Ex Vivo Preparations of the Intact Vomeronasal Organ and Accessory Olfactory Bulb
Summary
The mouse accessory olfactory bulb (AOB) has been difficult to study in the context of sensory coding. Here, we demonstrate a dissection that produces an ex vivo preparation in which AOB neurons remain functionally connected to their peripheral inputs, facilitating research into information processing of mouse pheromones and kairomones.
Abstract
The mouse accessory olfactory system (AOS) is a specialized sensory pathway for detecting nonvolatile social odors, pheromones, and kairomones. The first neural circuit in the AOS pathway, called the accessory olfactory bulb (AOB), plays an important role in establishing sex-typical behaviors such as territorial aggression and mating. This small (<1 mm3) circuit possesses the capacity to distinguish unique behavioral states, such as sex, strain, and stress from chemosensory cues in the secretions and excretions of conspecifics. While the compact organization of this system presents unique opportunities for recording from large portions of the circuit simultaneously, investigation of sensory processing in the AOB remains challenging, largely due to its experimentally disadvantageous location in the brain. Here, we demonstrate a multi-stage dissection that removes the intact AOB inside a single hemisphere of the anterior mouse skull, leaving connections to both the peripheral vomeronasal sensory neurons (VSNs) and local neuronal circuitry intact. The procedure exposes the AOB surface to direct visual inspection, facilitating electrophysiological and optical recordings from AOB circuit elements in the absence of anesthetics. Upon inserting a thin cannula into the vomeronasal organ (VNO), which houses the VSNs, one can directly expose the periphery to social odors and pheromones while recording downstream activity in the AOB. This procedure enables controlled inquiries into AOS information processing, which can shed light on mechanisms linking pheromone exposure to changes in behavior.
Introduction
Sensory processing in the mammalian brain typically spans multiple reciprocally-connected neuronal circuits, each of which extracts particular features from sensory input. In sensory pathways, early information processing is vital for normal perception and behavior. In the accessory olfactory system (AOS), the accessory olfactory bulb (AOB) is the principal neural circuit linking the sensory periphery to downstream structures that dictate hormonal balance1,2, aggression3, and arousal4. As such, information processing within this circuit is strongly linked to changes in animal behavior.
The accessory olfactory bulb is located in mice and rats at the dorsal/caudal/posterior aspect of the main olfactory bulb (MOB) beneath the dense, vascularized rhinal sinus. The AOB receives afferent innervation from axons of peripheral vomeronasal sensory neurons (VSNs) that reside in the vomeronasal organ (VNO), a small blind-ended tube in the anterior snout just above the soft palate. These axons traverse the delicate sheet of septal tissue at the medial boundary of the nasal passages. Several studies have probed AOB neural responses to sources of AOS odors (such as mouse urine) in vivo using anesthetized mice5-7 or freely-exploring animals8. The heroic anesthetized in vivo studies involved (a) tracheotomies to ensure deep anesthesia and prevent the aspiration of liquid stimuli5-7, (b) stimulation of the sympathetic cervical ganglion6 or direct cannulation of the vomeronasal organ5,7 to introduce nonvolatile odors and (c) craniotomies with or without frontal lobe ablations to allow electrode advancement into the AOB6. Awake/behaving studies8-10 involved surgical implantation of a microdrive. In sum, these experimental paradigms are powerful, but extremely difficult and often require anesthesia.
Interestingly, several studies attempted to maintain sensory structures and downstream neural circuits alive outside the body (ex vivo) with some success11-15. Because the connections between the VNO and AOB remain ipsilateral, and because the midline septal tissue can be exposed to oxygenated superfusate in a single hemisphere, we sought to develop such a single-hemisphere ex vivo approach to isolate these structures while maintaining their functional connectivity. We recently succeeded in achieving this goal16. This preparation keeps both the VNO and AOB alive and functionally connected for at least 4 – 6 hr because both the axons (along the midline soft septal tissue) and AOB are relatively shallow <600 µm features that are accessible to superfused oxygenated artificial cerebrospinal fluid (aCSF). This VNO-AOB ex vivo preparation allows introduction of controlled stimuli into the VNO via a thin cannula, and direct visual access to the small AOB for targeted electrode placement and/or live fluorescence microscopy. This method is advantageous if one desires to study these circuits in the absence of anesthetics. Because this approach severs centrifugal connections, it is not well suited to inquiries into centrifugal modulation of AOB function. The VNO-AOB ex vivo preparation is difficult to learn, but once achieved produces a reliable platform upon which to investigate circuit organization, information processing, and neural plasticity in this powerful sensory circuit.
Protocol
All experiments were carried out in accordance with protocols approved by the UT Southwestern Institutional Animal Care and Use Committee, and were chosen so as to minimize stress, discomfort, and pain experienced by the experimental animals.
1. Dissection Chamber
A custom dissection chamber and small, thin plastic plank are required for best results (Figure 1). Construct or obtain such a chamber in advance of attempting this protocol.
2. Dissection Solutions
- Prepare 1 L of standard artificial cerebrospinal fluid (aCSF) for use in Steps 3.22-5.4. Add NaCl 125 mM, KCl 2.5 mM, CaCl2 2 mM, MgCl2 1 mM, NaHCO3 25 mM, NaH2PO4 1.25 mM, myo-inositol 3 mM, sodium pyruvate 2 mM, sodium ascorbate 0.4 mM, and glucose 25 mM to 1 L of pyrogen free water.
- Prepare the initial dissection aCSF (200 ml) for use in steps 3.3-3.22. Take 200 ml of Standard aCSF and raise the MgCl2 concentration by 9 mM by adding 0.366 g of MgCl2 hexahydrate.
- Prepare standard Ringer’s solution to carry stimuli to the VNO through the cannula for use in steps 4.11-5.4 containing NaCl 115 mM, KCl 5 mM, CaCl2, 2 mM, MgCl2, 2 mM, NaHCO3 25 mM, HEPES 10 mM, glucose 10 mM.
- Bubble the 0.8 L of standard aCSF and 0.5 L of Ringer’s with 95% O2, 5% CO2 gas in a 37 °C water bath for a minimum of 15 min until use.
- Bubble the 200 ml of initial dissection aCSF with 95% O2, 5% CO2 gas on ice for 15-20 min until it reaches 4 °C.
3. Primary Dissection
- Prepare the dissection area. Place three 35 mm Petri dishes, oxygenated aCSF, decapitation scissors, curved dissecting scissors, straight dissecting scissors, Adson forceps, a #11 scalpel blade and handle and a razor blade on ice.
- After oxygenated aCSF has cooled to 4 °C, deeply anesthetize the animal in a desiccation chamber using 2-5 ml Isothesia (99.9% isofluorane). Perform tail and foot pinch tests to ensure the animal is deeply anesthetized.
- Once deep anesthesia is confirmed, decapitate the animal and immediately place the head in a Petri dish filled with the chilled dissection aCSF. NOTE: Immerse the tissue in chilled dissection aCSF to keep it cool throughout the procedure when not in use.
- Remove the lower aspect (ventral pharynx, lower jaw, and tongue) with a curved scissors.
- Using Adson forceps deglove (peel) the superficial skin and muscle attachments from the skull. Peel the scalp from caudal to rostral until the only site of attachment is over the front teeth. Cut the tissue at the attachment point using a curved scissors. Remove the eyes by grasping it with forceps and pull it away from the preparation. Near the end of the degloving procedure, avoid excess force on the delicate nasal cartilage by using the Adson forceps to free muscle attachments from the upper jaw bones. NOTE: Detach the scalp from the caudal aspect of the skull with straight scissors.
- With the straight scissors, cut down the midline of the skull from caudal to rostral until the scissor tips are a few mm into the frontal bones (just prior to the rhinal sinus).
- Remove the parietal and frontal bones of the skull using Adson forceps. NOTE: Pieces of the frontal bone will often remain attached to the nasal bones caudal to the rhinal sinus. Remove these pieces with care taken not to contact the olfactory bulbs.
- Use the scalpel to make a coronal cut through the frontal lobe 2 mm caudal to the sinuses and then remove the brain caudal to the cut. Ensure the tip of the scalpel contacts the bony contour of the ventral skull to completely sever the posterior tissue. NOTE: This enables removal of majority of the brain without putting mechanical stress on the lateral olfactory tracts or olfactory bulbs.
- Remove the exposed caudal aspects of the ventral skull with straight scissors. Leave the sinuses and remaining nervous tissue.
- Remove the zygomatic arch and facial muscles on the anatomical right side of the skull using straight scissors
- Turn the snout over (ventral side up) to expose the roof of the mouth.
- Cut and remove the palate immediately caudal to the incisors. Do this by inserting the tip of the scalpel blade under the palate parallel to the roof of the mouth and then move the blade towards the incisors.
- Grasp the freed (rostral) edge of the palate with Adson forceps and remove by pulling caudally.
- On the anatomical left side of the skull, use the scalpel blade to extend the palatine foramen caudally, starting from the small foramen near the molars. Achieve this by inserting the tip of the scalpel blade into the void near the molars and rotating the wrist. NOTE: Use extreme caution to only use the tip of the blade – a deep cut can damage the septal tissue.
- Using the tip of the scalpel blade, cut from the palatine foramen through the incisors, starting rostral to the anatomical left vomeronasal organ. Use multiple scoring cuts. Do not damage the septal tissue and vomeronasal nerves by inserting the blade too deep.
- Turn the tissue so the dorsal skull is facing up and then orient the straight razor blade vertically (parallel to the midline) at the caudal edge of the preparation.
- At the ventral edge of the tissue, touch the cutting edge of the razor blade immediately to the left of the midline. Gently rock the blade and move along the left palatine foramen (cut region introduced in Step 3.14).
- At the dorsal edge of the tissue, place the cutting edge of the razor immediately to the left of the midline (less than 1 mm).
- Keeping the blade parallel to the midline, rock the razor blade slowly with pressure towards the anterior of the tissue. Notice resistance at the cribriform plate. Stop immediately after breaking through this resistance. NOTE: At this point, the right hemisphere is loosened from the left hemisphere. The only remaining connection is between the hemispheres along the dorsal snout anterior to the cribriform plate.
- Separate the hemispheres by grasping them near the caudal sides with gloved fingers and gently rotating them laterally and anteriorly. NOTE: This step is a source of experimental variability, especially in mice with deviated or brittle snouts (e.g., older mice). Also, while attempting to rotate the hemispheres away from each other, if strong resistance is encountered score the dorsal snout (anterior to the cribriform plate) to weaken the connective tissues. While doing so, take extreme caution not to insert the scalpel deeply, which can damage the septal tissue and vomeronasal nerves.
- Apply a small amount (50-100 µl) of tissue glue to the plank and gently place the lateral edge of the right hemisphere onto the glue using the Adson forceps. Remove excess moisture from the lateral side of the preparation using a paper towel to prevent premature glue polymerization and loose adhesion to the plank. NOTE: Apply a few drops of the chilled dissection aCSF on the sample to accelerate the glue polymerization after tissue is placed onto the glue. This ensures that the tissue remains tightly held to the plank.
- Place a small amount (3-5 mm diameter, 2 mm deep) of vacuum grease in the middle of the secondary dissection chamber and place the side of the plank opposite the tissue firmly against the grease. Immediately fill the dissection chamber with chilled dissection aCSF, transfer to the fine dissection area and begin perfusion with oxygenated standard aCSF. Orient the plank so that the olfactory bulb is directly in the stream of freshly oxygenated standard aCSF.
4. Secondary Dissection
- Remove any visible contralateral (left) olfactory bulb or cortical tissue from the preparation using fine forceps. Pinch together the fine forceps (forming a semi blunt point), and then place it at the dorsal and caudal portion of the tissue near the midline. Gently run the pinched forceps along the midline, peeling the tissue away from the right hemisphere slowly in anterior motion. Remove all of the contralateral tissue, including the contralateral olfactory bulb tissue. Take extreme care not to stab through the tissue, which can cause damage to the AOB or vomeronasal nerve.
- Observe the white dura mater along the dorsal/medial surface of the olfactory bulb. Tear this dura mater along the dorsal/caudal edge of the olfactory bulb by grasping at the whitest portion of the dura with two pairs of fine forceps and pull them away from that point. The dura is tightly wrapped around the olfactory bulbs, and can easily itself slice through tissue while it is being removed. Avoid damage to the medial surface of the olfactory bulb and the AOB itself. NOTE: If the dura resists tearing easily at this point, introduce a small cut with fine spring tip dissection scissors to facilitate separation.
- Slowly and carefully peel away the frontal cortex with fine forceps without damaging the underlying olfactory bulbs. Observe a collection of blood vessels appearing as a semi-transparent net immediately caudal to the AOB. Remove this net with fine forceps to facilitate electrode penetration in electrophysiological experiments. Grasp the net with forceps as it separates from the AOB during frontal cortex retraction and remove it by pulling caudally and medially, being careful not to touch the AOB. Once the frontal cortex has been separated from the olfactory bulbs, remove it by cutting with forceps ventral and posterior to the AOB. NOTE: Take extreme care with net removal, for if it is done too roughly the glomerular layer can become damaged.
- In the exposed contralateral nasal cavity, remove any remaining contralateral septal tissue (flimsy yellowish sheet containing blood vessels) using the fine forceps. Grasp the tissue at the caudal/dorsal region (near the septal bone) and then pull/lift the tissue towards the anterior side. NOTE: The contralateral septal tissue can be inadvertently removed during the hemisection step (Step 3.20). In such case, observe a white, avascular septal cartilage and the slightly transparent, vascularized septal bone. If this is the case, Step 4.4 can be skipped.
- Carefully remove the contralateral VNO by running a single point of the fine forceps between the septal cartilage and the VNO “wing” (a thin piece of bony tissue that runs along the dorsal edge of both VNOs.
- Following the wing detachment from the cartilage, move the tip of forceps ventrally into the contralateral VNO near the midline. Repeat this step at several locations along the rostral/caudal axis of the contralateral VNO to loosen it, enabling it to be removed by grasping with forceps and lifting away.
- Prepare to remove the septal cartilage by making a cut at the ventral/caudal edge (where it narrows to a white avascular strip running along the ventral edge of the septal bone) with fine forceps.
- Remove the septal cartilage by pinching together the fine forceps to make a semi blunt point. Identify the cut made in Step 4.6, insert the pinched forceps behind (lateral) to that cut, and then slowly lift the cartilage away from the septal tissue and proceeding rostrally. NOTE: Do not disrupt the underlying septal tissue. As the cartilage is lifted, observe the underlying sheet of septal tissue stretch due to loose adhesions to the cartilage. A periodic, gentle, and stable movement of the pinched fine forceps along this site of loose adhesion can greatly facilitate successful separation of the two structures.
- Use a pair of #3 forceps with a bent tip to remove the septal bone. Approach the septal bone at an angle nearly parallel to the plane of the septal bone. Insert the curved tine between the septal tissue and septal bone. Grasp the bone with the forceps and gently move the bone back and forth until it cracks near the cribriform plate. NOTE: In some cases, the septal bone will resist breaking near the cribriform plate. In this case, use fine forceps to gently score the septal bone along the dorsal/ventral axis just anterior to the cribriform plate, then repeat Step 4.8.
- Remove the white cartilage just rostral to the VNO by pinching with one pair of fine forceps at the entrance and pulling rostrally with another pair of fine forceps.
- Attach the polyimide cannula to a fast perfusion device and start the flow of Ringer’s solution (0.1-0.3 ml/min) through the polyimide cannula. NOTE: Measure flow rate with an in-line, small volume flow meter. Use of computer controlled pneumatic stimulus switching device reduces flow rate changes during stimulation. Compare neural responses to test stimuli with responses to a mock stimulus (control Ringer’s solution) to ensure responses are stimulus specific.
- Orient the cannula parallel to the VNO entrance. When the cannula is satisfactorily parallel to the VNO entrance, watch as the outlet pressure causes the VNO to “inflate,” leading to evacuation of the blood vessel. Immediately insert the cannula into the VNO, keeping the angle of the cannula constant so that it remains parallel to the VNO opening. NOTE: It can be helpful to hold the cannula with one pair of fine forceps against the plastic plank and use another pair of fine forceps to angle the end of the cannula slightly upwards. If one inadvertently places the cannula behind the VNO, the outflow can also cause the VNO blood vessel to evacuate, leading to the impression a proper cannulation was performed. Proper location can be confirmed by incorporating a dye in the Ringer’s solution. When the cannula is properly placed, the dye should only exit via the VNO entrance. Additionally, one can confirm proper placement by ensuring that the cannula is easily visible through the semi-transparent medial aspect of the VNO.
- Move the plastic plank slowly until the AOB is in the direct flow of the standard aCSF, being careful not to dislodge the cannula from the VNO. The dissection is complete and assessment of physiological function can commence immediately. Step 5 briefly describes one approach for making such an assessment.
5. Evaluation
- Penetrate the surface of the AOB with a 2-4 MΩ borosilicate glass microelectrode attached to an extracellular amplifier and oscilloscope.
- Slowly advance the microelectrode to a depth of 100-200 µm from the surface at a rate of 50 µm/min. NOTE: At this tissue depth, one will encounter occasional spontaneous action potential waveforms exemplified by sharp, brief (~1-2 msec) deflections in the microelectrode voltage.
- Upon encountering an action potential waveform, stop advancing the microelectrode.
- Observe and/or record the action potential rate while changing the stimulus delivery solution from control Ringer’s to a known activator of VSN activity (e.g., Ringer’s solution containing 50 mM KCl). If the dissection was successful, a stimulus-dependent change in action potential rate or local field potential can be observed. NOTE: Not all AOB neurons will respond to every stimulus with an increase in firing rate (including Ringer’s solution containing 50 mM KCl). A stimulus-dependent decrease in firing rate also indicates functional connectivity and a successful dissection. In the event that two or more neurons are encountered that are unresponsive to VNO stimulation, or if no action potentials are observed after multiple electrode penetrations, this suggests an unsuccessful dissection. If this is the case, a new dissection can be initiated at Step 3.
Representative Results
Achieving success with this preparation takes extensive practice, and has several steps at which it can fail. One should expect to require many attempts before achieving success. The custom dissection chamber is required for successful completion of this protocol, and should be obtained prior to starting the later stages of the dissection. The chamber design presented in Figure 1 is sufficient for this purpose, and can be made of relatively inexpensive plastics with minimal machining demands. If one lacks the capacity to produce such a chamber, local or online machining companies can be consulted and can construct the chamber for a fee.
A large source of variability across preparations is the density and brittleness of the bones of the skull and snout of the experimental animals. During the hemisection steps (Steps 3.14-3.20), the goal is to isolate both vomeronasal organs along with the ipsilateral septal tissue and ipsilateral accessory olfactory bulb. However, it is not uncommon for the septal tissue to remain attached to the contralateral hemisphere, especially near attachment points to the dorsal bones of the snout. Figure 2 shows an example of an effective and ineffective hemisection.
Other common dissection errors arise during the fine dissection, during which the axons of the vomeronasal nerve can be damaged in the septal tissue near the vomeronasal organ (Figures 3A and 3B) or near the AOB (Figures 3C and 3D). These and similar events will result in incomplete connectivity between VSNs and the AOB, rendering the preparations unusable.
The final obstacle to the preparation’s success is the VNO cannulation step (Step 4.11). Proper placement of the VNO cannula will result in pressurization of the VNO lumen, causing immediate expulsion of residual blood from the vomeronasal pump. The cannula should be clearly visible beneath the relatively transparent vomeronasal epithelium and the appearance of the cannula should be uniform along its length inside the VNO. Upon successful completion of the procedure, one can verify functional connectivity using a physiological assay such as single unit electrophysiological recordings of neural activity in response to VNO stimulation with a known source of VSN odorants (Figure 4).
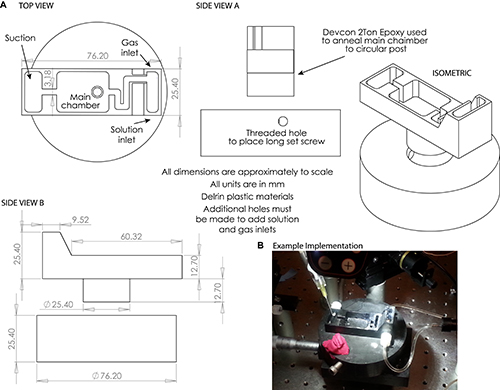
Figure 1. A) Engineering drawings of the custom dissection chamber used in Steps 3.22-5.4. NOTE: Small holes may be drilled to accommodate solution and gassing inlets and outlets as desired. B) Example photograph of dissection chamber used in this protocol. Please click here to view a larger version of this figure.

Figure 2. A, B) Representative images of successful (A) and unsuccessful (B) preparations after the hemisection stage (Step 3.20). The VNO, septal tissues, and olfactory bulbs (OBs) must all be maintained from the ipsilateral hemisphere (in this case, the right (bottom) hemisphere. If nasal turbinates are visible on the ipsilateral hemisphere, the dissection has failed. Tick marks are 1 mm apart. Please click here to view a larger version of this figure.

Figure 3. A, B) Representative images of intact and damaged septal tissue between the VNO and AOB. In B, the dissecting forceps made inadvertent contact with the tissue, damaging the VN axon tracts. C, D) Intact and damaged AOBs. In D, removal of the dura surrounding the ipsilateral OB resulted in severing the vomeronasal nerve near the AOB, causing a visible blob of tissue near the medial AOB. Scale bars are 1 mm in length.

Figure 4. A) Example raster plot of action potentials recorded from an AOB neuron (extracellular single unit recording) in a successful preparation. Traces in black show no change in firing rate when control Ringer’s solution was delivered to the VNO for 5 sec (gray box). Traces in red show the response of the same neuron to VNO stimulation with BALB/c female mouse urine diluted 100-fold into Ringer’s saline. B) Peri-stimulus time histogram of the same responses in A. Error bars represent standard errors of the mean.
Discussion
The VNO-AOB ex vivo preparation described in this protocol is a useful alternative to anesthetized in vivo5-7 and acute live slice17 experiments of AOB function. Unlike acute AOB slice experiments, which also expose circuit elements for electrophysiological and optical recordings, this preparation retains all sensory afferents and intra-AOB connections. Although this can also be said of anesthetized in vivo approaches, the presence of anesthetics necessarily alters neural function, namely excitatory/inhibitory balance, which is crucial for information processing in this and other olfactory circuits18. Because the preparation avoids the use of anesthetics, and because the AOB surface is completely exposed to the superfusate, it is amenable to pharmacological inquiries into local neural processing.
There are, of course, some limitations of this preparation. There is evidence for gradual degeneration of the deepest AOB layers, namely the deep portions of the internal cellular layer, which houses many inhibitory granule cells16. Additionally, centrifugal inputs are severed during the dissection, eliminating them from active participation in AOB function. Cannulation of the VNO bypasses the normal action of the vomeronasal pump, and flushes away the endogenous fluids present in the VNO lumen.
Critical steps to achieving success with this method are the delicate hemisection (Step 3.20) and VNO cannulation (Step 4.11). One can expect to require extensive practice to reliably produce functionally-connected ex vivo preparations. The secondary dissection chamber utilized here can be used to assess functional connectivity using single-electrode recordings during stimulation of the VNO with sources of mouse pheromones (such as dilute urine).13,15 Modifications of the secondary dissection chamber can be made that allow the preparation to rotated to angles suited for other purposes, such as multiphoton imaging.
The many technical hurdles to studying the living AOB have long posed a barrier to our understanding of social odor and pheromone sensory processing. By dissecting away the earliest neural components of this sensory pathway in this dissection protocol, one is able to gain experimental access to these hard-to-study circuits. We have utilized this preparation for detailed inquiries into vomeronasal sensory processing19 and sensory mapping (Hammen et al., unpublished). This technique will be useful for future studies into sensory processing in the AOB, especially those requiring optical access to the tissue (e.g., multiphoton imaging and optogenetics). The benefits of this approach complement those of in vivo approaches, and improve our toolkit for investigating the neural mechanisms of vomeronasal-mediated social and reproductive behaviors.
Disclosures
The authors have nothing to disclose.
Acknowledgements
This research was supported by R00 DC011780 (JPM: NINDS, NIH), F30 DC011673 (GFH: NINDS, NIH) and UT Southwestern startup funds (JPM).
Materials
| Straight Scissors | Fine Science Tools | 14002-14 | |
| Fine Scissors-Straight | Fine Science Tools | 14060-10 | |
| Fine Scissors-Curved | Fine Science Tools | 14061-10 | |
| Adson Forceps | Fine Science Tools | 11006-12 | |
| #3 Scalpel Handle | Fine Science Tools | 10003-12 | |
| #11 Scalpel Blades | Fisher Scientific | 3120030 | |
| Straight Carbon Steel Razor Blades | Fisher Scientific | 12-640 | |
| 35 mm Petri Dish | Fisher Scientific | 08-772-21 | |
| Dissection Chamber | Custom | N/A | See Fig. 1 |
| Delrin plastic plank 0.6 cm x 1.5 cm x 0.1 cm | Custom | N/A | |
| Dow Corning Silicon Vacuum grease | Fisher Scientific | 146355D | |
| #5 Forceps, Student | Fine Science Tools | 91150-20 | |
| #5 Forceps, Biologie Tip | Fine Science Tools | 11295-10 | |
| #5 Forceps, Student | Fine Science Tools | 91150-20 | |
| Vannas Spring Scissors | Fine Science Tools | 15000-08 | |
| 0.0045" Polyimide Tubing | A-M Systems | 823400 | |
| 1/16" Male Luer | Cole-Parmer | EW-45505-00 | |
| 1/16" Tubing | Fisher Scientific | 14-171-129 | |
| Two ton epoxy | Grainger | 5E157 | |
| ValveBank Pressurized Perfusion Kit | AutoMate Scientific | 09-16 | |
| ValveLink digital/manual controller | AutoMate Scientific | 01-18 | |
| NaCl | Sigma-Aldrich | various | |
| KCl | Sigma-Aldrich | various | |
| CaCl2 dihydrate | Sigma-Aldrich | various | |
| MgCl2 hexahydrate | Sigma-Aldrich | various | |
| NaHCO3 | Sigma-Aldrich | various | |
| NaH2PO4 | Sigma-Aldrich | various | |
| myo-inositol | Sigma-Aldrich | various | |
| Na-pyruvate | Sigma-Aldrich | various | |
| Na-ascorbate | Sigma-Aldrich | various | |
| HEPES buffer | Sigma-Aldrich | various | |
| glucose | Sigma-Aldrich | various |
References
- Bruce, H. M. An exteroceptive block to pregnancy in the mouse. Nature. 184, 105 (1959).
- Bellringer, J. F., Pratt, H. P., Keverne, E. B. Involvement of the vomeronasal organ and prolactin in pheromonal induction of delayed implantation in mice. J Reprod Fertil. 59, 223-228 (1980).
- Bean, N. J. Modulation of agonistic behavior by the dual olfactory system in male mice. Physiol Behav. 29, 433-437 (1982).
- Meredith, M. Vomeronasal organ removal before sexual experience impairs male hamster mating behavior. Physiol Behav. 36, 737-743 (1986).
- Hendrickson, R. C., Krauthamer, S., Essenberg, J. M., Holy, T. E. Inhibition shapes sex selectivity in the mouse accessory olfactory bulb. J Neurosci. 28, 12523-12534 (2008).
- Ben-Shaul, Y., Katz, L. C., Mooney, R., Dulac, C. In vivo vomeronasal stimulation reveals sensory encoding of conspecific and allospecific cues by the mouse accessory olfactory bulb. Proc Natl Acad Sci U S A. 107, (2010).
- Tolokh, I. I., Fu, X., Holy, T. E. Reliable sex and strain discrimination in the mouse vomeronasal organ and accessory olfactory bulb. The Journal of neuroscience : the official journal of the Society for Neuroscience. 33, 13903-13913 (2013).
- Luo, M., Fee, M. S., Katz, L. C. Encoding pheromonal signals in the accessory olfactory bulb of behaving mice. Science. 299, 1196-1201 (2003).
- Binns, K. E., Brennan, P. A. Changes in electrophysiological activity in the accessory olfactory bulb and medial amygdala associated with mate recognition in mice. Eur J Neurosci. 21, 2529-2537 (2005).
- Leszkowicz, E., et al. Noradrenaline-induced enhancement of oscillatory local field potentials in the mouse accessory olfactory bulb does not depend on disinhibition of mitral cells. Eur J Neurosci. 35, 1433-1445 (2012).
- Ames, A., Gurian, B. S. Electrical Recordings from Isolated Mammalian Retina Mounted as a Membrane. Arch Ophthalmol. 70, 837-841 (1963).
- Flock, A. F., Strelioff, D. Studies on hair cells in isolated coils from the guinea pig cochlea. Hear Res. 15, 11-18 (1984).
- Woodbury, C. J., Ritter, A. M., Koerber, H. R. Central anatomy of individual rapidly adapting low-threshold mechanoreceptors innervating the ‘hairy’ skin of newborn mice: early maturation of hair follicle afferents. J Comp Neurol. 436, 304-323 (2001).
- Llinas, R., Muhlethaler, M. An electrophysiological study of the in vitro, perfused brain stem-cerebellum of adult guinea-pig. The Journal of physiology. 404, 215-240 (1988).
- Riviere, S., Challet, L., Fluegge, D., Spehr, M., Rodriguez, I. Formyl peptide receptor-like proteins are a novel family of vomeronasal chemosensors. Nature. 459, 574-577 (2009).
- Meeks, J. P., Holy, T. E. An ex vivo preparation of the intact mouse vomeronasal organ and accessory olfactory bulb. J Neurosci Methods. 177, 440-447 (2009).
- Leinders-Zufall, T., et al. Ultrasensitive pheromone detection by mammalian vomeronasal neurons. Nature. 405, 792-796 (2000).
- Kato, H. K., Chu, M. W., Isaacson, J. S., Komiyama, T. Dynamic sensory representations in the olfactory bulb: modulation by wakefulness and experience. 76, 962-975 (2012).
- Meeks, J. P., Arnson, H. A., Holy, T. E. Representation and transformation of sensory information in the mouse accessory olfactory system. Nature. 13, 723-730 (2010).
