

Summary
Adjunto, divulgamos la síntesis y la cristalización de 3,5-lutidina N -óxido de deshidratan un protocolo simple que difiere de la síntesis clásica de la piridina N-óxido. Este protocolo utiliza el material de partida diferente e implica menos tiempo de reacción para dar una nueva estructura supramolecular solvatados, que cristaliza en evaporación lenta.
Abstract
La síntesis de 3,5-lutidina N -óxido de deshidratar, 1, fue alcanzado en la ruta de síntesis del 2-amino-piridina-3,5-ácido dicarboxílico. Ochiai primero utilizó la metodología de piridinas no sustituido en 1957 en un proceso de 12 h, pero cristales convenientes de rayos x no se obtuvieron. El anillo substituido en la metodología presentada aquí claramente influenciada la adición de moléculas de agua en la unidad asimétrica, que le confiere una diversa fuerza nucleófila en 1. El compuesto del cristal adecuado rayos x 1 fue posible debido a la estabilización de la carga negativa en el oxígeno por la presencia de dos moléculas de agua donde los átomos de hidrógeno donan carga positiva en el anillo; Estas moléculas de agua sirven también para construir una interacción supramolecular. Las moléculas hidratadas posible para el sistema alcalino que se llega ajustando el pH a 10. Lo importante, la doble metil sustituido anillo y un tiempo de reacción de 5 h, es un método más versátil y con más aplicaciones químicas para inserciones futuras anillo.
Introduction
En la actualidad, los científicos del mundo han estado invirtiendo recursos en el desarrollo de nuevas rutas sintéticas para la funcionalización de grupos aromáticos, que son conocidos por frente baja reactividad reacciones de adición1,2, 3. Piridina, donde un átomo de nitrógeno sustituye un átomo de carbono, presenta una reactividad química similar al análogo anillos compuestos exclusivamente de átomos de carbono3, y generalmente sufre un mecanismo de sustitución en lugar de adición. N-óxidos son característicos por la presencia de un donante enlace entre nitrógeno y oxígeno, formado por la superposición de la par enlazante de electrones en el nitrógeno con un orbital vacío en el átomo de oxígeno3. Particularmente, piridina N-óxidos son bases de Lewis, porque su parte N O puede actuar como donante de electrón, y puede combinar con ácidos de Lewis formando los correspondientes pares ácido-base de Lewis. Esta propiedad tiene una consecuencia esencial de química, porque puede aumentar el nucleophilicity de los ácidos de Lewis hacia electrophiles potenciales y así permitirles reaccionar bajo condiciones donde normalmente la reacción no se produciría. Probablemente el uso más frecuente de dichos compuestos es en varias reacciones de oxidación donde actúan como oxidantes4. Piridina N-óxidos y muchos de sus derivados funcionalizados de anillo son recurrentes moléculas de agentes farmacológicos y biológicamente activa5, y se ha establecido una distribución espacial claro por diferentes herramientas espectroscópicas para algunos de ellos de6,7. En la investigación sobre instalar diferentes grupos sobre el anillo de piridina, los científicos han probado varias metodologías para producir un método fácil y convencional, ya que isoxazolines requiere una cantidad catalítica de base como DBU en ebullición xileno para formar 6- sustituye 2-aminopiridina N-óxidos8,9. Una variedad de derivados de piridina fueron convertidos en sus correspondientes N-óxidos en presencia de una cantidad catalítica de manganeso tetrakis(2,6-diclorophenyl) porfirina y amonio acetato CH2Cl2/CH3 CN8,10. Otras piridinas son oxidados a sus óxidos en H2O2 en presencia de cantidades catalíticas de methyltrioxorhenium8,11, o por la adición de exceso dimethyldioxirane CH2Cl2 a 0 ° C, que conduce al correspondiente N-óxidos8,12,13,14. Bis (trimetilsilil) peróxido en presencia de trioxorhenium en CH2Cl2 se ha utilizado para la síntesis de la piridina N-óxidos8,11. La síntesis de aminopiridina N-óxidos que implica acilación con ácido de Caro (ácido peroxomonosulfúrico) también ha sido reportado8. Sin embargo, la metodología aquí registrados, que utiliza parte de la metodología reportada por Ochiai1, ofrece muy buenos resultados con el uso de reactivos más baratos y accesibles, H2O2 y ácido acético glacial. Esta práctica es más conveniente para el uso en preparaciones de gran escala que actúan sobre aminas terciarias, produce buenos rendimientos en una reacción que requiere sólo 30% peróxido de hidrógeno y ácido acético glacial en una temperatura entre 70-80 ° C y utiliza un proceso de purificación está disponible en más laboratorios de síntesis como destilación, sin el uso de catalizador o más caros reactivos1. La literatura reporta que otras metodologías también con frecuencia implican plazos de 10-24 h y temperaturas superiores a 100 ° C 4,8, y la producción de cristales bien formados para el análisis de rayos x se divulga raramente.
Reactivo, varios N -óxido derivados se utilizan para activar adecuadamente el anillo lutidina, de una forma nucleófila o electrófila. El factor nucleofílico o electrofílico es afectado por los sustitutos. Con el anillo de piridina siendo los grupos electrón-retirarse, el principal factor es la característica nucleófila1. Gratis N-compuestos de óxido son raramente aislados cristales adecuados para análisis de rayos x debido a la carga deslocalizada en el anillo aromático. Sin embargo, el factor de la solvatación es fundamental para estabilizar la densidad negativa del oxígeno15.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. reacción
- En una campana de humos un frasco abierto redondo de 100 mL con 0,5 mol (29,8 mL) de ácido acético glacial y añada 0.051 mol (mL 5,82) de 3,5-Dimetilpiridina y 5 mL de H2O2 (35%). Mantener la reacción de la mezcla bajo agitación magnética constante, a una temperatura interna de 80 ° C durante 5 h.
- Después del tiempo de reacción, enfriar el matraz a 24 ° C con hielo (do no exponga el ácido acético de los gases en el hielo) y conéctelo a una unidad de destilación a alto vacío para 90-120 min para eliminar el exceso de ácido acético.
PRECAUCIÓN: No utilice el material caliente. Espere hasta que el vidrio alcanza una temperatura manejable. Esto también evitará entrar en la parte superior de la unidad de destilación de vapores. - Agregar agua destilada (10 mL) dos veces para asegurar la eliminación de cualquier rastro de ácido acético y la mezcla se concentran tanto como sea posible.
2. basicidad ajuste y extracción
- Disolver en agua bi destilada aislado producto viscoso y transparente y utilizar un potenciómetro para ajustar el pH a 10 con puro sólido Na2CO3.
- Coloque cuidadosamente la solución en un embudo de separación de 250 mL y extracto 5 veces con 250 mL de CHCl3 para mejorar el rendimiento. Recuperar la capa orgánica y seca sobre Na sólida2tan4 30 minutos máximo, que contendrá el producto. Si es necesario, vuelva a extraer la fase acuosa con la cantidad deseada de CHCl3.
PRECAUCIÓN: CHCl3 puede provocar somnolencia y vértigo; manejar con cuidado y dentro de una campana de humos. - Eliminar el disolvente a presión reducida con una unidad de destilación a alto vacío, hasta la formación de un polvo cristalino de color beige claro muy higroscópico (70%).
3. cristalización proceso
- Disolver 4,3 g de polvo cristalino en 50 mL de éter dietílico de la cromatografía líquida realizada alta fría (HPLC) grado. Filtro de vacío la solución para eliminar cualquier rastro de material sólido de partida o incluso el polvo. Vierte el filtrado en un placa de Petri de vidrio, dejando que lentamente se evaporan a 4 ° C en un refrigerador de laboratorio.
- Asegúrese de que después de dos días, se obtienen cristales descoloridos claro. Luego se mide el punto de fusión, que debe estar en el rango de K. 310-311
4. Análisis de 3,5-lutidina N -óxido de deshidratar
- Quitar los cristales que se forman, de forma prismática y descolorido, por decantación de las paredes del frasco para su posterior análisis de rayos x. Si no inmediatamente, mantenga los cristales en éter dietílico para evitar la hidratación del cristalino.
- Disolver 0.010 g de N -óxido de 3,5-lutidina deshidratar en 0,4 mL de CDCl3 H NMR1 y C13 análisis para demostrar la efectividad del procedimiento.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
El protocolo es esencialmente una extensión de técnica1 de Ochiai. Sin embargo, se aplican temperaturas más bajas y menos tiempo. Este sencillo método puede utilizarse para obtener un ligando versátil, que es una piridina sustituido N-derivados de óxido. Para confirmar la formación de 1, NMR 1H y 13C análisis son las preferidas para probar la efectividad del procedimiento.
El cambio químico muestra la formación de 1. La señal en 2,28 ppm (partes por millones) corresponde a los seis hidrógenos equivalentes de los dos grupos metilo en las posiciones 3 y 5, que perciben el campo magnético en menos proporción que el magnético permanente. Hay dos conjuntos de septuplets: uno pertenece al protón en la posición c en 7.9, que dobles el tamaño de la otra señal a 6.9 que pertenece al protón en la posición a. figura 1 muestra los cambios químicos provocados por la presencia de la bonde de átomo de oxígeno d al átomo del nitrógeno del anillo de piridina. El átomo de oxígeno es retirada de electro y los átomos de hidrógeno más cercanos al átomo de oxígeno (c y a) Mostrar el desplazamiento a una frecuencia más alta que la de los hidrógenos del metilo (b).
El mismo proceso se traza para el espectro de 13C NMR, figura 2, donde las señales de los carbonos más cercanos al átomo de oxígeno (c y a) Mostrar la separación en frecuencia entre las señales de Δc = 1.300 Hz y Δun = 200 Hz. Una vez más, los carbonos del metilo no muestran ningún cambio. El espectro IR se puede utilizar para ver el éxito del método así.
El diagrama ORTEP, figura 4, muestra la presencia de dos moléculas de agua que rodea la molécula asimétrica. Se cree que estas moléculas son para estabilizar el enlace N-O. En casos similares, se ha descrito para la piridina N-óxidos y óxidos aromáticos relacionados. Hay una importante estabilización de tipo π O→N back-donación, reflejada en un orden de enlace calculado mayor que 1 y un número de pares solitarios de electrones en el átomo de O inferior a 36.
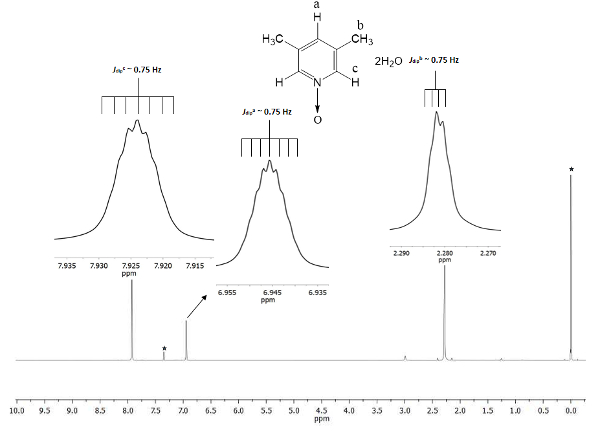
Figura 1 . TMS hace referencia CDCl3 espectro de 1H NMR de 500 MHz de 1. Integraciones de los desplazamientos químicos de las tres señales de acuerdo con tres tipos diferentes de átomos de hidrógeno presentes en lutine N-óxido. Haga clic aquí para ver una versión más grande de esta figura.
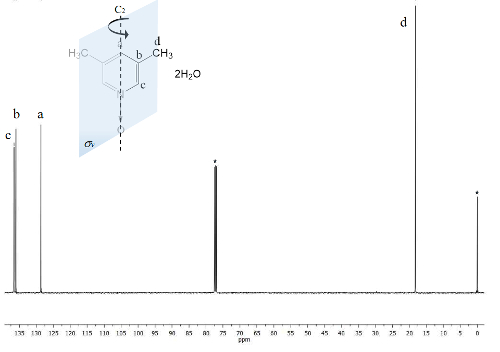
Figura 2 . TMS hace referencia CDCl3 espectro de 13C NMR de 100 MHz de 1. Se observan tres señales para los cinco carbonos aromáticos y uno para los dos grupos metilo. Haga clic aquí para ver una versión más grande de esta figura.
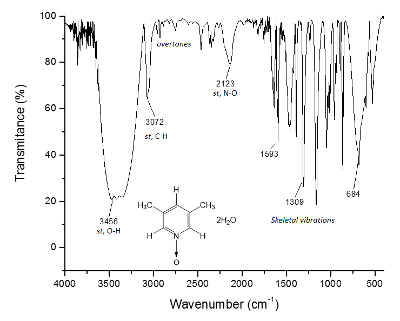
Figura 3 . Espectro de IR de 1. Enlaces O-H, por encima de los 3.300 cm-1, son responsables de la formación de la estructura supramolecular y la formación de cristales. Haga clic aquí para ver una versión más grande de esta figura.

Figura 4 . Diagrama ORTEP de donde dos moléculas de formas de H2O puente de hidrógeno con el oxígeno de lutidina, conduciendo sus átomos de hidrógeno hacia el átomo de oxígeno de 1. Esta figura ha sido modificada desde Merino García et al. 12 Haga clic aquí para ver una versión más grande de esta figura.

Figura 5 . Imágenes de difracción de rayos x adecuada cristales de 1 en éter dietílico (arriba) y al aire libre (parte inferior). Uno de estos cristales fue confirmada en el difractómetro de rayos x y mostró un camino de difracción de rayos x, que estaba traduciendo y refinado por programas computacionales especiales en una estructura cristalina y molecular24,25, 2627,de,28. Haga clic aquí para ver una versión más grande de esta figura.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
El protocolo presentado aquí es un método convencional para enlazar un átomo de oxígeno el átomo de nitrógeno de 3, 5-lutidina como un método de funcionalización de sustratos. Esta técnica también es establecida para producir cristales deshidratados adecuados (figura 5, fotografías tomadas con una cámara Sony DSC-HX300 Cyber-shot) de rayos x. Como estamos preocupados, no muchos informes han descrito la producción de tales cristales16. Muchos compuestos crecen cristales ideal para análisis de rayos x cuando son quelados por varios metales17,18,19,20. Una vez que se forma el polvo cristalino, es importante extraer de sus licores madre utilizando un matraz Kitasato y un embudo de Buchner. Utilizar mangueras de goma, el matraz Kitasato se conecta a una línea de vacío y en la parte superior se coloca el Embudo Buchner con un papel de filtro. Una vez encendido el vacío, se humedece el papel filtro con una pequeña cantidad de solvente de la cual el producto cristalizado. Esto evita que el polvo cristalino de filtrado en el embudo de Buchner, por el efecto del vacío. Después de asegurar el papel de filtro, la solución que contiene el polvo cristalino se agita para asegurar que todo el polvo cristalino se filtra, y ninguno permanece en el fondo del matraz. La solución se vierte rápidamente en el embudo de Buchner. El polvo cristalino obtenido se deja por unos 10 minutos en el papel de filtro y entonces el vacío se apaga y el polvo cristalino se desprende el papel almacenado en un frasco de cristal opaco, etiqueta con su código y mantenido a 4 ° C hasta su posterior análisis. El líquido filtrado se vierte en un vaso de Petri, dejándola a la evaporación lentamente a 4 ° C para mejorar la formación de cristales adecuados para análisis de rayos x.
Es importante hacer notar que este protocolo usa disolventes y los materiales que son fácilmente obtenibles y generalmente se encuentran en cualquier laboratorio de investigación. El ajuste de pH mediante la adición de Na2CO3 y la agitación magnética constante son fundamentales para el rendimiento del producto final. Sin embargo, es importante prestar atención especial cuidado en todas las etapas de proceso, especialmente en la etapa de extracción donde ningún rastro de material de partida debe estar presente para permitir la formación de polvo cristalino y posteriormente los cristales. Por lo tanto, esta etapa de extracción/purificación puede controlarse mediante espectroscopia NMR o IR para asegurar la calidad del producto.
Para asegurar la reproducibilidad de este protocolo, NMR es una excelente herramienta. Incluso detalles son visibles en el espectro. Todas las señales se muestran como inserciones en la figura 1. Estos recuadros representan claramente una división, es decir, la multiplicidad, de todas las señales. Por ejemplo, lo protones b (dipJb ~ Hz 0,75) muestran cuatro picos en la cresta de la señal, con una separación entre ellos constante mareado (Δpico pico) de ~ 0,0075 ppm. Ppm 0,0075 puede transformarse en energía usando la siguiente ecuación21
 Ecuación 1
Ecuación 1

La transformación se recomienda porque las señales de desarrollo vienen de la interacción dipolar y espacial entre los núcleos de tres hidrógenos del grupo metilo, y aunque son más que los 4 enlaces sencillos con protones c y a, son capaces de percibir su momento magnético dipolar interacciones22. Además, la sigma libre rotación en el grupo metilo de la vinculación permite la interacción de protón-protón de super hiperfina sea visible en la multiplicidad de la señal. Septuplets de protones una y c a 6,9 y 7,9 ppm, respectivamente se derivan de un mismo fenómeno de naturaleza dipolar. En estos casos, los protones una y c puede distinguir los protones del grupo metilo para la misma dinámica de rotación. Por último, como era de esperar, el calculado Jdip para a, b, y c tienen el mismo valor, apenas de ~ Hz 0,75. Estas cantidades de las interacciones confirman el arreglo espacial de los núcleos de hidrógeno a lo largo de la anisotropía magnética.
Por otro lado, la simetría C2v 1 hace carbonos equivalentes23. El espectro de 13C, figura 2, muestra la típica señal de grupos metilo Unidos a anillos aromáticos, carbonos d a 18 ppm. Por otra parte, una señal en 129 ppm es visible en esta región debido a la menos electronegative elemento influenciado carbono una. A altas frecuencias la señal de los núcleos de átomos de carbono más expuestas al campo magnético se presentó en 137 ppm22.
La metodología presentada es muy útil para la síntesis de la piridina N-óxidos, proporcionar buenos rendimientos, en un tiempo razonable con las condiciones de reacción suaves y reactivos accesibles fáciles y baratos, que no requieren catalizadores adicionales. Estas condiciones pueden utilizarse para la comunidad científica y educativa para obtener una amplia gama de piridinas N-óxidos como precursores de otras moléculas de interés. La metodología adecuada da la oportunidad de adquirir las herramientas básicas conceptuales y experimentales en laboratorios educativos para estudiantes, demostrando una acertada síntesis de compuestos y la dicha de ver la formación de cristales. Sin embargo, es importante destacar que, como cualquier reacción química, es necesario tomar todas las precauciones, ya que generalmente los reactivos usados son peligrosos.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Todos los autores no declaran conflicto de intereses.
Acknowledgments
El presente trabajo ha sido apoyado por Vicerrectoría de Investigación y Estudios de Posgrado de la BUAP, divulgación de la ciencia y no de proyectos. REOY NAT14, 15, 16-G. HEAS-NAT17. RMG agradece a CONACyT (México) para la beca 417887.
Materials
| Name | Company | Catalog Number | Comments |
| 3,5-lutidine | Sigma-Aldrich | L4206-500ML | |
| Glacial acetic acid | Fermont | 3015 | |
| Hidrogen peroxide (35%) | Sigma-Aldrich | 349887-500ML | |
| Na2CO3 anhydrous | Productos Químicos Monterrey | 1792 | |
| Na2SO4 anhydrous | Alfa reactivos | 25051-C | |
| CHCl3 | Fermont | 6205 | |
| Ethyl eter | Mercury Chemist | QME0309 | |
| Distilled water | Comercializadora Química Poblana | not-existent |
References
- Ochiai, E. Recent Japanese work on the chemistry of pyridine 1-oxide and related compounds. J. Org. Chem. 18 (5), 534-551 (1953).
- Solomons, T. W. G. Organic Chemistry 2nd Edition. , John Wiley & Sons. 1110 (1976).
- Albini, A., Pietra, S. Heterocyclic N-Oxides. , CRC Press. ISBN: 0849345529 328 (1991).
- Koukal, P., Ulc, J., Necas, D., Kotora, Heterocyclic N.-Oxides. Topics in Heterocyclic Chemistry. 53, 29-58 (2017).
- Wen-Man, Z., Jian-Jun, D., Xu, J., Jun, X., Huan-Jian, X. Visible-Light-Induced C2 alkylation of pyridine N.-oxides. J. Org. Chem. 82 (4), 2059-2066 (2017).
- Merino García, M. R., Ríos-Merino, F. J., Bernès, S., Reyes-Ortega, Y. Crystal structure of 3,5-dimethylpyridine N-oxide dihydrate. Acta Cryst. 72 (12), 1687-1690 (2016).
- Sarma, R., Karmakar, A., Baruah, J. B. N-Oxides in Metal-Containing Multicomponent Molecular Complexes. Inorg. Chem. 47 (3), 763-765 (2008).
- Youssif, S. Recent trends in the chemistry of pyridine N-oxides. ARKIVOC. 2001, 242-268 (2001).
- Chucholowski, A. W., Uhlendorf, S. Base catalyzed rearrangement of 5-cyanomethyl-2-isoxazolines; novel pathway for the formation of 2-aminopyridine N-oxides. Tetrahedron Lett. 31 (14), 1949-1952 (1990).
- Thellend, A., Battioni, P., Sanderson, W., Mansuy, D. Oxidation of N-Heterocycles by H2O2 Catalyzed by a Mn-Porphyrin: An Easy Access to N-Oxides Under Mild Conditions. Synthesis. 1997 (12), 1387-1388 (1997).
- Copéret, C., Adolfson, H., Tinh-Alfredo, V. K. h, Yudin, A. K., Sharpless, K. B. A simple and Efficient Method for the Preparation of Pyridine N-Oxides. J. Org. Chem. 63 (5), 1740-1741 (1998).
- Ferrer, M., Sánchez-Baeza, F., Messeguer, A. On the preparation of amine N-oxides by using dioxiranes. Tetrahedron. 53 (46), 15877-15888 (1997).
- Adam, W., Briviba, K., Duschek, F., Golsch, D., Kiefer, W., Sies, H. Formation of singlet oxygen in the deoxygenation of heteroarene N-oxides by dimethyldioxirane. J. Chem. Soc. Chem. Commun. 1995 (18), 1831-1832 (1995).
- Murray, R. W., Singh, M. A Facile One-Step Synthesis of C-Arylnitrones Using Dimethyldioxirane. J.Org.Chem. 55 (9), 2954-2957 (1990).
- Kim, S. W., Um, T., Shin, S. Brønsted acid-catalyzed α-halogenation of ynamides from halogenated solvents and pyridine-N-oxides. Chem. Commun. 53 (18), 2733-2736 (2017).
- Campeau, L., Rousseaux, R., Fagnou, K. A solution to the 2-pyridyl organometallic cross-coupling problem: regioselective catalytic direct arylation of pyridine N-oxides. J. Am. Chem. Soc. 127 (51), 18020-18021 (2005).
- Gang, L., et al. Metal-free methylation of a pyridine N-oxide C-H bond by using peroxides. Org. Biomol. Chem. 13 (46), 11184-11188 (2015).
- May, D., Nyman,, Hampden-Smith, M. J., Duesler, E. N. Synthesis, characterization, and reactivity of group 12 metal thiocarboxylates M(SOCR)2Lut2[M) Cd, Zn; R ) CH3, C(CH3)3; Lut ) 3,5-Dimethylpyridine (Lutidine)]. Inorg. Chem. 36 (10), 2218-2224 (1997).
- Cho, S. H., Hwang, S. J., Chang, S. Palladium-Catalyzed C-H Functionalization of Pyridine N-Oxides: Highly Selective Alkenylation and Direct Arylation with Unactivated Arenes. J. Am. Chem. Soc. 130 (29), 9254-9256 (2008).
- Ide, Y., et al. Spin-crossover between high-spin (S = 5/2) and low-spin (S = 1/2) states in six-coordinate iron(III) porphyrin complexes having two pyridine-N. oxide derivatives. Dalton Trans. 46 (1), 242-249 (2017).
- Drago, R. S. Physical Methods in Chemistry. , Saunders College Publishing USA. 750 (1977).
- Cervantes-Mejía, V., et al. Branched Polyamines Functionalized with Proposed Reaction Pathways Based on 1H-NMR, Atomic Absorption and IR Spectroscopies. American Journal of Analytical Chemistry. 5 (16), 1090-1101 (2014).
- Huheey, J. E., Keiter, E. A., Keiter, R. L. Inorganic Chemistry: Principles of Structure and Reactivity, 4th Edition. , Oxford University Press. Mexico. ISBN: 9706131620 1023 (1997).
- Rigaku, CrysAlisPRO. , (2013).
- Sheldrick, G. M. SHELXT - Integrated space-group and crystal-structure determination. Acta Cryst. 71 (1), 3-8 (2015).
- Sheldrick, G. M. Crystal structure refinement with SHELXL. Acta Cryst. 71 (1), 3-8 (2015).
- Sheldrick, G. M. A short history of SHELX. Acta Cryst. 64 (1), 112-122 (2008).
- Macrae, C. F., et al. Mercury CSD 2.0 - new features for the visualization and investigation of crystal structures. J. Appl. Cryst. 41 (2), 466-470 (2008).
- ChemBioDraw Ultra 13. , PerkinElmer. (2013).
