NOTE: Figure 1 illustrates the complete experimental design.
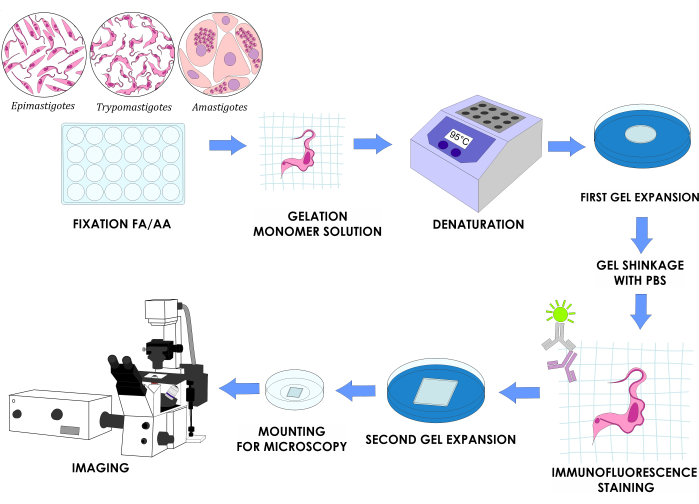
Figure 1: U-ExM workflow for three in vitro life cycle stages of T. cruzi. Please click here to view a larger version of this figure.
1. Preparation of the poly-D-lysine-coated coverslips
- Place a 10 cm x 10 cm square of sealing film in a Petri dish. Wash the coverslips by bathing them in absolute ethanol in a 35 mm glass Petri dish.
- Remove the coverslips with tweezers from the ethanol bath and drain the excess liquid with tissue paper. Place the coverslips over the sealing film.
- Absorb the rest of the ethanol with microscopy-grade paper. Add a 0.1% v/v solution of poly-D-lysine on the center of the coverslip and spread it with the tip to cover approximately 80% of its surface. Close the Petri dish and incubate for 1 h at 37 °C.
NOTE: For 22 mm2 coverslips, use 200 µL of poly-D-lysine solution; for 12 mm round coverslips, use 100 µL of poly-D-lysine solution. - Wash the coverslips with ultra-pure water three times. Use aspiration with a vacuum to remove the water between each wash. Keep at 4 °C for up to 1 week.
2. Solution preparation
- Prepare stock solutions of 38% (w/w) sodium acrylate (SA).
- Slowly add 19 g of SA to 31 mL of nuclease-free water while stirring.
NOTE: This solution is very viscous; pay attention when pipetting. - Once the SA is completely dissolved, store it in a sterile container. Keep it at 4 °C and change it every 6 months.
NOTE: SA sometimes shows signs of contamination with sodium polyacrylate depending on the brand, which is noticeable when preparing the stock solution as it turns yellowish and cloudy. Use it cautiously if this is the case.
- Slowly add 19 g of SA to 31 mL of nuclease-free water while stirring.
- Prepare a stock solution of 40% (w/v) acrylamide (AA) and a stock solution of 2% (w/v) N, N'-methylene bisacrylamide (BIS). Dissolve each compound in ultrapure water and filter with a 0.22 µm sterile syringe filter. Store in a sterile container at 4 °C.
CAUTION: AA and BIS are highly toxic substances. Work under a fume hood and use proper protection elements (gloves, protective clothing, mask, and safety glasses). - Prepare the protein crosslinking prevention (CP) solution by mixing 38 µL of a 37% formaldehyde (FA) solution with 50 µL of the 40% acrylamide stock solution (step 2.2) in 912 µL of phosphate-buffered saline (PBS; Table 1) to obtain a final concentration of 1.4% formaldehyde and 2% acrylamide.
NOTE: Always prepare the CP solution freshly and be extremely precise when pipetting. For example, use a P1000 pipette to take 900 µL of PBS and a P20 pipette to take the remaining 12 µL of PBS. - Prepare the monomeric solution
NOTE: Do not use the solution immediately after being prepared; store at -20 °C at least 24 h before being used.- Mix 500 µL of the 38% SA solution (step 2.1), 250 µL of the 40% AA solution, and 50 µL of the BIS solution (step 2.2), as well as 100 µL of 10x PBS (Table 1).
- Aliquot this final volume of 900 µL in 10x 1.5 mL microcentrifuge tubes with 90 µL of monomeric solution each. Store at -20 °C for up to 2 weeks.
NOTE: The SA solution may stay on the tip when pipetting because it is very viscous; be careful and dispense everything.
- Prepare the denaturing solution
- Mix 114.3 mL of a 350 mM sodium dodecyl sulfate (SDS) solution prepared in ultrapure water with 10 mL of a 4 M sodium chloride solution prepared in ultrapure water. Add 12 g of Tris while stirring in a 250 mL beaker.
CAUTION: SDS is highly toxic; use it under a fume hood wearing gloves, protective clothing, a mask, and safety glasses. - Adjust the pH to 9 with a concentrated solution of hydrochloric acid. Make up to 200 mL with ultrapure water and store in a sterile flask at 4 °C.
- Mix 114.3 mL of a 350 mM sodium dodecyl sulfate (SDS) solution prepared in ultrapure water with 10 mL of a 4 M sodium chloride solution prepared in ultrapure water. Add 12 g of Tris while stirring in a 250 mL beaker.
- Prepare a 10% ammonium persulfate (APS) solution and a 10% TEMED solution.
- Dissolve 0.1 g of APS in 1 mL of ultrapure water.
- Prepare 1 mL of a 10% TEMED solution in ultrapure water.
- Prepare 100 µL aliquots of both solutions in 1.5 mL microcentrifuge sterile tubes and store them at -20 °C for up to 1 month.
- Prepare the paraformaldehyde solution.
- Dissolve 2 g of paraformaldehyde in 40 mL of PBS while stirring at 60 °C. Add 1 M NaOH dropwise until the solution turns from white to colorless.
CAUTION: Formaldehyde is highly toxic; use it under a fume hood wearing gloves, protective clothing, a mask, and safety glasses. - Cool the solution at room temperature and adjust the pH with NaOH to 7.2 in a final volume of 50 mL. Filter with a 0.22 µm sterile syringe filter and store in a sterile container.
- Prepare aliquots of 1 mL in 1.5 mL sterile microcentrifuge tubes and store at -20 °C.
- Dissolve 2 g of paraformaldehyde in 40 mL of PBS while stirring at 60 °C. Add 1 M NaOH dropwise until the solution turns from white to colorless.
- Prepare the paraformaldehyde/glutaraldehyde solution.
- Add 0.2 g of paraformaldehyde in 3.5 mL of ultrapure water and 50 µL of 16 M NaOH solution. Heat the solution to 60 °C to dissolve the paraformaldehyde.
- Cool down and add 300 µL of 70% glutaraldehyde. Bring up the volume to 5 mL with ultrapure water and finally to 10 mL with PBS.
CAUTION: Paraformaldehyde and glutaraldehyde are highly toxic; use them under a fume hood wearing gloves, protective clothing, a mask, and safety glasses. - Prepare aliquots of 1 mL in 1.5 mL sterile microcentrifuge tubes and store at -20 °C.
3. Preparation of the parasite cultures
- Grow T. cruzi epimastigotes.
- Use an axenic culture in a T-25 flask (25 cm2 growth area) and maintain the cultures in the logarithmic phase by sub-culturing every 48-72 h in liver infusion tryptose (LIT) medium with 10% fetal calf serum (FCS; Table 1).
- Ensure the cap is securely closed and keep the culture flask vertically at 28 °C for incubation. Monitor the parasite growth by cell counting in a Neubauer chamber during each subculture.
NOTE: For the Dm28c strain used in this study, the concentration of epimastigotes in log-phase cultures is between 1-5 x 107 parasites/mL. - Prepare a suspension of 2 x 106 epimastigotes/mL from a log-phase culture in LIT medium supplemented with 10% FCS. Centrifuge the suspension at 5,000 x g for 10 min at room temperature (RT). Wash with PBS once or twice and resuspend in 200 µL of PBS.
- Adhere to the round 12 mm coverslip previously coated with poly-D-lysine (section 1). Incubate at RT for 15-20 min. Continue to the crosslinking prevention step (section 4).
NOTE: Alternatively, fix the epimastigotes with cold methanol for 7 min or paraformaldehyde/glutaraldehyde solution (step 2.8) for 10 min at RT prior to adherence of the parasites to the coverslip. It is possible to store the fixed parasites at 4 °C for up to 1 week.
- Obtain the amastigotes from infected Vero cells.
- Lay a sterile 12 mm round coverslip on the bottom of a 24-well tissue culture plate. Prepare a suspension of 2 x 105 Vero cells/mL in Dulbecco's modified Eagle medium (DMEM) supplemented with 2% fetal calf serum (FCS). Seed 500 µL of the suspension per well.
NOTE: This study uses DMEM with 2% FCS to allow the cells to grow slower. - Incubate overnight (ON; 12-16 h) at 37 °C and 5% CO2 to ensure cell attachment.
- Following incubation, wash the cells twice using 500 µL of sterile PBS. Add T. cruzi trypomastigotes into the cells at a multiplicity of infection (MOI) of 10 in 100 µL of DMEM with 2% FCS per well, corresponding to one million trypomastigotes per well. Incubate at 37 °C and 5% CO2 for 6 h.
- After incubation, rinse the plates twice with PBS. Add 500 µL of DMEM supplemented with 2% FCS. At this point, continue to the crosslinking prevention step (section 4).
NOTE: Intracytoplasmic amastigotes will be visible through an inverted optical microscope 2 days post-infection (Supplementary Figure 1). Alternatively, fix the trypomastigotes with cold methanol for 7 min at -20 °C, or paraformaldehyde/glutaraldehyde for 10 min at RT, prior to adherence of the parasites to the coverslip.
- Lay a sterile 12 mm round coverslip on the bottom of a 24-well tissue culture plate. Prepare a suspension of 2 x 105 Vero cells/mL in Dulbecco's modified Eagle medium (DMEM) supplemented with 2% fetal calf serum (FCS). Seed 500 µL of the suspension per well.
- Obtain trypomastigotes from infected Vero cells.
- Collect the supernatant of a Vero cell monolayer (30%-40% confluence) infected with trypomastigotes (MOI 1:10; ON incubation) 4 days post-infection.
NOTE: For a T-25 flask, the initial concentration of Vero cells used is 800,000 cells, and for a T-75 flask is two million cells. - Determine the trypomastigote concentration using a Neubauer chamber for cell counting. Centrifuge 4 x 106 trypomastigotes at 7,000 x g at RT for 10 min.
- Rinse with PBS twice and resuspend in 200 µL of PBS. Apply to a round, 12 mm coverslip coated with poly-D-lysine (section 1). Incubate for 10-15 min at RT. Proceed with the crosslinking prevention step (section 4).
NOTE: Alternatively, fix the trypomastigotes with cold methanol for 7 min at -20 °C or paraformaldehyde/glutaraldehyde for 10 min at RT prior to adherence of the parasites to the coverslip.
- Collect the supernatant of a Vero cell monolayer (30%-40% confluence) infected with trypomastigotes (MOI 1:10; ON incubation) 4 days post-infection.
4. Performing crosslinking prevention (DAY 1)
- Submerge the 12 mm coverslip with the adhered parasites or infected cells (facing upward) in a 24-well plate with 0.5 mL of CP solution (step 2.3) in each well.
- Fill the empty wells with water to reduce evaporation. Seal the plate with a sealing film. Incubate for 5 h at 37 °C. This step can be extended up to an ON incubation at 4 °C.
NOTE: Always submerge the coverslip in the solution; do not pipette the fixative solution over the coverslips.
5. Performing gelation of the sample
- Assemble a humid chamber in a Petri dish with a sealing film on top of tissue paper (Figure 2A). Add water to the tissue paper and incubate at -20 °C for 20 min to cool down. Thaw a TEMED and an APS aliquot on ice for 20 min (step 2.6).
NOTE: Do not freeze-thaw the APS more than three times. - Lay the cool, humid chamber on ice (Figure 2A). Take the 24-well plate prepared in section 4 out of the incubator. Aspirate the CP solution with a 3 mL Pasteur pipette, leaving some solution, otherwise the coverslips will be hard to remove.
- Remove the 12 mm coverslips from the fixative solution with tweezers and lay them on tissue paper with the parasites facing up.
NOTE: It is helpful to use a sterile needle to lift the coverslip and then hold it with the tweezers. - To an aliquot of 90 µL of monomeric solution (step 2.4), add 5 µL of TEMED and 5 µL of APS previously thawed. Mix with a vortex mixer for no more than 2-3 s; it is not necessary to close the tube with the lid.
NOTE: Always add TEMED first and APS last. The addition of APS first and TEMED last to the monomeric solution makes the gelation process faster, giving no time to manipulate it. - Quickly make one drop of 35 µL over the sealing film of the humid chamber for each coverslip. Immediately pick up the coverslip with tweezers and lay over the drop (Figure 2B) with the parasites facing down.
NOTE: Make a maximum of two coverslips at a time; it is crucial not to delay this step because the solution polymerizes very quickly. - Incubate the humid chamber for 5 min on ice and then for 1 h at 37 °C. Turn on a heating block at 95 °C to ensure the correct temperature for the next step.

Figure 2: Gelation step details. (A) Assembly of the humid chamber. (B) Dropping the coverslips onto the monomer solution with TEMED and APS for gelation. (C) Schematic representation of the gel assembled for imaging. Please click here to view a larger version of this figure.
6. Denaturing the gelified samples and performing the isotropic expansion
- Remove the denaturation solution (step 2.5) from 4 °C. If it is precipitated, put it in a hot water bath until it dissolves completely.
- Add 2 mL of the denaturing solution to each well of a 6-well plate. Transfer the coverslips from step 5.3 to the plate with the denaturation solution. Incubate for 15 min at RT with gentle shaking so the gel detaches from the coverslip.
- Carefully transfer the gel (removing it from the 12 mm coverslip) with a metal spatula into a 1.5 mL sterile microcentrifuge tube with 1 mL of denaturing solution. Use cap locks to secure the tubes. Incubate for 1 h and 30 min at 95 °C in a heating block.
NOTE: Gels start to expand during this step; be gentle when transferring the gel to the 1.5 mL microcentrifuge tube.
CAUTION: After the incubation, the tube containing the gel is at 95 °C, which can be dangerous. Use protective gloves and let the tubes cool down before handling them to avoid burns and projections. - Perform the first round of expansion
- Aspirate the denaturing solution from the 1.5 mL microcentrifuge tube with the gel with a P1000 pipette. Transfer the gel from the 1.5 mL microcentrifuge tube to a Petri dish with 10 mL of ultrapure water for 30 min using a small spatula.
- Change the ultrapure water using a disposable 3 mL Pasteur pipette. Incubate ON at RT.
NOTE: Be gentle with the gels because, after the first 30 min of incubation, they became fragile. - Change the ultrapure water using a disposable 3 mL Pasteur pipette one more time.
NOTE: Three water incubations of 30 min each are sufficient, but for practicality, it is better to leave the second incubation ON.
7. Performing fluorescence labeling of the target proteins (DAY 2)
- Remove the water from the Petri dish with the gel with a disposable 3 mL Pasteur pipette. Measure the diameter of the gel with a caliper to calculate the expansion (between four and five times).
- Wash twice with 10 mL of PBS for 15 min. Cut the gel with a razor blade into squares of approximately 10 mm x 10 mm in the center of the circular gel. Use one square per condition to be tested.
NOTE: The gel shrinks after the PBS incubations. Gels can be stored in PBS at 4 °C for up to 1 week. - Transfer each square to a 12-well plate and incubate with 500 µL of primary antibody diluted in 2% PBS-bovine serum albumin (BSA) for 2 h and 30 min at 37 °C with shaking.
NOTE: Alternative antibody incubation can be performed ON at 4 °C. The minimum antibody volume that can be used is 300 µL in a 24-well plate. As a general rule, use twice the concentration of antibodies used for conventional immunolabeling. - Wash three times with 2 mL of PBS with 0.1% polysorbate 20 for 10 min while shaking in a 6-well plate.
- Transfer the gel to a 12-well plate and incubate with 500 µL of secondary antibody in PBS with DAPI and 10 µg/mL NHS-ester conjugated to the desired fluorophore for 2 h and 30 min at 37 °C with gentle shaking.
NOTE: As a general rule, use twice the concentration of antibodies used for conventional immunolabeling. Alternatively, this incubation can be performed ON at 4 °C. - Wash three times with 2 mL of PBS with 0.1% polysorbate 20 for 10 min while shaking in a 6-well plate.
- Transfer the gel to a Petri dish with ultrapure water. Incubate for 30 min. Change the water twice with a disposable 3 mL Pasteur pipette, as done in step 6.4.
8. Imaging and image processing (DAY 3)
- Remove the water from the Petri dishes with a disposable 3 mL Pasteur pipette and measure the gel diameter with a caliper to calculate the expansion factor.
- Cut a small piece of ~10 mm x 10 mm with a razor blade and place it on a 35 mm glass bottom dish.
NOTE: As an alternative, place the gel between a slide and a coverslip (without poly-D-lysine). The slide should have two smaller slide pieces cut with a diamond knife attached on the sides to form a chamber about the thickness of the gel. - Check the orientation at 10x or 20x magnification. In order to focus properly, ensure that the parasites are facing the coverslip; otherwise, one has to turn the gel around and check again (Figure 2C).
- Once the proper orientation is found, dry the remaining gel with microscopy paper. Cover the gel with a poly-D-lysine-coated coverslip.
- Add a small drop of ultrapure water to the gel to visualize with a confocal microscope.
NOTE: To ensure proper imaging, it is crucial to maintain the gel orientation throughout the process so that the side of the gel containing cells near its surface is facing the glass-bottom dish at the end. When using 35 mm dishes, adding poly-D-lysine to the coverslip placed in the bottom reduces the gel shifting during the image acquisition. - Image acquisition parameters
- Image the samples in a confocal microscope (Table of Materials) using a 63x oil immersion 1.4 numerical aperture (NA) objective.
- Acquire Z stacks, the width of each z step, and an exposure time per pixel, to be determined empirically depending on the sample, the signal intensity, and optimization of acquisition times. Use the scan zoom for effective magnification if desired.
- Open the Z stack using image processing software (Table of materials). For each channel, group the stacked images using the Group Z-project option. Select the maximum intensity projection.
- To merge images, use the Merge Channels tool and select the color of each channel. Add a scale bar using the Scale Bar tool in the processing software.
NOTE: Alternatively, one can color each channel as desired (see examples in the representative results).
If the protocol has been properly executed (Figure 1), samples will be visible as a planar and translucent gel that can be expanded up to a factor of 4-4.5x in water (Figure 3A). This expansion provided an effective resolution of about 70 nm, which may vary depending on the final expansion factor and imaging system employed. After the second expansion process and image acquisition in a confocal microscope, we were able to observe expansion factors of around 4.5. To quantify this expansion, we measured the gels before and after step 7.1. Also, we labeled epimastigotes with α-tubulin antibodies in non-expanded and expanded parasites (Figure 3B).
When staining with cytoskeletal markers like anti-tubulin antibodies, we observed-in epimastigotes, trypomastigotes, and amastigotes-the correct localization of this protein in the subpellicular corset of microtubules and the flagellar axoneme, highlighting this technique's value in three in vitro life cycle stages of T. cruzi (Figure 3C). Also, the condensation state of the chromatin in the nucleus can be clearly distinguished when staining with DAPI (inset in Figure 3C).
To exemplify this optimized protocol for cytoskeletal proteins, we immunolocalized α-tubulin and the paraflagellar rod (PFR) in epimastigotes and overlapped it with pan-proteome labeling (Figure 3D). In the third panel of Figure 3D, we can observe a basal body stained with anti α-tubulin/Fluorescein isothiocyanate (FITC) that has divided (marked with arrows); this is the first step in the cell division of epimastigotes.
Finally, it is worth mentioning that when performing pan-proteome labeling, one can identify different parasite structures, such as the nucleus, flagella, kinetoplast, and flagellar pocket, among other organelles in all life cycle stages (Figure 3E).

Figure 3: Expanded gel with the parasites and edited confocal images. (A) Expanded gel measured with a caliper. (B) Non-expanded and expanded epimastigotes stained with anti-α-tubulin antibodies (grayscale; Alexa 555). Scale bar: 10 µm. (C) Epimastigotes, trypomastigotes, and amastigotes stained with anti-α-tubulin antibodies (magenta; Alexa 555) and DAPI (cyan; kDNA and nuclear DNA). Scale bar: 10 µm. Inset: zoom to a nucleus stained with DAPI. (D) Expanded epimastigotes stained with anti-α-tubulin antibodies or anti-PFR (magenta; FITC) and NHS-ester conjugated to Atto 594 (grayscale). Scale bar: 10 µm. Arrows indicate a duplicated basal body in the flagellar pocket area. (E) Epimastigotes, trypomastigotes, and uninfected and infected cells with amastigotes stained with NHS-ester conjugated to Atto 594 (grayscale). Scale bar: 10 µm. Images B–E were acquired with a confocal microscope using a 63x oil immersion 1.4 (NA) objective. Lasers used: 552 nm solid-state laser (20 mW), 405 nm diode laser (50 mW), and 488 nm solid-state laser (20 mW). Please click here to view a larger version of this figure.
Supplementary Figure 1: Visualization of intracytoplasmic amastigotes. (A) Inverted optical microscope 48 h post-infection of a Vero cell monolayer with Dm28c trypomastigotes; 40x objective. (B) Direct microscope; 60x oil objective. Abbreviations: N: nucleus of the Vero cell (marked with a dotted line). Red arrow: amastigotes. Please click here to download this File.
Table 1: Recipes for LIT medium, hemin, and PBS. Please click here to download this Table.
