse models of tumor development. In Figure 2, we set out to determine if cyclin D1 expression was increased in enlarged spleens in a spontaneous mouse model of lymphoma development. Splenic tissue samples were fixed in paraformaldehyde, embedded in paraffin, sectioned, stained using an anti-cyclin D1 antibody (diluted 1:200 in blocking buffer), and then the sections were imaged at 20X magnification. Cyclin D1 expressing cells are indicated by the reddish-brown color against the blue tissue background. These results suggest that cyclin D1 expression was increased in enlarged spleens, indicating a correlation between cancer development and cyclin D1 expression in this model.

Figure 2: Splenic Cyclin D1 Expression in a Spontaneous Double Transgenic (DTG) Mouse Model of Lymphoma. An image of splenic tissue stained with an anti-Cyclin D1 primary antibody, counterstained with methyl green, and visualized using a biotinylated secondary antibody and ABC reagent activated with DAB substrate. The reddish-brown color represents locations where the antibody has bound indicating the presence of Cyclin D1 expressing tumor cells within the structure of splenic tissue that has been counterstained blue. Please click here to view a larger version of this figure.