Source: Alexander S. Gold1, Tonya M. Colpitts1
1 Department of Microbiology, Boston University School of Medicine, National Emerging Infections Diseases Laboratories, Boston, MA
Transduction is a form of genetic exchange between bacteria that utilizes bacteriophages, or phages, a class of virus that infects exclusively prokaryotic organisms. This form of DNA transfer, from one bacterium to another by way of a phage, was discovered in 1951 by Norton Zinder and Joshua Ledererg, who termed the process "transduction" (1). Bacteriophages were first discovered in 1915 by British bacteriologist Frederick Twort, then independently discovered again in 1917 by French-Canadian microbiologist Felix d'Herelle (2). Since then, the structure and function of these phages have been widely characterized (3), dividing these phages into two classes. The first of these classes are the lytic phages which upon infection multiply within the host bacterium, disrupting the bacterial metabolism, lysing the cell, and releasing progeny phage (4). As a result of this anti-bacterial activity and the increasing prevalence of antibiotic-resistant bacteria, these lytic phages have recently proved useful as a substitute treatment for antibiotics. The second of these classes are the lysogenic phages which can either multiply within the host via the lytic cycle or enter a quiescent state in which their genome is integrated into that of the host (Figure 1), a process known as lysogeny, with the ability for phage production to be induced in multiple later generations (4).
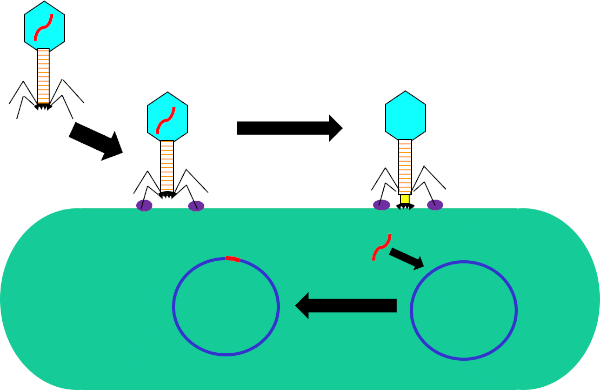
Figure 1: Infection of host cell by bacteriophage. Adsorption by the phage to the bacterial cell wall via interactions between the tail fibers and receptor (purple). Once on the cell surface, the phage is irreversibly attached to the bacterial cell using the base plate (black) which is moved to the cell wall by the contractile sheath (yellow). Phage genome (red) then enters the cell and integrates into the host cell genome.
While bacterial transduction is a naturally occurring process, using modern technology it has been manipulated for the transfer of genes into bacteria in the laboratory setting. By inserting genes of interest into the genome of a lysogenic phage, such as phage, one is able to transfer these genes into the genomes of bacteria and consequently express them within these cells. While other methods of gene transfer, such as transformation, use a plasmid for gene transfer and expression, the insertion of the phage genome into that of the recipient bacterium not only has the potential to confer new traits to this bacterium, but also allows for naturally occurring mutations and other factors of the cellular environment to alter the function of the transferred gene.
Compared to other methods of horizontal gene transfer, such as conjugation, transduction is fairly flexible in the criteria required for donor and recipient cells. Any genetic element that can fit inside the genome of the phage being used can be transferred from any strain of donor bacteria to any strain of recipient bacteria as long as both are permissive to the phage, requiring the expression of necessary phage receptors on the cell surfaces. Once this gene is moved out of the donor genome and packaged into the phage, it can be transferred to the recipient. Following transduction, it is necessary to select for recipient bacteria that contain the gene of interest have to be selected for. This could be done by use of a genetic marker, such as a FLAG-tag or polyhistidine-tag, to mark the gene of interest, or the intrinsic function of the gene, in the case of genes that encode for antibiotic resistance. Additionally, PCR could be used to further confirm successful transduction. By using primers for a region within the gene of interest and comparing signal to a positive control, bacteria that has the gene of interest, and a negative control, bacteria that underwent the same steps as the transduction reaction with no phage. While bacterial transduction is a useful tool in molecular biology, it has and continues to play an important role in the evolution of bacteria, particularly with regard to the recent rise of antibiotic resistance.
In this experiment, bacterial transduction was used to transfer the gene encoding for resistance to the antibiotic ampicillin from the W3110 strain of E.coli to the J53 strain via the P1 bacteriophage (5). This experiment consisted of two main steps. First, the preparation of P1 phage containing the ampicillin resistance gene from the donor strain. Second, the transfer of this gene to the recipient strain by transduction with the P1 phage (Figure 1). Once carried out, the successful transfer of the ampicillin resistance gene could be determined by qPCR (Figure 2). If transduction was successful, the J53 strain of E. coli would be resistant to ampicillin, and the gene conferring this resistance detectable by qPCR. If unsuccessful, there would be no detection of the ampicillin resistance gene and ampicillin would still function as an effective antibiotic against the J53 strain.
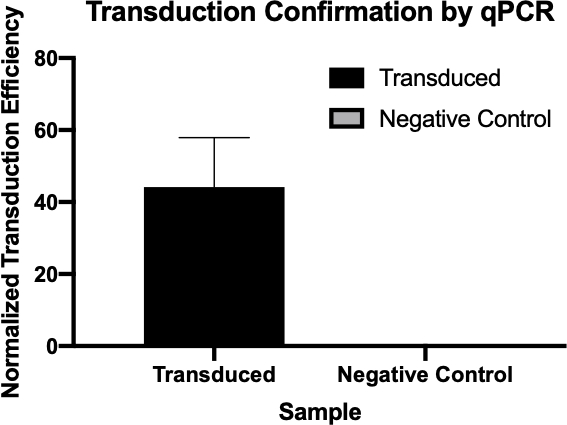
Figure 2: The confirmation of successful transduction by qPCR. By comparing the Cq values detected for the gene of interest from the transduction reaction and the negative control reaction, and normalizing these values against a housekeeping gene, one was able to confirm that bacterial transduction was successful.