

Summary
Qui, segnaliamo la sintesi e la cristallizzazione di 3,5-lutidine N -ossido di disidratare da un semplice protocollo che differisce dalla classica sintesi della piridina N-ossido. Questo protocollo utilizza materiale di partenza differente e comporta meno tempo di reazione per produrre una nuova struttura supramolecolare solvatato, che cristallizza sotto lenta evaporazione.
Abstract
La sintesi di 3,5-lutidine N -ossido di disidratare, 1, è stata realizzata nell'itinerario della sintesi dell'acido 2-ammino-piridina-3,5-dicarbossilico. Ochiai prima usato la metodologia per non-sostituiti piridine nel 1957 in un processo di 12 h, ma sono stato ottenuto senza cristalli adatto ai raggi x. L'anello sostituito utilizzato nella metodologia presentata qui chiaramente influenzato l'aggiunta di molecole d'acqua nell'unità asimmetrica, che conferisce una forza nucleofila diversa in 1. I raggi x cristallo adatto composto 1 era possibile dovuto la stabilizzazione della carica negativa nell'ossigeno dalla presenza di due molecole d'acqua dove gli atomi di idrogeno donano carica positiva sul ring; tali molecole di acqua servono bene per costruire un'interazione supramolecolare. Le molecole idratate potrebbero essere possibile per il sistema alcalino che si raggiunge regolando il pH a 10. D'importanza, il metile doppio sostituito con anello e un tempo di reazione di 5h, lo rende un metodo più versatile e con applicazioni chimiche più ampia per gli inserimenti futuri anello.
Introduction
Al giorno d'oggi, gli scienziati di tutto il mondo hanno investito risorse nello sviluppo di nuove vie di sintesi per la funzionalizzazione dei gruppi aromatici, che sono conosciuti per la parte anteriore bassa reattività per aggiunta reazioni1,2, 3. Piridina, dove un atomo di azoto sostituisce un atomo di carbonio, presenta una reattività chimica simile analogico anelli è composto unicamente da atomi di carbonio3e subisce solitamente un meccanismo di sostituzione anziché aggiunta. N-ossidi si distinguono per la presenza di un legame di donatore tra azoto e ossigeno formata dalla sovrapposizione di nonbonding coppia di elettroni sull'azoto con un orbitale vuoto sul atomo di ossigeno3. In particolare, piridina N-ossido è basi di Lewis, perché loro frazione di N-O può agire come un donatore di elettroni, e si può combinare con acidi di Lewis, formando il corrispondente coppie acido-base di Lewis. Questa proprietà ha una conseguenza chimica essenziale, perché può aumentare la nucleofilicità degli acidi di Lewis verso potenziali elettrofili e quindi permettere loro di reagire in condizioni dove normalmente la reazione non si verificherebbe. Probabilmente l'uso più frequente di tali composti è nelle varie reazioni di ossidazione, dove agiscono come ossidanti4. Piridina N-ossidi e molti dei loro derivati funzionalizzati anello sono molecole ricorrenti di agenti biologicamente attivi e farmacologici5, e una distribuzione chiara spaziale di diversi strumenti spettroscopici è stata stabilita per alcuni di loro6,7. Nella ricerca sul collegamento di gruppi diversi per l'anello piridinico, gli scienziati hanno testato varie metodologie per produrre un metodo facile e convenzionale, dal isossazoline richiede una quantità catalitica di base quali DBU in ebollizione xilene per formare 6- sostituito-2-aminopyridine N-ossidi8,9. Una varietà di derivati della piridina sono stati convertiti in loro corrispondenti N-ossidi in presenza di una quantità catalitica di manganese tetrakis(2,6-diclorophenyl) porfirina e ammonio acetato CH2Cl2/CH3 CN8,10. Altri piridine sono ossidati a loro ossidi utilizzando H2O2 in presenza di quantità catalitiche di methyltrioxorhenium8,11, o con l'aggiunta di dimethyldioxirane in eccesso in CH2Cl2 a 0 ° C, che conduce al corrispondente N-ossidi8,12,13,14. Bis (trimetilsilile) perossido in presenza di trioxorhenium CH2Cl2 è stato utilizzato per la sintesi della piridina N-ossidi8,11. La sintesi di aminopiridina N-ossidi che coinvolge acilazione utilizzando (acido peroxomonosulfuric) di Caro è stato anche segnalato8. Tuttavia, la metodologia segnalato qui, e che utilizza parte della metodologia riportata da Ochiai1, fornisce risultati molto buoni con l'uso di reagenti più convenienti e accessibili, H2O2 e acido acetico glaciale. Questa pratica è più adatto ad uso nelle preparazioni su larga scala che agiscono sulle ammine terziarie, produce buone rese in una reazione che richiede solo 30% di perossido di idrogeno e acido acetico glaciale in una temperatura compresa tra 70-80 ° C e utilizza un processo di purificazione che è disponibile nella maggior parte dei laboratori di sintesi come la distillazione, senza l'uso del catalizzatore o più costosi reagenti1. La letteratura segnala che altre metodologie anche frequentemente coinvolgono tempo cornici da 10-24 h e temperature superiori a 100 ° C 4,8, e la resa di cristalli ben formati per analisi dei raggi x è segnalata raramente.
Reattivo, vari N -ossido derivati vengono utilizzati per attivare adeguatamente l'anello lutidine, in maniera elettrofila o nucleofila. Il fattore elettrofilo o nucleofilo risente i sostituenti. Con l'anello di piridina essendo i gruppi elettron-attrattori, il fattore principale è il nucleofilo caratteristica1. Il gratuito N-ossido composti sono raramente isolate come cristalli adatti per analisi di raggi x dovuto la carica delocalizzata sull'anello aromatico. Tuttavia, il fattore di solvatazione è fondamentale per stabilizzare la densità negativa dell' ossigeno15.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. reazione
- Mettere in una cappa un pallone rotondo aperto 100 mL con 0,5 mol (29,8 mL) di acido acetico glaciale e aggiungere 0,051 mol (5,82 mL) di 3,5-dimethylpyridine e 5 mL di H2O2 (35%). Mantenere la reazione di miscela sotto costante agitazione magnetica, ad una temperatura interna di 80 ° C per 5 h.
- Dopo il tempo di reazione, raffreddare il pallone a 24 ° C con ghiaccio (do non esporre l'acido acetico di gas per il ghiaccio) e collegarlo a un'unità di distillazione sotto vuoto ad alta per 90-120 min per rimuovere l'eccesso di acido acetico.
Attenzione: Non utilizzare materiale caldo. Attendere fino a quando il vetro raggiunge una temperatura gestibile. Questo eviterà anche vapori entrando parte superiore dell'unità di distillazione. - Aggiungere acqua distillata acqua (10 mL) due volte a garantire la rimozione di qualsiasi traccia di acido acetico e di concentrare la miscela molto possibile.
2. estrazione e regolazione basicità
- Sciogliere in acqua bi-distillata prodotto viscoso e trasparente isolato e utilizzare un potenziometro per regolare il pH a 10 con puro solido Na2CO3.
- Posizionare con cura la soluzione in un imbuto separatore mL 250 ed estrarlo 5 volte con 250 mL di CHCl3 per migliorare il rendimento. Recuperare lo strato organico e asciugarlo sopra tinta Na2così4 per 30 min massimo, che conterrà il prodotto. Se necessario, estrarre nuovamente la fase acquosa con la quantità desiderata di CHCl3.
Attenzione: CHCl3 può provocare sonnolenza e vertigini; maneggiare con cura e all'interno di una cappa aspirante. - Rimuovere il solvente sotto pressione ridotta con un'unità di distillazione sotto vuoto ad alta, fino alla formazione di una polvere cristallina chiaro beige molto igroscopica (70%).
3. della cristallizzazione
- Sciogliere 4,3 g di polvere cristallina in 50 mL di etere etilico grado di freddo alta eseguita cromatografia liquida (HPLC). Filtro del vuoto la soluzione per rimuovere qualsiasi traccia di materiale solido di partenza o anche polvere. Versare il filtrato in un bicchiere di Petri, lasciandola a rallentare evaporare a 4 ° C in un frigorifero di laboratorio.
- Assicurarsi che dopo due giorni, si ottengono cristalli limpido incolore. Quindi misurare il punto di fusione, che dovrebbe essere nel range di K. 310-311
4. analisi di 3,5-Lutidine N -ossido di disidratare
- Rimuovere i cristalli che si formano, di forma prismatica e incolore, per decantazione dalle pareti del pallone per ulteriore analisi ai raggi x. Se non immediatamente utilizzato, è possibile mantenere i cristalli in etere etilico per evitare idratazione di cristallo.
- Sciogliere 0,010 g di 3,5-lutidine N -ossido di disidratare in 0,4 mL di CDCl3 per eseguire NMR H1 e C13 analisi per dimostrare l'efficacia della procedura.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Il protocollo è essenzialmente un'estensione di Ochiai tecnica1. Tuttavia, vengono applicate le temperature più basse e meno tempo. Questo semplice metodo può essere utilizzato per ottenere un ligando versatile, che è un sostitutivo piridina N-ossido derivato. Per confermare la formazione di 1, NMR 1H e 13C analisi sono preferiti per testare l'efficacia della procedura.
Lo spostamento chimico dimostra la formazione di 1. Il segnale a 2,28 ppm (parti per milione) corrisponde a sei atomi di idrogeno equivalente dei due gruppi metilici nelle posizioni 3 e 5, che percepiscono il campo magnetico in minore proporzione che il magnetico permanente. Ci sono due insiemi di settine: uno appartiene al protone nella posizione c 7.9, che raddoppia le dimensioni dell'altro segnale a 6,9 che appartiene al protone nella posizione a. Figura 1 Mostra gli spostamenti chimici provocati dalla presenza della bonde di atomo di ossigeno d per l'atomo di azoto dell'anello di piridina. L'atomo di ossigeno è electro-ritiro e gli atomi di idrogeno più vicino per l'atomo di ossigeno (c e) Visualizza lo spostamento a una frequenza più alta di quella per l'atomi di idrogeno di metile (b).
Lo stesso processo viene tracciato per lo spettro di 13C NMR, Figura 2, dove i segnali per il carbonio più vicino per l'atomo di ossigeno (c e un) Visualizza la frequenza di separazione tra i loro segnali di Δc = 1.300 Hz e Δun = 200 Hz. Ancora una volta, i carboni di metile non mostrano alcun cambiamento. Spettro IR può essere utilizzato per vedere il successo del metodo pure.
Il diagramma ORTEP, Figura 4, dimostra la presenza di due molecole di acqua che circondano la molecola asimmetrica. Queste molecole sono credute per stabilizzare il legame N-O. In simili casi, è stata descritta per la piridina N-ossido e relativi ossidi aromatici. Vi è una significativa stabilizzazione π-tipo O→N retro-donazione, riflessa in un ordine di legame calcolato superiore a 1 e un numero di coppie singole di elettroni dell'atomo O inferiore a 36.
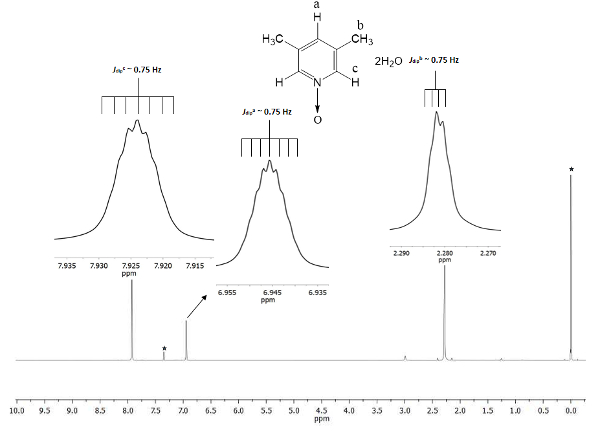
Figura 1 . TMS a cui fa riferimento CDCl3 500 MHz NMR 1H spettro di 1. Le integrazioni e gli spostamenti chimici dei tre segnali d'accordo con tre diversi tipi di atomi di idrogeno presenti in lutine N-ossido. Clicca qui per visualizzare una versione più grande di questa figura.
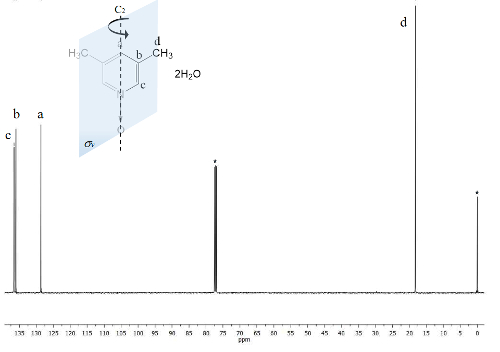
Figura 2 . TMS a cui fa riferimento CDCl3 100 MHz NMR 13C spettro della 1. Tre segnali sono osservati per i cinque atomi di carbonio aromatici e uno per i due gruppi metilici. Clicca qui per visualizzare una versione più grande di questa figura.
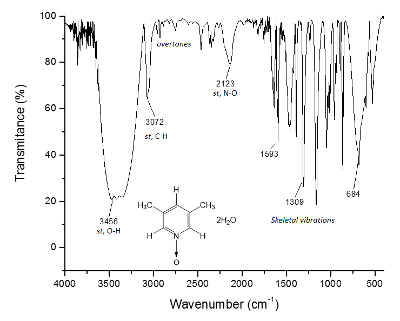
Figura 3 . Spettro IR del 1. Legami O-H, di sopra di 3.300 cm-1, sono responsabili per la formazione della struttura supramolecolare e la formazione di cristalli. Clicca qui per visualizzare una versione più grande di questa figura.

Figura 4 . Diagramma ORTEP di cui due molecole di forme di H2O ponte legami idrogeno con l'ossigeno di lutidine, guidando i loro atomi di idrogeno verso l'atomo di ossigeno 1. Questa figura è stata modificata da Merino García et al. 12 Clicca qui per visualizzare una versione più grande di questa figura.

Figura 5 . Immagini di diffrazione di raggi x adatto cristalli di 1 in etere etilico (in alto) e all'aria aperta (in basso). Uno di questi cristalli è stato confermato nel diffrattometro a raggi x e ha mostrato un percorso di diffrazione di raggi x, che è stato popolazionale e raffinato dai programmi speciali computazionale in una struttura cristallina e molecolare24,25, 26,27,28. Clicca qui per visualizzare una versione più grande di questa figura.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Il protocollo presentato qui è un metodo convenzionale per collegare un atomo di ossigeno all'atomo di azoto di 3,5-lutidine come un metodo di funzionalizzazione dei substrati. Questa tecnica è anche ben consolidata per produrre cristalli disidratati adatti di raggi x (Figura 5, le foto scattate con una fotocamera Sony DSC-HX300 Cyber-shot). Per quanto siamo preoccupati, non molti rapporti hanno descritto la produzione di tali cristalli16. Molti composti cresceranno cristalli ideale per analisi dei raggi x, quando essi sono chelati con vari metalli17,18,19,20. Una volta che la polvere cristallina è formata, è importante per estrarlo dalle loro madri utilizzando una boccetta di Kitasato e un imbuto di Buchner. Utilizzando tubi di gomma, la boccetta di Kitasato è connesso a una linea del vuoto e su di esso l'imbuto di Buchner viene inserito con una carta da filtro. Una volta che il vuoto è stato acceso, la carta da filtro è inumidita con una piccola quantità di solvente da cui il prodotto cristallizzato. Ciò impedisce che la polvere cristallina gocciolando nell'imbuto Buchner dall'effetto vuoto. Dopo essersi assicurato la carta da filtro, la soluzione contenente la polvere cristallina è scosso per garantire che tutta la polvere cristallina viene filtrata, e nessuno rimane nella parte inferiore del pallone. La soluzione è rapidamente versata sopra l'imbuto di Buchner. La polvere cristallina ottenuta viene lasciata per circa 10 minuti su carta da filtro e quindi il vuoto è spento e la polvere cristallina è staccato dalla carta e memorizzati in un flaconcino di vetro opaco, etichettati con il suo codice e conservato a 4 ° C fino ulteriore analisi. Il liquido filtrato viene versato in un bicchiere di Petri, lasciando agli lentamente evaporazione a 4 ° C per migliorare la formazione di cristalli adeguate per l'analisi di raggi x.
È importante notare che questo protocollo utilizza solventi e materiali che sono facilmente ottenibili e generalmente si trovano in qualsiasi laboratorio di ricerca. La regolazione del pH con l'aggiunta di Na2CO3 e l'agitazione magnetica coerente sono fondamentali per la resa del prodotto finale. Tuttavia, è importante prestare attenzione prestare la massima attenzione in tutte le fasi del processo, soprattutto nella fase di estrazione dove nessuna traccia di materiale di partenza deve essere presente per permettere la formazione di polvere cristallina e, successivamente, cristalli. Così, questa fase di estrazione/purificazione può essere monitorata mediante spettroscopia NMR o IR per garantire la qualità del prodotto.
Per garantire la riproducibilità del presente protocollo, NMR è un ottimo strumento. Anche piccoli dettagli sono visibili nello spettro. Tutti i segnali vengono visualizzati come inserti in Figura 1. Questi inserti raffigurano chiaramente una scissione, vale a dire molteplicità, di tutti i segnali. Per esempio, la b di protoni (Jdipb ~ 0.75 Hz) mostrano quattro picchi sulla cresta del segnale, con una separazione fra loro (Δpicco-picco) di nausea costante di ~ 0,0075 ppm. Il ppm 0,0075 si trasformi in energia utilizzando la seguente equazione21
 Equazione 1
Equazione 1

La trasformazione è consigliata poiché i segnali dispiegarsi provengono dall'interazione spaziale dipolare tra i nuclei di tre atomi di idrogeno del gruppo metilico, e anche se sono oltre i 4 singoli legami con protoni c e a, essi sono in grado di percepire loro interazioni dipolari magnetica slancio22. Inoltre, il sigma libero rotazione del gruppo metilico di legame consente interazione iperfine super protone-protone sia visibile nella molteplicità del segnale. Settine di protoni a e c a 6,9 e 7,9 ppm, rispettivamente sono derivati dallo stesso fenomeno di natura dipolare. In questi casi, protoni a e c possono differenziare i protoni nel gruppo metilico per la stessa rotazione dinamica. Infine, come previsto, il calcolato Jtuffo per a, b, e c hanno a malapena lo stesso valore, ~ 0.75 Hz. Queste quantità delle interazioni confermano la disposizione spaziale di nuclei di idrogeno in tutta l'anisotropia magnetica.
D'altra parte, la simmetria C2v 1 rende equivalente carboni23. Lo spettro di 13C, Figura 2, Mostra il tipico segnale per gruppi metilici agganciate agli anelli aromatici, carboni d 18 ppm. Inoltre, un segnale a 129 ppm è visibile in questa regione a causa il meno elettronegativo carbonio elemento influenzato un. Alle alte frequenze il segnale per i nuclei di atomi di carbonio più esposti al campo magnetico sono presentati alle 137 ppm22.
La metodologia presentata è molto utile per la sintesi della piridina N-ossidi, fornendo buoni rendimenti, in un tempo ragionevole con condizioni di reazione morbido e reagenti accessibili a buon mercato e facili, che non richiedono ulteriori catalizzatori. Queste condizioni consente di ottenere una vasta gamma di piridine N-ossidi come precursori di altre molecole di interesse per la comunità scientifica e didattica. La metodologia adeguata dà l'opportunità di acquisire strumenti di base concettuali e sperimentali in laboratori didattici per gli studenti, dimostrando una riuscita sintesi di composti e la felicità di vedere la formazione di cristalli. Tuttavia, è importante sottolineare che, come qualsiasi reazione chimica, è necessario prendere tutte le precauzioni, poiché generalmente i reagenti impiegati sono pericolosi.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Tutti gli autori non dichiarano alcun conflitto di interessi.
Acknowledgments
Il presente lavoro è stato supportato da Vicerrectoría de Investigación y Estudios de Posgrado da BUAP, divulgazione della scienza e progetti n. REOY-NAT14, 15, 16-G. EMEL-NAT17. RMG grazie CONACyT (Messico) per la borsa di studio 417887.
Materials
| Name | Company | Catalog Number | Comments |
| 3,5-lutidine | Sigma-Aldrich | L4206-500ML | |
| Glacial acetic acid | Fermont | 3015 | |
| Hidrogen peroxide (35%) | Sigma-Aldrich | 349887-500ML | |
| Na2CO3 anhydrous | Productos Químicos Monterrey | 1792 | |
| Na2SO4 anhydrous | Alfa reactivos | 25051-C | |
| CHCl3 | Fermont | 6205 | |
| Ethyl eter | Mercury Chemist | QME0309 | |
| Distilled water | Comercializadora Química Poblana | not-existent |
References
- Ochiai, E. Recent Japanese work on the chemistry of pyridine 1-oxide and related compounds. J. Org. Chem. 18 (5), 534-551 (1953).
- Solomons, T. W. G. Organic Chemistry 2nd Edition. , John Wiley & Sons. 1110 (1976).
- Albini, A., Pietra, S. Heterocyclic N-Oxides. , CRC Press. ISBN: 0849345529 328 (1991).
- Koukal, P., Ulc, J., Necas, D., Kotora, Heterocyclic N.-Oxides. Topics in Heterocyclic Chemistry. 53, 29-58 (2017).
- Wen-Man, Z., Jian-Jun, D., Xu, J., Jun, X., Huan-Jian, X. Visible-Light-Induced C2 alkylation of pyridine N.-oxides. J. Org. Chem. 82 (4), 2059-2066 (2017).
- Merino García, M. R., Ríos-Merino, F. J., Bernès, S., Reyes-Ortega, Y. Crystal structure of 3,5-dimethylpyridine N-oxide dihydrate. Acta Cryst. 72 (12), 1687-1690 (2016).
- Sarma, R., Karmakar, A., Baruah, J. B. N-Oxides in Metal-Containing Multicomponent Molecular Complexes. Inorg. Chem. 47 (3), 763-765 (2008).
- Youssif, S. Recent trends in the chemistry of pyridine N-oxides. ARKIVOC. 2001, 242-268 (2001).
- Chucholowski, A. W., Uhlendorf, S. Base catalyzed rearrangement of 5-cyanomethyl-2-isoxazolines; novel pathway for the formation of 2-aminopyridine N-oxides. Tetrahedron Lett. 31 (14), 1949-1952 (1990).
- Thellend, A., Battioni, P., Sanderson, W., Mansuy, D. Oxidation of N-Heterocycles by H2O2 Catalyzed by a Mn-Porphyrin: An Easy Access to N-Oxides Under Mild Conditions. Synthesis. 1997 (12), 1387-1388 (1997).
- Copéret, C., Adolfson, H., Tinh-Alfredo, V. K. h, Yudin, A. K., Sharpless, K. B. A simple and Efficient Method for the Preparation of Pyridine N-Oxides. J. Org. Chem. 63 (5), 1740-1741 (1998).
- Ferrer, M., Sánchez-Baeza, F., Messeguer, A. On the preparation of amine N-oxides by using dioxiranes. Tetrahedron. 53 (46), 15877-15888 (1997).
- Adam, W., Briviba, K., Duschek, F., Golsch, D., Kiefer, W., Sies, H. Formation of singlet oxygen in the deoxygenation of heteroarene N-oxides by dimethyldioxirane. J. Chem. Soc. Chem. Commun. 1995 (18), 1831-1832 (1995).
- Murray, R. W., Singh, M. A Facile One-Step Synthesis of C-Arylnitrones Using Dimethyldioxirane. J.Org.Chem. 55 (9), 2954-2957 (1990).
- Kim, S. W., Um, T., Shin, S. Brønsted acid-catalyzed α-halogenation of ynamides from halogenated solvents and pyridine-N-oxides. Chem. Commun. 53 (18), 2733-2736 (2017).
- Campeau, L., Rousseaux, R., Fagnou, K. A solution to the 2-pyridyl organometallic cross-coupling problem: regioselective catalytic direct arylation of pyridine N-oxides. J. Am. Chem. Soc. 127 (51), 18020-18021 (2005).
- Gang, L., et al. Metal-free methylation of a pyridine N-oxide C-H bond by using peroxides. Org. Biomol. Chem. 13 (46), 11184-11188 (2015).
- May, D., Nyman,, Hampden-Smith, M. J., Duesler, E. N. Synthesis, characterization, and reactivity of group 12 metal thiocarboxylates M(SOCR)2Lut2[M) Cd, Zn; R ) CH3, C(CH3)3; Lut ) 3,5-Dimethylpyridine (Lutidine)]. Inorg. Chem. 36 (10), 2218-2224 (1997).
- Cho, S. H., Hwang, S. J., Chang, S. Palladium-Catalyzed C-H Functionalization of Pyridine N-Oxides: Highly Selective Alkenylation and Direct Arylation with Unactivated Arenes. J. Am. Chem. Soc. 130 (29), 9254-9256 (2008).
- Ide, Y., et al. Spin-crossover between high-spin (S = 5/2) and low-spin (S = 1/2) states in six-coordinate iron(III) porphyrin complexes having two pyridine-N. oxide derivatives. Dalton Trans. 46 (1), 242-249 (2017).
- Drago, R. S. Physical Methods in Chemistry. , Saunders College Publishing USA. 750 (1977).
- Cervantes-Mejía, V., et al. Branched Polyamines Functionalized with Proposed Reaction Pathways Based on 1H-NMR, Atomic Absorption and IR Spectroscopies. American Journal of Analytical Chemistry. 5 (16), 1090-1101 (2014).
- Huheey, J. E., Keiter, E. A., Keiter, R. L. Inorganic Chemistry: Principles of Structure and Reactivity, 4th Edition. , Oxford University Press. Mexico. ISBN: 9706131620 1023 (1997).
- Rigaku, CrysAlisPRO. , (2013).
- Sheldrick, G. M. SHELXT - Integrated space-group and crystal-structure determination. Acta Cryst. 71 (1), 3-8 (2015).
- Sheldrick, G. M. Crystal structure refinement with SHELXL. Acta Cryst. 71 (1), 3-8 (2015).
- Sheldrick, G. M. A short history of SHELX. Acta Cryst. 64 (1), 112-122 (2008).
- Macrae, C. F., et al. Mercury CSD 2.0 - new features for the visualization and investigation of crystal structures. J. Appl. Cryst. 41 (2), 466-470 (2008).
- ChemBioDraw Ultra 13. , PerkinElmer. (2013).
