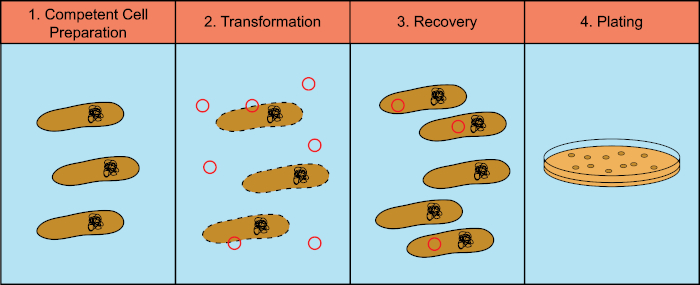
Bacteria are remarkably adaptable and one mechanism which facilitates this adaptation is their ability to take in external DNA molecules. One type of DNA that bacteria can uptake is called a plasmid, a circular piece of DNA that frequently contains useful information, such as antibiotic resistance genes. The process of bacteria being modified by new genetic information incorporated from an external source is referred to as transformation. Transformation can easily be performed in the laboratory using Escherichia coli, or E. coli.
In order to be transformed, E. coli cells must first be made competent, which means capable of taking in DNA molecules from their environment. The protocol for accomplishing this is surprisingly simple, a short incubation of the cells in a calcium chloride solution. This incubation causes the cells to become permeable to DNA molecules. After the cells are pelleted by centrifugation, the supernatant is removed. The plasmid DNA is now added to the competent cells. After incubating the cells with DNA, the mix is briefly heated to 42 degrees Celsius, followed by rapid cooling on ice. This heat shock causes the DNA to be transferred across the cell’s wall and membranes. The cells are then incubated in fresh media. Then, the bacteria are placed at 37 degrees to allow them to reseal their membranes and express resistant proteins.
Those cells which have taken in the plasmids will faithfully copy the DNA and pass it to their progeny and express any proteins that might be encoded by it, including antibiotic resistance mediators. Those resistance genes can be used as selectable markers to identify bacteria which have been successfully transformed because cells that have not taken up the plasmid will not express the resistance gene product. This means that when the cells are plated on a solid medium which contains the appropriate antibiotic, only cells that have taken up the plasmid will grow. Transformation of the cells in a growing colony can be further confirmed by culturing those cells in liquid media overnight to increase the yield before extracting the DNA from the sample. Once the DNA is isolated, a diagnostic restriction enzyme digest can be carried out. Because restriction enzymes cut DNA in predictable locations, running these digests on a gel should show a predictable pattern if the desired plasmid was successfully transformed. For example, if pUC19 is prepared and cut with the restriction enzyme HindIII, a single band of 2686 nucleotides should be seen on the gel.
In this lab, you will transform E. coli strain DH-5 Alpha with pUC19, and then confirm the successful transformation by DNA gel electrophoresis.
Before starting the procedure, put on the appropriate personal protective equipment, including a lab coat and gloves. Next, sterilize the workspace with 70% ethanol.
Now, prepare chemically competent cells by depositing a loopfull of bacteria onto a sterile LB agar plate and streaking the bacteria with a new loop. Then, incubate the plate at 37 degrees Celsius overnight. The next day, sterilize the bench top with 70% ethanol again, and remove the plate from the incubator.
Inoculate a single, well-isolated colony into 3 milliliters of LB broth in a tube with a sterile loop. Then, grow the culture at 37 degrees Celsius overnight, with shaking at 210 RPM. The next day, measure the optical density of the overnight culture with a spectrophotometer. Then, add 100 milliliters of LB broth to a one-liter flask, and inoculate it with the overnight culture at an optical density of 0. 01. Now, incubate the culture at 37 degrees Celsius with shaking, and check the OD600 every 15 to 20 minutes until the culture reaches mid-exponential growth phase.
After approximately three hours, transfer 50 milliliters of the culture to two ice-cold polypropylene bottles. Then, place the bottles back on ice for 20 minutes to cool. Next, recover the cells via centrifugation. Discard the supernatants and place the bottles upside down on a paper towel. Next, resuspend the bacterial pellet in five milliliters of ice-cold calcium chloride magnesium chloride solution and swirl carefully until the pellet has dissolved completely. Then, add another 25 milliliters of the solution to the dissolved bacterial pellet. Resuspend the other bacterial pellet as previously demonstrated. After this, repeat the centrifugation, and remove the supernatants.
If the competent cells are going to be directly transformed, resuspend each bacterial pellet in two milliliters of an ice-cold 0.1 molar calcium chloride solution by swirling the tubes carefully. To begin the transformation procedure, transfer 50 microliters of competent cells to two labeled 1.5 milliliter polypropylene tubes. Then, add one microliter of pUC19 plasmid DNA to one of the tubes. Mix gently, avoiding bubble formation, and incubate both tubes for 30 minutes on ice. After incubation, transfer the tubes to a heat block and incubate at 42 degrees Celsius for 45 seconds. Immediately transfer the tubes to ice, and incubate for two minutes. Now, add 950 microliters of SOC media to each tube and incubate them for one hour at 37 degrees Celsius to allow the bacteria to recover, and express the antibiotic resistant marker encoded in the plasmid.
To make a 1 to 100 dilution, add 990 microliters of SOC media and 10 microliters of cell suspension to a 1.5 milliliter tube. Then, make a 1 to 10 dilution by adding 900 microliters of SOC media and 100 microliters of cell suspension to a 1.5 milliliter tube. Next, plate 100 microliters of the diluted cell suspensions and 100 microliters of the negative control, onto separate selective plates containing ampicillin using a spreader and incubate the plates at 37 degrees Celsius for 12 to 16 hours. After incubation, count the colony-forming units, or CFUs, per plate, obtained through transformation, and record these data. To verify that the transformants have the pUC19 plasmid, pick a single, well-isolated colony from a plate with a sterile loop, and introduce it to a tube containing 3 milliliters of LB broth. Then, incubate the culture at 37 degrees Celsius with shaking, overnight. The next day, use a DNA mini prep kit to isolate DNA from 3 milliliters of the culture, according to the manufacturer’s instructions. After completing the DNA mini prep, digest the 1 microgram of purified pUC19 with a restriction enzyme at 37 degrees Celsius for 1 hour. Now, load 20 microliters of a molecular weight ladder, 1 microgram of digested plasmid DNA, and 1 microgram of undigested plasmid DNA into consecutive wells of a 1% agarose gel containing 1 microgram per milliliter ethidium bromide. Then, run the gel for 1 hour at 95 volts. Finally, visualize the gel with a UV illuminator.
In this experiment, E. coli DH5 Alpha chemically competent cells were prepared using an adaptation of the calcium chloride procedure, and then transformed with the plasmid pUC19 to determine transformation efficiency. To calculate the transformation efficiency, use the recorded CFU counts for the 1 in 100 and 1 in 10 dilutions, and any other dilutions with CFU counts between 30 and 300. First, the recorded CFU count, 246 in this example, is divided by the amount of DNA, .0001 micrograms here, that was plated. Then, this number is divided by the dilution factor used to give the transformation efficiency in CFUs per microgram. In this example, a 1 to 10 dilution was used and 100 microliters of a 1 milliliter solution was plated, giving a final dilution factor of 0.01. In the undigested plasmid lane, the circular DNA may appear as two or three different bands of varying brightness. This is because the circular, uncut DNA may exist in several different conformation states, such as supercoiled, open circle, or more linear, and each of these move through the gel at different rates. Analysis of the recovered plasmid DNA digestion indicated that the plasmid used has an expected size of pUC19 DNA, 2,686 base pairs.