Fonte: Natalia Martin1, Andrew J. Van Alst1, Rhiannon M. LeVeque1e Victor J. DiRita1
1 Dipartimento di Microbiologia e Genetica Molecolare, Michigan State University
I batteri hanno la capacità di scambiare materiale genetico (acido desossiribonucleico, DNA) in un processo noto come trasferimento genico orizzontale. L’incorporazione di DNA esogeno fornisce un meccanismo attraverso il quale i batteri possono acquisire nuovi tratti genetici che consentono loro di adattarsi alle mutevoli condizioni ambientali, come la presenza di antibiotici o anticorpi (1) o molecole presenti negli habitat naturali (2). Esistono tre meccanismi di trasferimento genico orizzontale: trasformazione, trasduzione e coniugazione (3). Qui ci concentreremo sulla trasformazione, la capacità dei batteri di prendere il DNA libero dall’ambiente. In laboratorio, il processo di trasformazione ha quattro fasi generali: 1) Preparazione di cellule competenti, 2) Incubazione di cellule competenti con DNA, 3) Recupero di cellule e 4) Placcatura delle cellule per la crescita dei trasformanti (Figura 1).
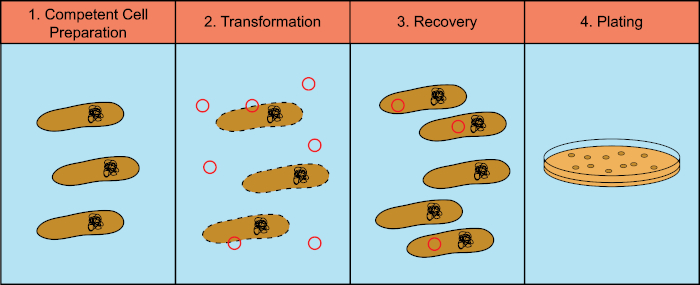
Figura 1: Fasi generali del processo di trasformazione. Il processo di trasformazione ha quattro fasi generali: 1) Preparazione delle cellule competenti, 2) Incubazione con DNA, 3) Recupero delle cellule e 4) Cellule placcate per la crescita dei trasformanti.
Affinché si verifichi la trasformazione, i batteri riceventi devono essere in uno stato noto come competenza. Alcuni batteri hanno la capacità di diventare naturalmente competenti in risposta a determinate condizioni ambientali. Tuttavia, molti altri batteri non diventano competenti naturalmente, o le condizioni per questo processo sono ancora sconosciute. La capacità di introdurre il DNA nei batteri ha una serie di applicazioni di ricerca: generare più copie di una molecola di DNA di interesse, esprimere grandi quantità di proteine, come componente nelle procedure di clonazione e altri. A causa del valore della trasformazione in biologia molecolare, ci sono diversi protocolli volti a rendere le cellule artificialmente competenti quando le condizioni per la competenza naturale sono sconosciute. Due metodi principali sono utilizzati per preparare cellule artificialmente competenti: 1) attraverso il trattamento chimico delle cellule e 2) esponendo le cellule a impulsi elettrici (elettroporazione). Il primo utilizza diverse sostanze chimiche a seconda della procedura per creare attrazione tra il DNA e la superficie cellulare, mentre il secondo utilizza campi elettrici per generare pori nella membrana cellulare batterica attraverso i quali le molecole di DNA possono entrare. L’approccio più efficiente per la competenza chimica è l’incubazione con cationi bivalenti, in particolare calcio (Ca2+)(4,5) La competenza indotta dal calcio è la procedura che verrà descritta qui (6). Questo metodo viene utilizzato principalmente per la trasformazione dei batteri Gram-negativi e questo sarà il focus di questo protocollo.
La procedura di trasformazione chimica comporta una serie di passaggi in cui le cellule sono esposte a cationi per indurre competenza chimica. Questi passaggi sono successivamente seguiti da un cambiamento di temperatura – shock termico – che favorisce l’assorbimento di DNA estraneo da parte della cellula competente (7). Gli involucri cellulari batterici sono caricati negativamente. Nei batteri Gram-negativi come l’Escherichia coli,la membrana esterna è caricata negativamente a causa della presenza di lipopolisaccaride (LPS) (8). Ciò si traduce in repulsione delle molecole di DNA caricate negativamente in modo simile. Nell’induzione della competenza chimica, gli ioni calcio caricati positivamente neutralizzano questa repulsione di carica consentendo l’assorbanza del DNA sulla superficie cellulare (9). Il trattamento del calcio e l’incubazione con dna vengono effettuati su ghiaccio. Successivamente, viene eseguita un’incubazione a temperature più elevate (42°C), lo shock termico. Questo squilibrio di temperatura favorisce ulteriormente l’assorbimento del DNA. Le cellule batteriche devono essere nella fase di crescita media-esponenziale per resistere al trattamento di shock termico; in altre fasi di crescita le cellule batteriche sono troppo sensibili al calore con conseguente perdita di vitalità che diminuisce significativamente l’efficienza di trasformazione.
Diverse fonti di DNA possono essere utilizzate per la trasformazione. Tipicamente, i plasmidi, piccole molecole di DNA circolari a doppio filamento, vengono utilizzati per la trasformazione nella maggior parte delle procedure di laboratorio in E. coli. Affinché i plasmidi possano essere mantenuti nella cellula batterica dopo la trasformazione, devono contenere un’origine di replicazione. Ciò consente loro di essere replicati nella cellula batterica indipendentemente dal cromosoma batterico. Non tutte le cellule batteriche vengono trasformate durante la procedura di trasformazione. Pertanto, la trasformazione produce una miscela di cellule trasformate e cellule non trasformate. Per distinguere tra queste due popolazioni, viene utilizzato un metodo di selezione per identificare le cellule che hanno acquisito il plasmide. I plasmidi di solito contengono marcatori selezionabili, che sono geni che codificano un tratto che conferisce un vantaggio per la crescita (cioè resistenza a un antibiotico o a una sostanza chimica o salvataggio da un’auxotrofia di crescita). Dopo la trasformazione, le cellule batteriche vengono placcate su mezzi selettivi, che consentono solo la crescita delle cellule trasformate. Nel caso di cellule trasformate con un plasmide che conferisce resistenza a un determinato antibiotico, il mezzo selettivo sarà un mezzo di crescita contenente quell’antibiotico. Diversi metodi possono essere utilizzati per confermare che le colonie coltivate nei mezzi selettivi sono trasformanti (cioè hanno incorporato il plasmide). Ad esempio, i plasmidi possono essere recuperati da queste cellule utilizzando metodi di preparazione del plasmide (10) e digeriti per confermare le dimensioni del plasmide. In alternativa, la PCR della colonia può essere utilizzata per confermare la presenza del plasmide di interesse (11).
Lo scopo di questo esperimento è quello di preparare cellule chimicamente competenti di E. coli DH5α, utilizzando un adattamento della procedura del cloruro di calcio (12), e di trasformarle con il plasmide pUC19 per determinare l’efficienza di trasformazione. Il ceppo di E. coli DH5α è un ceppo comunemente usato nelle applicazioni di biologia molecolare. A causa del suo genotipo, in particolare recA1 e endA1, questo ceppo consente una maggiore stabilità dell’inserto e migliora la qualità del DNA plasmidico nei preparati successivi. Poiché l’efficienza di trasformazione diminuisce con l’aumentare delle dimensioni del DNA, il plasmide pUC19 è stato utilizzato in questo protocollo a causa delle sue piccole dimensioni (2686 bp) (vedi https://www.mobitec.com/cms/products/bio/04_vector_sys/standard_cloning_vectors.html per una mappa vettoriale). pUC19 conferisce resistenza all’ampicillina e, quindi, questo era l’antibiotico utilizzato per la selezione.