

Summary
Neste documento, nós relatamos a síntese e a cristalização de 3,5-lutidine N-óxido de desidratam por um protocolo simples que difere a síntese clássica de piridina N-óxido. Este protocolo utiliza diferentes matérias-primas e envolve menos tempo de reação para produzir uma nova estrutura solvated supramolecular, que cristaliza sob evaporação lenta.
Abstract
A síntese de 3,5-lutidine N-óxido de desidratar, 1, foi alcançado na rota de síntese do ácido 2-amino-piridina-3,5-dicarboxílico. Ochiai primeiro usado a metodologia para pyridines não-substituídos em 1957, em um processo de 12 h, mas sem cristais adequados de raios-x foram obtidos. O anel substituído utilizado na metodologia apresentada aqui claramente influenciado a adição de moléculas de água na unidade assimétrica, o que confere uma força diferente nucleofílica em 1. O raio-x cristal apropriado composto 1 foi possível devido a estabilização da carga negativa no oxigênio pela presença de duas moléculas de água, onde os átomos de hidrogênio doar uma carga positiva no ringue; tais moléculas de água servem bem para construir uma interação supramolecular. As moléculas hidratadas podem ser possíveis para o sistema alcalino que é alcançado ajustando o pH para 10. Importante, a dupla metil substituído anel e um tempo de reação de 5h, torna um método mais versátil e com aplicações químicas mais amplas para inserções de futuro anel.
Introduction
Hoje em dia, cientistas ao redor do globo tem vindo a investir recursos para o desenvolvimento de novas rotas sintéticas para o functionalization de grupos aromáticos, que são conhecidos por frente de baixa reatividade para adição reações1,2, 3. Piridina, onde um átomo de nitrogênio substitui um átomo de carbono, apresenta uma reatividade química semelhante a analógica anéis compostos unicamente por átomos de carbono3, e ele geralmente sofre um mecanismo de substituição, em vez de adição. N-óxidos são distintos pela presença de um vínculo de doador entre nitrogênio e oxigênio formado pela sobreposição do pares par de elétrons no nitrogênio com um orbital vazio do átomo de oxigênio3. Particularmente, piridina N-óxidos são bases de Lewis, porque sua fracção N-O pode agir como um dador de electrões, e eles podem combinar com ácidos de Lewis, formando os pares de ácido-base de Lewis correspondentes. Esta propriedade tem uma consequência química essencial, porque pode aumentar o nucleophilicity dos ácidos de Lewis para eletrófilos potenciais e, portanto, permitir-lhes reagir em condições onde normalmente a reação não ocorreria. Provavelmente o uso mais frequente de tais compostos é em várias reações de oxidação, onde atuam como oxidantes4. Piridina N-óxidos e muitos dos seus derivados acrescida de anel são moléculas recorrentes de agentes biologicamente ativos e farmacológicos5, e estabeleceu-se uma distribuição espacial clara por diferentes ferramentas espectroscópicas para alguns deles6,7. Em pesquisas sobre diferentes grupos de anexar para o anel de piridina, cientistas testaram várias metodologias para produzir um método fácil e convencional, desde que isoxazolines requer uma quantidade catalítica de base como o DBU em xileno a ferver para formar 6- substituído-2-aminopiridina N-óxidos8,9. Uma variedade de derivados de piridina foram convertidas em suas correspondentes N-óxidos na presença de uma quantidade catalítica de manganês tetrakis(2,6-diclorophenyl) acetato de porfirina e amônio em CH2Cl2/CH3 CN8,10. Outros pyridines são oxidados para seus óxidos usando H2O2 na presença de quantidades catalíticas de methyltrioxorhenium8,11, ou pela adição de excesso dimethyldioxirane no CH2Cl2 a 0 ° C, o que leva à correspondente N-óxidos8,12,13,14. Bis (trimetilsilil) peróxido na presença de trioxorhenium no CH2Cl2 tem sido usado para a síntese da piridina N-óxidos8,11. A síntese de aminopiridina N-óxidos envolvendo acilação usando ácido de Caro (ácido peroxomonosulfuric) também tem sido relataram8. No entanto, a metodologia aqui relatados, e que usa parte da metodologia relatada por oliveira1, fornece resultados muito bons com o uso de reagentes mais baratos e acessíveis, H2O2 e ácido acético glacial. Esta prática é mais adequada para uso em preparações de grande escala que agem em aminas terciárias, produz bons rendimentos em uma reação que requer apenas 30% de peróxido de hidrogênio e ácido acético em uma temperatura entre 70-80 ° C, e ele usa um processo de purificação que está disponível na maioria dos laboratórios de síntese como destilação, sem o uso de catalisador ou mais caro reagentes1. A literatura relata que outras metodologias também frequentemente envolvem prazos de 10-24 h e temperaturas acima de 100 ° C 4,8, e o rendimento de cristais bem formados para análises de raio-x é raramente relatado.
Reativa, vários derivados de N -óxido são usados para ativar adequadamente o anel de lutidine, de qualquer forma nucleofílica ou eletrofílica. O fator nucleofílico ou eletrofílico é afetado por substituintes. Com o anel de piridina, sendo os grupos elétron-retirando, o principal fator é a característica nucleofílica1. A enciclopédia N-óxido compostos são raramente isolados como cristais adequados para análise de raios-x devido a carga deslocalizada no anel aromático. No entanto, o fator de solvatação é fundamental para estabilizar a densidade negativa do oxigênio15.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. reação
- Coloque em uma coifa um frasco aberto redondo 100 mL com 0,5 mol (29,8 mL) de ácido acético glacial e adicionar 0.051 mol (5,82 mL) de 3,5-dimethylpyridine e 5 mL de H2O2 (35%). Manter a reação de mistura sob agitação magnética constante, a uma temperatura interna 80 ° C por 5h.
- Após o tempo de reação, esfriar o frasco de 24 ° C com gelo (do não exponha o ácido acético de gases para o gelo) e ligá-lo a uma unidade de destilação de vácuo elevado para 90-120 min para remover o excesso de ácido acético.
Atenção: Não utilize material quente. Espere até que a vidraria atinja uma temperatura controlável. Isto também irá evitar vapores entrando no top da unidade de destilação. - Adicione água destilada água (10 mL) duas vezes para garantir a remoção de qualquer vestígio de ácido acético e dedicar-se a mistura muito possível.
2. extração e ajuste basicidade
- Dissolver em água bi-destilado do produto viscoso e transparente isolado e usar um potenciômetro para ajustar o pH para 10 com puro sólido Na2CO3.
- Coloque cuidadosamente a solução em um funil de separação de 250 mL e extraí-lo 5 vezes com 250 mL de CHCl3 para melhorar o rendimento. Recuperar a camada orgânica e secá-la mais sólida Na2então4 por 30 min no máximo, que irá conter o produto. Se necessário, extrair novamente a fase aquosa com a quantidade desejada de CHCl3.
Cuidado: CHCl3 pode provocar sonolência e vertigens; manipular com cuidado e dentro uma coifa. - Remova o solvente sob pressão reduzida com uma unidade de destilação a vácuo elevado, até a formação de um pó cristalino bege claro muito higroscópico (70%).
3. processo de cristalização
- Dissolva 4,3 g do pó cristalino em 50 mL de éter dietílico de frio a cromatografia líquida de alta (resolução HPLC). A solução para remover qualquer vestígio de material sólido de partida ou até mesmo poeira do filtro de vácuo. Coloque o filtrado num copo de Petri, deixando-a retardar a evaporar a 4 ° C, em um frigorífico de laboratório.
- Certifique-se de que, depois de dois dias, cristais incolores claro são obtidos. Medindo-se então o ponto de fusão, que deve estar no intervalo de K. 310-311
4. análise de 3,5-Lutidine N -óxido desidratando
- Remova os cristais que se formam, de forma prismática e incolor, por decantação das paredes do balão, para posterior análise de raios-x. Se não imediatamente usado, manter os cristais em éter dietílico para evitar hidratação de cristal.
- Dissolva 0,010 g de 3,5-lutidine N-óxido de desidratar em 0,4 mL de CDCl3 para executar NMR H1 e C13 análise para provar a eficácia do procedimento.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
O protocolo é essencialmente uma extensão do Ochiai técnica1. No entanto, são aplicadas a temperatura mais baixa e menos tempo. Este método simples pode ser usado para obter um ligante versátil, que é uma piridina substituída N-óxido de derivados. Para confirmar a formação de 1, NMR 1H e 13C análise são preferidos para testar a eficácia do procedimento.
A mudança química demonstra a formação de 1. O sinal em 2,28 ppm (partes por milhão) corresponde os seis hidrogênios equivalentes dos dois grupos metil na posição 3 e 5, que percebe o campo magnético em menor proporção do que a permanente magnética. Há dois conjuntos de septuplets: pertence o próton na posição c em 7,9, que dobra o tamanho do outro sinal em 6,9 que pertence o próton o e.u.a. posição Figura 1 mostra as mudanças químicas provocadas pela presença do bonde do átomo de oxigênio d para o átomo de nitrogênio do anel de piridina. O átomo de oxigênio é electro-retirada e os átomos de hidrogênio mais perto para o átomo de oxigênio (c e a) Mostrar deslocamento para uma frequência mais elevada do que para os hidrogênios de metilo (b).
O mesmo processo é plotado para o spectrum NMR de 13C, Figura 2, onde os sinais para os carbonos mais perto para o átomo de oxigênio (c e um) mostram a separação de frequência entre os seus sinais de Δc = 1.300 Hz e Δum = 200 Hz. Mais uma vez, os carbonos de metila não mostram alterações. O espectro de IR pode ser usado para ver o sucesso do método também.
O diagrama ORTEP, Figura 4, demonstra a presença de duas moléculas de água ao redor da molécula assimétrica. Estas moléculas são acreditadas para estabilizar a ligação N-O. Em casos semelhantes, tem sido descrito por piridina N-óxido e óxidos aromáticos relacionados. Há uma significativa estabilização π-tipo O→N costas-doação, refletido em uma ordem de ligação calculado maior do que 1 e um número de pares de elétrons solitários no átomo de O inferior a 36.
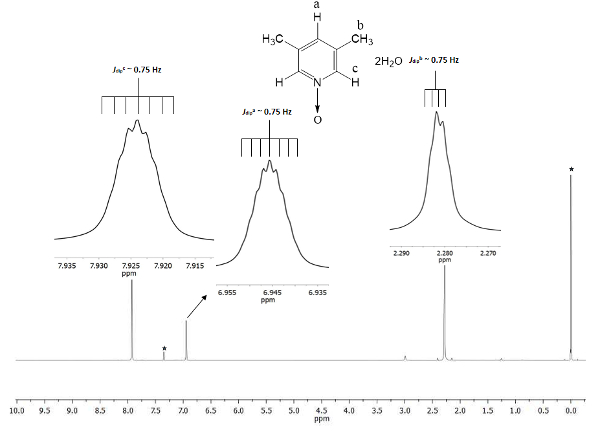
Figura 1 . TMS referenciado CDCl3 espectro 1H de 500MHz NMR de 1. As integrações e mudanças químicas dos três sinais de acordo com três tipos diferentes de átomos de hidrogênio presentes no lutine N-óxido. Clique aqui para ver uma versão maior desta figura.
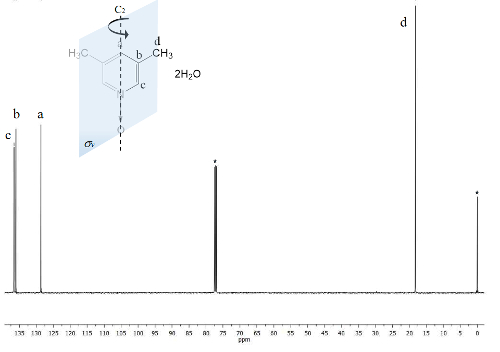
Figura 2 . TMS referenciado CDCl3 espectro de 13C 100 MHz NMR de 1. Três sinais são observados para os cinco carbonos aromáticos e um para os dois grupos metilo. Clique aqui para ver uma versão maior desta figura.
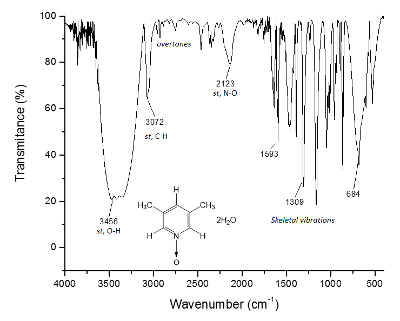
Figura 3 . Espectro de IR de 1. O O-H-títulos, acima dos 3.300 cm-1, são responsáveis para a formação de estrutura supramolecular e a formação de cristais. Clique aqui para ver uma versão maior desta figura.

Figura 4 . Diagrama ORTEP de onde duas moléculas de H2O formulários ponte de hidrogênio com o oxigênio do lutidine, dirigindo seus átomos de hidrogênio para o átomo de oxigênio de 1. Esta figura foi modificada de Merino García et al . 12 Clique aqui para ver uma versão maior desta figura.

Figura 5 . Fotos de cristais de difração de raio x apropriado de 1 em éter dietílico (topo) e no open air (parte inferior). Dentre estes cristais foi confirmado no Difratômetro de raios-x e mostrou um caminho de difração de raios-x, que foi a companhia de Jesus era e refinada por programas computacionais especiais em uma estrutura molecular e cristalina24,25, 26de27,,28. Clique aqui para ver uma versão maior desta figura.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
O protocolo aqui apresentado é um método convencional de ligação de um átomo de oxigênio para o átomo de nitrogênio do 3,5-lutidine como um método de functionalization de substratos. Esta técnica também é bem estabelecida para produzir cristais desidratados apropriados (Figura 5, fotos tiradas com uma câmera Sony DSC-HX300 Cyber-shot) de raio-x. Tanto quanto estamos preocupados, não muitos relatórios descreveram a produção de tais cristais16. Muitos compostos crescem cristais ideais para análise de raios-x quando eles são quelatados por vários metais17,18,19,20. Uma vez que o pó cristalino é formado, é preciso extraí-lo de seus licores-mãe usando um balão Shibasaburo e um funil de Buchner. Usando mangueiras de borracha, o balão Shibasaburo é conectado a uma linha de vácuo e por cima o funil de Buchner é colocado com um papel de filtro. Uma vez que o vácuo tem sido ativado, o papel de filtro é umedecido com uma pequena quantidade de solvente a partir do qual o produto cristalizado. Isso evita que o pó cristalino dentro do funil de Buchner que gotejam pelo efeito do vácuo. Depois de garantir o papel de filtro, a solução contendo o pó cristalino é abalada para garantir que todo o pó cristalino é filtrado, e nada permanece no fundo do frasco. A solução rapidamente é derramada sobre o funil de Buchner. O pó cristalino obtido é deixado por cerca de 10 min sobre o papel de filtro e então o vácuo está desligado e o pó cristalino é retirado o papel e armazenado em um frasco de vidro opaco, rotulado com o seu código e mantido a 4 ° C até uma análise mais aprofundada. O líquido filtrado é vertido em um copo de Petri, deixando-se lentamente a evaporação a 4 ° C, para melhorar a formação de cristais adequados para análise de raios-x.
É importante notar que este protocolo utiliza solventes e materiais que são facilmente obtidos e geralmente são encontrados em qualquer laboratório de pesquisa. O ajuste do pH pela adição Na2CO3 e a agitação magnética consistente são essenciais para o rendimento do produto final. No entanto, é importante prestar atenção extra cuidadosa em todas as etapas do processo, especialmente na fase de extração, onde nenhum vestígio de material começar deve estar presente para permitir a formação de pó cristalino e, posteriormente, cristais. Assim, nesta fase de extração/purificação pode ser monitorada por espectroscopia NMR ou IR para garantir a qualidade do produto.
Para garantir a reprodutibilidade do presente protocolo, NMR é uma excelente ferramenta. Detalhes ainda são visíveis no espectro. Todos os sinais são mostrados como inserções na Figura 1. Estas inserções mostram claramente uma divisão, multiplicidade, ou seja, de todos os sinais. Por exemplo, o b de prótons (Jmergulhob ~ 0,75 Hz) Mostrar quatro picos na crista do sinal, com uma separação entre eles (Δpico-pico) constante enjoado de ~ 0,0075 ppm. O ppm 0,0075 podem ser transformado em energia utilizando a seguinte equação21
 Equação 1
Equação 1

A transformação é recomendada, pois os sinais desdobramento vem a dipolar interação espacial entre os núcleos de três hidrogênios do grupo metila, e mesmo que eles são mais do que a 4 ligações simples com prótons c e a, eles são capazes de perceber seu momento magnético dipolar interações22. Adicionalmente, a sigma livre rotação do grupo metil de ligação permite interação próton-próton hyperfine super ser visível na multiplicidade do sinal. Os septuplets de prótons um e c a ppm 6.9 e 7.9, respectivamente são derivados do mesmo fenômeno de natureza dipolar. Nestes casos, os prótons a e c pode diferenciar os prótons do grupo metila para a mesma dinâmica de rotação. Por último, como esperado, calculado Jmergulho para a, b, e c tem quase o mesmo valor, ~ 0,75 Hz. Essas quantidades das interações confirmam o arranjo espacial de núcleos de hidrogênio em toda a anisotropia magnética.
Por outro lado, a simetria de C2v 1 torna equivalente carbonos23. O espectro de 13C, Figura 2, mostra o sinal típico de grupos metil ligados aos anéis aromáticos, d carbonos 18 ppm. Além disso, um sinal em 129 ppm é visível nesta região devido a menos eletronegativo elemento influenciado carbono um. Em altas frequências do sinal para os núcleos de carbonos mais expostos ao campo magnético são apresentados em ppm 13722.
A metodologia apresentada é muito útil para a síntese da piridina N-óxidos, proporcionando bom rendimento, em um tempo razoável, com condições de reação suave e reagentes acessíveis baratos e fácil, que não exigem catalisadores adicionais. Essas condições podem ser usadas para a comunidade científica e educacional para obter uma ampla gama de pyridines N-óxidos como precursores de outras moléculas de interesse. A metodologia apropriada dá a oportunidade de adquirir ferramentas experimentais e conceituais básicas em laboratórios educacionais para estudantes, provando uma síntese bem sucedida de compostos e a felicidade de ver a formação de cristais. No entanto, é importante ressaltar que, como qualquer reação química, é necessário tomar todas as precauções, já que geralmente os reagentes utilizados são perigosos.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Todos os autores não declaram nenhum conflito de interesses.
Acknowledgments
O presente trabalho foi apoiado por Vicerrectoría de Investigación y Estudios de Posgrado de BUAP, divulgação da ciência e projetos no. REOY-NAT14, 15, 16-G. HEAS-NAT17. RMG Obrigado CONACyT (México) para bolsa de estudos 417887.
Materials
| Name | Company | Catalog Number | Comments |
| 3,5-lutidine | Sigma-Aldrich | L4206-500ML | |
| Glacial acetic acid | Fermont | 3015 | |
| Hidrogen peroxide (35%) | Sigma-Aldrich | 349887-500ML | |
| Na2CO3 anhydrous | Productos Químicos Monterrey | 1792 | |
| Na2SO4 anhydrous | Alfa reactivos | 25051-C | |
| CHCl3 | Fermont | 6205 | |
| Ethyl eter | Mercury Chemist | QME0309 | |
| Distilled water | Comercializadora Química Poblana | not-existent |
References
- Ochiai, E. Recent Japanese work on the chemistry of pyridine 1-oxide and related compounds. J. Org. Chem. 18 (5), 534-551 (1953).
- Solomons, T. W. G. Organic Chemistry 2nd Edition. , John Wiley & Sons. 1110 (1976).
- Albini, A., Pietra, S. Heterocyclic N-Oxides. , CRC Press. ISBN: 0849345529 328 (1991).
- Koukal, P., Ulc, J., Necas, D., Kotora, Heterocyclic N.-Oxides. Topics in Heterocyclic Chemistry. 53, 29-58 (2017).
- Wen-Man, Z., Jian-Jun, D., Xu, J., Jun, X., Huan-Jian, X. Visible-Light-Induced C2 alkylation of pyridine N.-oxides. J. Org. Chem. 82 (4), 2059-2066 (2017).
- Merino García, M. R., Ríos-Merino, F. J., Bernès, S., Reyes-Ortega, Y. Crystal structure of 3,5-dimethylpyridine N-oxide dihydrate. Acta Cryst. 72 (12), 1687-1690 (2016).
- Sarma, R., Karmakar, A., Baruah, J. B. N-Oxides in Metal-Containing Multicomponent Molecular Complexes. Inorg. Chem. 47 (3), 763-765 (2008).
- Youssif, S. Recent trends in the chemistry of pyridine N-oxides. ARKIVOC. 2001, 242-268 (2001).
- Chucholowski, A. W., Uhlendorf, S. Base catalyzed rearrangement of 5-cyanomethyl-2-isoxazolines; novel pathway for the formation of 2-aminopyridine N-oxides. Tetrahedron Lett. 31 (14), 1949-1952 (1990).
- Thellend, A., Battioni, P., Sanderson, W., Mansuy, D. Oxidation of N-Heterocycles by H2O2 Catalyzed by a Mn-Porphyrin: An Easy Access to N-Oxides Under Mild Conditions. Synthesis. 1997 (12), 1387-1388 (1997).
- Copéret, C., Adolfson, H., Tinh-Alfredo, V. K. h, Yudin, A. K., Sharpless, K. B. A simple and Efficient Method for the Preparation of Pyridine N-Oxides. J. Org. Chem. 63 (5), 1740-1741 (1998).
- Ferrer, M., Sánchez-Baeza, F., Messeguer, A. On the preparation of amine N-oxides by using dioxiranes. Tetrahedron. 53 (46), 15877-15888 (1997).
- Adam, W., Briviba, K., Duschek, F., Golsch, D., Kiefer, W., Sies, H. Formation of singlet oxygen in the deoxygenation of heteroarene N-oxides by dimethyldioxirane. J. Chem. Soc. Chem. Commun. 1995 (18), 1831-1832 (1995).
- Murray, R. W., Singh, M. A Facile One-Step Synthesis of C-Arylnitrones Using Dimethyldioxirane. J.Org.Chem. 55 (9), 2954-2957 (1990).
- Kim, S. W., Um, T., Shin, S. Brønsted acid-catalyzed α-halogenation of ynamides from halogenated solvents and pyridine-N-oxides. Chem. Commun. 53 (18), 2733-2736 (2017).
- Campeau, L., Rousseaux, R., Fagnou, K. A solution to the 2-pyridyl organometallic cross-coupling problem: regioselective catalytic direct arylation of pyridine N-oxides. J. Am. Chem. Soc. 127 (51), 18020-18021 (2005).
- Gang, L., et al. Metal-free methylation of a pyridine N-oxide C-H bond by using peroxides. Org. Biomol. Chem. 13 (46), 11184-11188 (2015).
- May, D., Nyman,, Hampden-Smith, M. J., Duesler, E. N. Synthesis, characterization, and reactivity of group 12 metal thiocarboxylates M(SOCR)2Lut2[M) Cd, Zn; R ) CH3, C(CH3)3; Lut ) 3,5-Dimethylpyridine (Lutidine)]. Inorg. Chem. 36 (10), 2218-2224 (1997).
- Cho, S. H., Hwang, S. J., Chang, S. Palladium-Catalyzed C-H Functionalization of Pyridine N-Oxides: Highly Selective Alkenylation and Direct Arylation with Unactivated Arenes. J. Am. Chem. Soc. 130 (29), 9254-9256 (2008).
- Ide, Y., et al. Spin-crossover between high-spin (S = 5/2) and low-spin (S = 1/2) states in six-coordinate iron(III) porphyrin complexes having two pyridine-N. oxide derivatives. Dalton Trans. 46 (1), 242-249 (2017).
- Drago, R. S. Physical Methods in Chemistry. , Saunders College Publishing USA. 750 (1977).
- Cervantes-Mejía, V., et al. Branched Polyamines Functionalized with Proposed Reaction Pathways Based on 1H-NMR, Atomic Absorption and IR Spectroscopies. American Journal of Analytical Chemistry. 5 (16), 1090-1101 (2014).
- Huheey, J. E., Keiter, E. A., Keiter, R. L. Inorganic Chemistry: Principles of Structure and Reactivity, 4th Edition. , Oxford University Press. Mexico. ISBN: 9706131620 1023 (1997).
- Rigaku, CrysAlisPRO. , (2013).
- Sheldrick, G. M. SHELXT - Integrated space-group and crystal-structure determination. Acta Cryst. 71 (1), 3-8 (2015).
- Sheldrick, G. M. Crystal structure refinement with SHELXL. Acta Cryst. 71 (1), 3-8 (2015).
- Sheldrick, G. M. A short history of SHELX. Acta Cryst. 64 (1), 112-122 (2008).
- Macrae, C. F., et al. Mercury CSD 2.0 - new features for the visualization and investigation of crystal structures. J. Appl. Cryst. 41 (2), 466-470 (2008).
- ChemBioDraw Ultra 13. , PerkinElmer. (2013).
