Fonte: Natalia Martin1, Andrew J. Van Alst1, Rhiannon M. LeVeque1, e Victor J. DiRita1
1 Departamento de Microbiologia e Genética Molecular, Universidade Estadual de Michigan
As bactérias têm a capacidade de trocar material genético (Ácido Desoxiribonucleico, DNA) em um processo conhecido como transferência de genes horizontais. A incorporação do DNA exógeno fornece um mecanismo pelo qual as bactérias podem adquirir novos traços genéticos que lhes permitem adaptar-se às mudanças nas condições ambientais, como a presença de antibióticos ou anticorpos (1) ou moléculas encontradas em habitats naturais (2). Existem três mecanismos de transferência genética horizontal: transformação, transdução e conjugação (3). Aqui vamos focar na transformação, na capacidade das bactérias de tirar DNA livre do meio ambiente. Em laboratório, o processo de transformação tem quatro etapas gerais: 1) Preparação de células competentes, 2) Incubação de células competentes com DNA, 3) Recuperação de células e 4) Revestimento das células para crescimento dos transformadores (Figura 1).
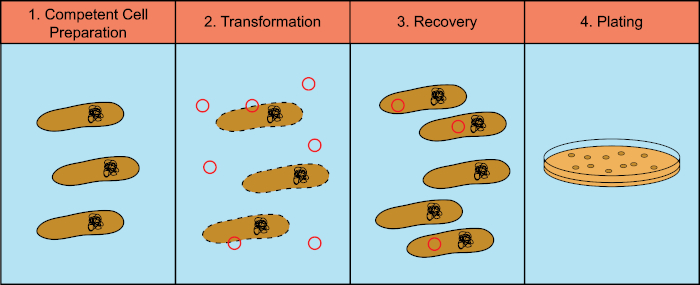
Figura 1: Etapas gerais do processo de transformação. O processo de transformação tem quatro etapas gerais: 1) Preparação de células competentes, 2) Incubação com DNA, 3) Recuperação das células e 4) Células de chapeamento para crescimento dos transformadores.
Para que a transformação ocorra, a bactéria receptora deve estar em um estado conhecido como competência. Algumas bactérias têm a capacidade de se tornarem naturalmente competentes em resposta a certas condições ambientais. No entanto, muitas outras bactérias não se tornam competentes naturalmente, ou as condições para esse processo ainda são desconhecidas. A capacidade de introduzir DNA em bactérias tem uma gama de aplicações de pesquisa: gerar múltiplas cópias de uma molécula de DNA de interesse, expressar grande quantidade de proteínas, como componente em procedimentos de clonagem, entre outros. Devido ao valor da transformação para a biologia molecular, existem vários protocolos que visam tornar as células artificialmente competentes quando as condições de competência natural são desconhecidas. Dois métodos principais são utilizados para preparar células artificialmente competentes: 1) através do tratamento químico das células e 2) expondo as células a pulsos elétricos (eletroporação). O primeiro usa diferentes produtos químicos dependendo do procedimento para criar atração entre o DNA e a superfície celular, enquanto o segundo usa campos elétricos para gerar poros na membrana celular bacteriana através da qual moléculas de DNA podem entrar. A abordagem mais eficiente para a competência química é a incubação com cátions divalent, mais notavelmente cálcio (Ca2+) (4,5) Competência induzida por cálcio é o procedimento que será descrito aqui (6). Este método é usado principalmente para a transformação de bactérias Gram-negativas, e esse será o foco deste protocolo.
O procedimento de transformação química envolve uma série de etapas em que as células são expostas a ceações para induzir a competência química. Essas etapas são posteriormente seguidas por uma mudança de temperatura – choque térmico – que favorece a absorção de DNA estranho pela célula competente (7). Os envelopes de células bacterianas são carregados negativamente. Em bactérias gram-negativas como escherichia coli,a membrana externa é carregada negativamente devido à presença de lipopólise (LPS) (8). Isso resulta em repulsa das moléculas de DNA igualmente carregadas negativamente. Na indução de competência química, íons de cálcio carregados positivamente neutralizam essa repulsão de carga permitindo a absorção de DNA na superfície celular (9). O tratamento de cálcio e a incubação com DNA são feitos no gelo. Posteriormente, é realizada uma incubação a temperaturas mais altas (42°C), o choque térmico. Esse desequilíbrio de temperatura favorece ainda mais a absorção de DNA. As células bacterianas precisam estar na fase de crescimento exponencial médio para suportar o tratamento de choque térmico; em outros estágios de crescimento, as células bacterianas são muito sensíveis ao calor, resultando em perda de viabilidade, o que diminui significativamente a eficiência da transformação.
Diferentes fontes de DNA podem ser usadas para transformação. Tipicamente, plasmídeos, pequenas moléculas circulares de DNA de dupla laga, são usados para transformação na maioria dos procedimentos laboratoriais em E. coli. Para que os plasmídeos sejam mantidos na célula bacteriana após a transformação, eles precisam conter uma origem da replicação. Isso permite que eles sejam replicados na célula bacteriana independentemente do cromossomo bacteriano. Nem todas as células bacterianas são transformadas durante o procedimento de transformação. Assim, a transformação produz uma mistura de células transformadas e células não transformadas. Para distinguir entre essas duas populações, é utilizado um método de seleção para identificar as células que adquiriram o plasmídeo. Plasmídeos geralmente contêm marcadores selecionáveis, que são genes codificando um traço que confere uma vantagem para o crescimento (ou seja, resistência a um antibiótico ou químico ou resgate de uma auxotrofia de crescimento). Após a transformação, as células bacterianas são banhadas em mídias seletivas, o que só permite o crescimento das células transformadas. No caso de células transformadas com uma resistência plasmid conferenciando a um dado antibiótico, a mídia seletiva será a mídia de crescimento contendo esse antibiótico. Vários métodos diferentes podem ser usados para confirmar que as colônias cultivadas nas mídias seletivas são transformadores (ou seja, incorporaram o plasmídeo). Por exemplo, plasmídeos podem ser recuperados dessas células usando métodos de preparação plasmídeos (10) e digeridos para confirmar o tamanho plasmídeo. Alternativamente, a COLÔNIA PCR pode ser usada para confirmar a presença do plasmídeo de interesse (11).
O objetivo deste experimento é preparar células quimicamente competentes E. coli DH5α, utilizando uma adaptação do procedimento de cloreto de cálcio (12), e transformá-las com o pUC19 plasmídeo para determinar a eficiência da transformação. A cepa E. coli DH5α é uma cepa comumente usada em aplicações de biologia molecular. Devido ao seu genótipo, especificamente o recA1 e o endA1,esta cepa permite maior estabilidade da inserção e melhorar a qualidade do DNA plasmídeo nas preparações subsequentes. Uma vez que a eficiência de transformação diminui com o aumento do tamanho do DNA, o plasmídeo pUC19 foi usado neste protocolo por causa de seu pequeno tamanho (2686 bp) (ver https://www.mobitec.com/cms/products/bio/04_vector_sys/standard_cloning_vectors.html para um mapa vetorial). pUC19 confere resistência à ampicilina e, portanto, este foi o antibiótico utilizado para seleção.