1. Preparation of Plasmid Sets Encoding AAV Capsid Genes
- To facilitate routine preparation of sufficient amounts of the various AAV capsid (cap) genes for subsequent DNA shuffling, initially subclone these genes into a common plasmid backbone. It is important to include identical flanking sequences of >20 nucleotides for later use as primer binding sites for nested PCR and for cloning (Fig. 2).
- Using appropriate primers (see Table for exemplary primers for AAV5), PCR amplify desired cap genes from commonly available AAV plasmids that typically contain the AAV2 rep gene next to the cap gene of choice. Because the PCR product will be used for standard cloning, ~1 μg of purified product is already sufficient, and any conventional PCR protocol can hence be used.
- Digest the purified PCR product (e.g., use agarose gel purification or a standard PCR purification kit) and recipient plasmid with restriction enzymes whose recognition sites are present in the primers used for cap amplification (1.1) as well as in the recipient plasmid. In our lab, we use Pac I and Asc I sites (Fig. 2) as they are absent in most AAVs.
2. DNase-based Cap Gene Fragmentation
- PCR amplify cap genes of choice from the plasmids generated in steps 1.1-1.3. One reaction as described below will yield ~3 μg of PCR product. Depending on the number of cap genes to be included in the library, this suffices for up to six shuffling reactions.
- For the PCR, set up a 50 μl reaction containing 200 ng cap plasmid, each primer at 2 μM final concentration, 10 μl 5x Hifi buffer and 1 μl Hifi polymerase. Start with 5 min at 95 °C and then run 40 cycles of 15 sec 94 °C, 30 sec 57 °C and 3 min 68 °C, followed by a final 10 min step at 72 °C. Purify the PCR products via gel or kit and then set up a controlled DNase digest to create cap gene fragments for re-assembly into chimeras.
- Therefore, equally mix the various cap PCR products to a total amount of 4 μg in 54 μl H2O. Add 6 μl DNase reaction buffer and 0.5 μl DNase I to the reaction, carefully flick three times, spin briefly and immediately put on a 25 °C heating block. Incubate between 1 and 2 min (set up multiple parallel reactions and terminate them in increments of 15 sec), then stop the reaction by adding 6 μl 25 mM EDTA and by briefly vortexing and incubating 10 min at 75 °C.
- Purify the cap fragments on a standard 1% agarose gel. Ideally, a smear should be visible between 100 and 500 base pairs. Since DNase I is a highly potent enzyme, proper handling and timing are critical at this step, and multiple variations in incubation time in step 2.3 might be needed for optimal results (Fig. 3). Purify the eluted DNA using a standard kit and determine its concentration.
3. DNA Family Shuffling
- First, re-assemble the cap fragments into full-length sequences via a PCR in which they self-prime based on partial homologies. Therefore, set up a 50 μl reaction with 500 ng purified fragments (step 2.4), 10 μl 10x Phusion buffer, 1 μl 10 mM dNTPs, 1.5 μl DMSO and 0.5 μl Phusion II polymerase. Incubate 30 sec at 98 °C and then run 40 cycles of 10 sec 98 °C, 30 sec 42 °C and 45 sec 72 °C, followed by a final 10 min step at 72 °C.
- In an ensuing second PCR, amplify the re-assembled cap genes for subsequent cloning, using primers that bind to the conserved flanking sequences (Fig. 2). Therefore, set up a 50 μl reaction containing 2 μl of the first PCR (step 3.1), each primer at a final concentration of 2 μM, 0.5 μl MgCl2, 10 μl 5x Hifi buffer and 1 μl Hifi polymerase. We recommend running 16-24 PCRs to ensure sufficiently high yields for subsequent cloning. Pool the PCRs and purify the full-length cap band (gel or kit).
- Digest the purified cap gene pool with Pac I and Asc I for cloning into a replication-competent AAV plasmid carrying AAV ITRs (inverted terminal repeats; replication and packaging signals) as well as the AAV2 rep gene. The latter should be followed by the same sites to accommodate the cap gene pool “in frame” (see Fig. 2 for details). To achieve complete digestion of the PCR product, incubate overnight with excess enzyme.
- Ligate the cap fragments and the appropriately cut AAV ITR/rep backbone at a 3:1 molar ratio. Make a 40 μl total volume mastermix (sufficient for 20 transformations) with a final DNA concentration of 50 ng/μl and incubate overnight at 16 °C.
- Transform ligation reaction by mixing (on ice) 2 μl with 30 μl electro-competent E. coli (grown from commercial cells and made competent using any standard protocol). Add into pre-cooled electroporation cuvettes (1 mm gap) on ice. Electroporate at 1.8 kV, 200 Ω and 25 μF. The time constant should be close to 5 ms. Immediately add 1 ml pre-warmed SOC medium and transfer to a 250 ml flask. 20 such electroporations will yield a library with a diversity of about 1×106 different clones.
- Add 1 volume pre-warmed SOC medium to the pooled transformations and shake at 37 °C and 180 rpm for 1 h. Then bring up the total volume to 800 ml with LB medium plus ampicillin (final concentration of 50 μg/ml) and incubate for another 16 h under the same conditions. Purify the library plasmid DNA using e.g. a Qiagen Mega prep kit.
Optional at step 3.6: To calculate the exact library diversity, plate an aliquot of the 800 ml solution (prior to the 16 h incubation) on LB-ampicillin plates (e.g., 10 μl on a 10 cm plate) and count colonies the next day. Also, to validate high shuffling efficiencies, sequence e.g. 24 clones and align them to the parental cap genes (see also Fig. 8). Finally, to confirm library vitality and high functional diversity, subclone randomly picked cap genes into an AAV helper plasmid and use them to produce and analyse recombinant vectors in small scale (Figs. 4-5) (see also step 5.4 below).
4. Production of Viral Library
- Seed 10 15 cm2 dishes of HEK293T cells (4.5×106 cells/dish) and 48 h later transfect with 220 μg AAV library and 220 μg adenoviral plasmid (required for AAV propagation). Therefore, pre-warm PEI (polyethylenimine) and 300 mM NaCl at 37 °C. Then mix 7.9 ml NaCl and DNA, and add H2O to a total volume of 15.8 ml. In a separate tube, mix 3.52 ml PEI, 7.9 ml NaCl and 4.38 ml H2O (all volumes for ten transfections). Combine the mixes (vortex) and incubate for 10 min at room temperature, before distributing the solution evenly across the dishes (3 ml per dish).
- After 48 h, scrape off the cells into the medium and spin them down at 1200 rpm for 15 min. Resuspend the cell pellet in 6 ml lysis buffer (50 mM Tris-HCl pH 8.5, 50 mM NaHCO3) and subject to 5 freeze-thaw cycles (-80/37 °C). Incubate with 50 U benzonase per ml for 1 h at 37 °C, before spinning down cell debris at 3750 rpm for 20 min.
- Prepare 15%, 25% and 40% dilutions (with 2.5 μl/ml phenolred) from a 60% iodixanol (OptiPrep in PBS-MK) stock in PBS-MK (1x PBS, 1 mM MgCl2, 2.5 mM KCl).
- Set up a gradient for AAV purification by using a Pasteur pipette to add 5 ml virus suspension into a Beckman Quick-Seal centrifuge tube (14×89 mm), followed by 1.5 ml each of 15%, 25% and 40% iodixanol solution. Top off the gradient with lysis buffer.
- Ultracentrifuge at 50.000 K for 2 h at 4 °C in a Beckman Ti70.1 rotor. Then clean the outside of the tube with 70% ethanol, stick a needle into the top of the tube for ventilation and draw 1.2 ml of the 40% iodixanol fraction using a needle. Take care to avoid drawing from the 25% fraction as it contains empty AAV capsids.
5. Screening and Selection
- At this point, one can either iteratively amplify the whole library in cultured cells or in animals until chimeric capsids exhibiting desired properties have emerged (Figs. 6-11).
- To select in cultured cells, co-infect various aliquots of the library (e.g., 1, 10 and 100 μl) and Adenovirus-5 to support AAV growth. Testing multiple variations of library and adenoviral helper is essential as library infectivity in a given cell type cannot be predicted. Harvest the cells after ~3 days, extract amplified AAV via freeze-thawing, inactivate Adenovirus for 30 min at 56 °C and re-infect new cells. Repeat up to 5 times until distinct capsids become enriched (validate by sequencing).
- For selection in animals, infect with the library and extract the desired tissue or cell type after ~1 week. Do not co-infect with Adenovirus as this will cause adverse toxicity in the animals. Rescue viral DNA via PCR using the same primers as before (Fig. 2), re-clone the cap pool, produce a fresh library and repeat until distinct capsids become enriched (validate by sequencing).
- The exact conditions (volume, titer, route) for selection in vivo will depend on the target tissue of interest. For liver, mice are typically infected with 1×1011 to 1×1012 viral particles in a total volume of 200 μl PBS via tail vein injection (IV). The limiting factor is usually the viral titer of the original preparation, because injection of 1×1012 AAV in 200 μl requires a titer of at least 5×1012/ml, which not all labs may routinely achieve. Hence, while a maximum titer is beneficial for the first infection (because the library has not yet been enriched for efficient capsids in a given tissue and may thus have a relatively low overall infectivity), we would recommend to use at least 1×1011 particles per mouse for liver selection.
- If multiple mice are available, it is very useful to inject different particle numbers akin to the selection in cultured cells, and to use several mice per group (and then pool the collected livers within each group) in order to minimize variability and to increase success rates, e.g. 3 mice at 1×1011 and 3 mice at 1×1012 particles.
- Since AAV is a non-pathogenic and replication-incompetent (without helpervirus) virus, there are no adverse reactions to look for in the absence of Adenovirus.
- For euthanization, animals were anesthesized using an isoflurane vaporizer and subsequently euthanized via cervical dislocation.
- The tissues (liver in this case) were harvested after the animals were confirmed euthanized. In general, there is no need also for other tissues to perfuse the animals or to harvest AAV-infected organs prior to death, since the AAV DNA is stable and can easily be rescued from frozen cells/tissues.
- To study single capsids (from the library (step 3.6) or after selection (steps 5.2-5.3)), produce AAV vectors encoding a reporter gene (e.g., gfp6) by following steps 4.1-4.5. Therefore, clone the cap gene of interest into a standard AAV helper plasmid2. At step 4.1, triple-transfect the cells with 14.7 μg each of AAV vector (encoding the reporter), AAV helper and adenoviral helper. Infect cultured cells or animals with the purified virus at different amounts and determine transduction efficiency e.g. via FACS or microscopy.
6. Representative Results
Use of the protocol outlined here typically results in viral libraries with a diversity of about 1×106 unique capsids which can then be screened for single particles displaying most or all desired properties on a given cell line or in animals. In the following, we will provide representative examples for results from such in vitro or in vivo screenings.
Prior to that, however, we consider it important to again point out the usefulness of analysing individual clones from the original plasmid library for their functionality and diversity (optional at step 3.6). This is because the latter two parameters are utmost critical prerequisites for the success of selection of the actual viral library that is made from the plasmid library. Therefore, one can randomly pick single shuffled cap genes and use them to produce recombinant AAV vectors (crude extracts or purified particles, depending on the desired degree of accuracy) expressing an easily detectable and quantifiable reporter gene. A typical assay to compare the different capsid variants is then fluorescence microscopy, as shown in the representative example in Fig. 4.
An alternative method is FACS-based measurement of fluorescent reporter gene expression which holds the extra benefits that it also allows for the determination of gene expression per cell, plus that it works for suspension cells. Fig. 5 shows a typical result from such a FACS-based analysis of crude AAV vector lysates in various cell types.
Because the above described selection of individual cap chimeras is random and restricted, it is of course not useful as an actual approach for enrichment of desired candidates. Instead, selection of the viral library in target cells or tissues in animals is more appropriate. As the conditions and parameters will vary with each application, we will only highlight a few representative guidelines and results.
The easiest selection is iterative library amplification on cultured cell lines or primary cells (step 5.2). Since AAV requires Adenovirus co-infection for its propagation, the target cells must be susceptible for Adenovirus. One can then grow and infect them e.g. in 6-well plates which hold sufficient cell numbers per well to ensure full coverage of the library. A second important notion is that the balance of AAV and Adenovirus is delicate; too much AAV will inhibit the Adenovirus, while an excess of the latter will kill the cells (unlike AAV, Adenovirus causes lytic infections) before the AAVs could replicate.
However, since library infectivity on a certain cell type is unknown, finding “good” AAV:Adenovirus ratios at which both can propagate requires parallel testing of various combinations of doses of the library and the adenoviral helper (e.g., 10:1, 1:1 and 1:10). A good measure for potent Adenovirus infection is the occurrence of cytopathic effects three days after virus inoculation, evidenced by cell rounding and detaching as seen in Fig. 6. Vice versa, a useful read-out for AAV infection and amplification is detection of capsid proteins by Western blotting, using the B1 antibody that recognizes a highly conserved AAV capsid epitope (Fig. 7)7.
An important decision is then which crude AAV extracts to pick for subsequent re-infection of fresh cells (step 5.2). Ideally, one will take those where the AAV capsid bands are least notable (Fig. 7) because these suggest a tight genotype-phenotype linkage. The latter describes the situation in which a genome encoding a certain capsid variant is actually packaged into the corresponding capsid. To achieve and maintain a tight genotype-phenotype linkage is key to successful selection of individual viral candidates as it in turn ensures that capsids with desired properties deliver the cognate genetic template into the cells during successive infection rounds. Therefore, we recommend to pick the crude extracts with moderate capsid expression for re-infection and to use minimal amounts. Together, these two measures will prevent overloading of the newly infected cells with different capsid/genome combinations which could otherwise perturb the genotype-phenotype linkage once the re-infected viruses start replicating and re-packaging their genomes into non-cognate capsids.
In addition, it is critical to monitor library diversity during repeated infections rounds by DNA sequencing. Ideally, one will notice shifts in the composition of individual clones indicative of successful selection, i.e., accumulation of distinct serotype fragments and losses of others, as exemplified in Fig. 8. In the perfect case, only a few or even a single clone will ultimately be detected which can then be analysed as described above. If, however, no shifts are observed after four or five passages, one should either increase the selection pressure (see Discussion) or consider that the cell line used may be inappropriate because it may be too susceptible to too many serotypes.
For library amplification in vivo (step 5.3), the same rules and considerations apply, with two critical differences: Firstly, one does not screen randomly selected clones in animals due to the associated costs and ethical considerations. Secondly, one will not co-infect with helper Adenovirus as it causes toxicities or fatalities in animals, plus the adenoviral tropism would restrict selection to cells susceptible to the helper virus. Instead, the AAV library is infused into the animals, rescued from the target cells/tissue by PCR (Fig. 9) and then re-cloned and re-packaged for a new infection round. As with selection in culture, this process is repeated until individual candidates have emerged.
Regardless of selection procedure, the final step is validation of the enriched capsid variants in appropriate systems. Figs. 10 and 11 show representative examples from our own previous selection of AAV chimeras that perform exceptionally well in tissue culture or in livers of mice. As seen in Fig. 10, one particular clone (AAV-DJ3) indeed outperforms a collection of eight natural AAV wildtypes in a broad range of cell lines. Finally, another clone selected in cultured hepatoma lines exhibits a higher tropism for the murine liver and accordingly less off-targeting when peripherally infused into adult mice than the potent AAV8 control vector tested in direct comparison (Fig. 11). Note that albeit being more specific for the liver, clone AAV-DN actually transduces this organ a bit less efficient than AAV8. In this respect, AAV-DN is a good representative example for the outcome of AAV evolution and selection where final candidates typically exhibit a number of desired properties but are not necessarily perfect in all aspects.
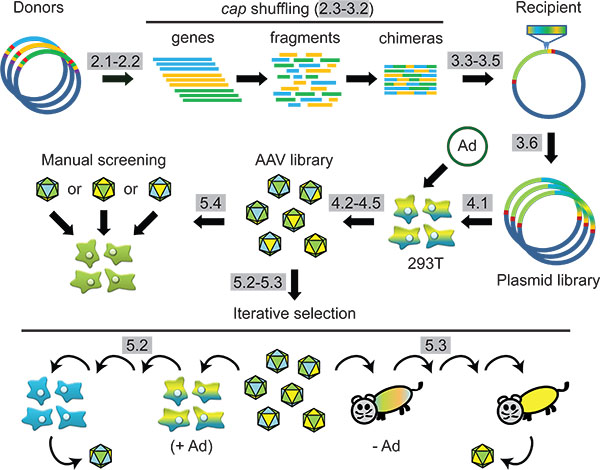
Figure 1. Scheme: Synthetic AAV capsid engineering via DNA family shuffling and subsequent selection in cells or in animals. Protocol steps are highlighted in grey.

Figure 2. AAV cap gene donor and recipient plasmids used in our lab for DNA family shuffling and library generation. Note that these are only representative examples, and that the exact sites and sequences can be customized. Our basic donor plasmid is derived from the commercially available pBlueScript II KS(+) vector which we engineered to contain the shown primer binding as well as restriction sites, depicted by arrows or triangles, respectively. We then cloned the cap genes of AAV serotypes 1-9 (amplified with primers capF/R) into this plasmid, to become flanked by Pac I and Asc I restriction sites. Primers T3 and T7 are used for the cap isolation in step 2.2, while later amplification of re-assembled chimeric sequences (step 3.2) can be accomplished using either primer pairs SAF/R or CUF/R, or both in a nested PCR. The replication-competent recipient plasmid carries AAV inverted terminal repeats (ITRs, replication and packaging signals) flanking the AAV2 rep gene under the control of the AAV p5 promoter. Pac I and Asc I restriction sites downstream of rep allow for “in frame” cloning of the pool of shuffled cap genes. The shown Lseq primer binding sites are useful for cap gene sequencing (step 3.6).

Figure 3. Example for DNase I digest of cap genes (AAV2, 8 and 9). Shown is an agarose gel electrophoresis analysis of the products of cap gene digests using the indicated different incubation times (in minutes:seconds). Lane U shows the undigested input cap fragment pool as control. Sizes of DNA marker bands in lanes M are in kilobases. In this example, ideal digests were obtained with incubation times of 1:45 or 2:00 min, which yielded the preferred predominant peak around 100 to 500 base pairs (yellow box).

Figure 4. Example for microscopy-based analysis of three human cell lines infected with five different recombinant Yfp-expressing AAV vectors (names on top) made with shuffled cap genes randomly selected from an original library based on AAV2, 8 and 9.

Figure 5. Example for FACS-based analysis of four cell types infected (at serial ten-fold dilutions) with 18 different recombinant Yfp-expressing AAV vectors (names on top, including those from Fig. 4) made with shuffled cap genes randomly selected from an original library (AAV2, 8 and 9). Yfp expression was color-coded for easier visualization. Depicted are percentages of transduced cells, whereby black always indicates 0% and white the highest number measured in each cell type. Clone B2 (red) exemplifies a clone with poor overall efficacy, as can be expected from unselected capsids. Note that FACS analysis is more sensitive and can also be used for suspension cells (such as SupT1) that are less amenable to microscopy. hu, human; mu, murine.

Figure 6. Typical appearance of cytopathic effects in cells (HeLa in this case) after productive co-infection with AAV and Adenovirus. Cells were either left uninfected (top left panel) or infected with the indicated amounts (particles per cell) of Adenovirus.

Figure 7. Detection of AAV capsid protein expression by Western blotting as a measure for library infection and amplification. The left blot shows cells co-infected with various volumes (in μl) of an AAV library and helper Adenovirus. The first two lanes are good examples for conditions yielding barely detectable AAV protein expression, indicating sufficient but not excessive AAV infection and amplification and hence the desired tight genotype-phenotype linkage. Thus, 0.1, 1 or 10 μl of these supernatants were used for re-infection of fresh cells (right blot). Cells in lane C were infected with AAV alone as a negative control (no detectable expression due to the absence of the helpervirus).

Figure 8. Comparisons of protein sequences (numbers are amino acids) of AAV clones from a library based on AAV2, 8 and 9 before and after selection. Sequences above the red line show the parental AAVs. Purple arrows indicate homologous recombination events. The red arrow marks a cross-over between AAV2 and AAV9 noted in all selected clones.

Figure 9. Rescue of successfully infected AAV clones from mouse tissues via PCR. In this example, AAV cap genes were PCR-amplified from mouse livers extracted one week after peripheral library infusion, using primer pair SAF/R (Fig. 2). Lane 1 shows the expected 2.2 kilobase band, while lane 2 is a non-template control. Following Pac I and Asc I restriction, the amplified fragments were re-cloned into the original recipient plasmid (Fig. 2) for subsequent production and re-infusion of a secondary library. Sizes of DNA marker bands in lanes M are indicated in kilobases.

Figure 10. Example for superior performance of an enriched AAV chimera in cultured cells. Clone AAV-DJ has been reported by us before3; it mostly represents a hybrid between AAV serotypes 2, 8 and 9, and has been selected from a library containing these three plus five additional serotypes in human liver cells in the presence of pooled human antisera. To compare its efficiency to eight natural AAV serotypes (indicated by 1-6, 8 and 9 on top), all cap genes were used to produce purified self-complementary Gfp-expressing vectors8,19. These were normalized to contain 2×109 vector genomes per ml and then used to transduce the shown cell lines at ten-fold serial dilutions. Three days later, Gfp-expressing cells were counted and infectious titers were determined by taking into account the dilution factor. In contrast to the code in Fig. 5, darker colors here indicate higher infectivities per particle number. As is evident, the selected AAV-DJ chimera outperforms all natural AAV wildtypes, exemplifying the success of the applied selection scheme. fibr, fibroblasts; ha, hamster; hu, human; mu, murine; si, simian.

Figure 11. Example for analysis of a selected AAV chimera in mouse liver. Clone AAV-DN was selected on murine hepatoma cells and then used to produce luciferase-expressing recombinant vectors8. Shown are representative mice (three per group) one week after peripheral infusion of equal doses of this vector or a control based on wildtype AAV8, one of the most potent known natural isolates in mouse liver9. Note that while the AAV-DN clone gives slightly less overall expression in the liver (panel (I)), it is more specific for this organ since it exhibits substantially less off-targeting in non-hepatic tissues once the liver expression levels have been adjusted via the imaging software (panel (II)).
