

Summary
Häri, rapporterar vi syntes och kristallisation av 3,5-lutidine N -oxid torkar ut av ett enkelt protokoll som skiljer sig från klassisk syntesen av pyridin N-oxid. Detta protokoll använder olika utgångsmaterial och innebär mindre reaktion tid att ge en ny solvatiserade supramolekylär struktur, som kristalliserar under långsam avdunstning.
Abstract
Syntesen av 3,5-lutidine N -oxid torka, 1, uppnåddes i vägen syntes av 2-amino-pyridin-3,5-dicarboxylic syra. Ochiai först används metoden för icke-substituerade pyridines 1957 i en 12 h process, men ingen röntgen lämplig kristaller erhölls. Substituerade ringen används i den metod som presenteras här tydligt influerad tillägg av vattenmolekyler i asymmetrisk enheten, som ger en annan nukleofil styrka i 1. X-ray lämplig crystal förening 1 var möjligt på grund av stabiliseringen av negativa laddningen i syre av närvaron av två vattenmolekyler där väteatomerna donera positiv laddning in i ringen; sådana vattenmolekyler fungera väl för att konstruera en supramolekylär interaktion. De hydratiserade molekylerna kan vara möjligt för alkaliska systemet som nås genom att justera pH till 10. Ännu viktigare, den dubbla metyl substituerade ring och en reaktionstid på 5 h, gör det en mer mångsidig metod och med bredare kemiska tillämpningar för framtida ring infogningar.
Introduction
Nuförtiden, forskare runt om i världen har varit att investera resurser i utvecklingen av nya syntetiska rutter för funktionalisering av aromatiska grupper, som är kända för låg reaktivitet främre till tillägg reaktioner1,2, 3. Pyridin, där en kväveatom ersätter en kolatom, presenterar en liknande kemisk reaktivitet till analoga ringar består enbart av kol atomer3, och det genomgår oftast en substitution mekanism i stället för tillägg. N-oxider är särskiljande av närvaron av en givare band mellan kväve och syre som bildas av överlappningen av nonbonding elektron par på kvävet med en tom orbital på syre atom3. Särskilt, pyridin N-oxider är Lewis baser, eftersom deras N-O-delen kan fungera som en elektron donator, och de kan kombinera med Lewis syror bildar de motsvarande Lewis syra-baspar. Detta boende har en grundläggande kemisk följd, eftersom det kan öka nucleophilicity av Lewis syror mot potentiella elektrofiler och därmed tillåta dem att reagera under förhållanden där normalt reaktionen skulle inte förekommer. Förmodligen är den mest frekventa användningen av sådana föreningar i olika oxidationsreaktioner där de agerar som oxidanter4. Pyridin N-oxider och många av deras ring-functionalized derivat är återkommande molekyler av biologiskt aktiva och farmakologiska agenter5, och en tydlig rumslig fördelning av olika spektroskopiska verktyg har fastställts för vissa av dem6,7. I forskning om fästa olika grupper till pyridin ringen, har forskare testat olika metoder för att producera en lätt och konventionell metod, eftersom isoxazolines kräver en katalytisk mängd bas såsom DBU i kokande xylen till formuläret 6- ersätta-2-aminopyridin N-oxider8,9. En mängd pyridin derivat omvandlades till deras motsvarande N-oxider i närvaro av en katalytisk mängden mangan tetrakis(2,6-diclorophenyl) porphyrin och ammonium acetat i CH2Cl2/CH3 KN-8,10. Andra pyridines oxideras till deras oxider som använder H2O2 i närvaro av katalytisk mängder methyltrioxorhenium8,11eller genom tillsats av överflödig dimethyldioxirane i CH2Cl2 vid 0 ° C, vilket leder till motsvarande N-oxider8,12,13,14. Bis (trimetylsilyl) peroxid i närvaro av trioxorhenium i CH2Cl2 har använts för syntesen av pyridin N-oxider8,11. Syntesen av aminopyridin N-oxider som involverar acylation med caros syra (peroxomonosulfuric acid) har också rapporterats8. Ändå, den metod som redovisas här, och som använder en del av den metod som rapporterats av Ochiai1, ger mycket bra resultat med hjälp av billigare och tillgänglig reagenser, H2O2 och isättika. Denna praxis är mer lämplig för användning i stor skala preparat som verkar på tertiära aminer, det ger god avkastning i en reaktion som kräver endast 30% väteperoxid och koncentrerad ättiksyra i en temperatur mellan 70-80 ° C och den använder en reningsprocess som finns i de flesta syntes laboratorier som destillation, utan användning av katalysator eller dyrare reagens1. Litteraturen rapporterar att andra metoder också ofta involvera tidsramar från 10-24 h och temperaturer över 100 ° C 4,8, och avkastningen av välformade kristaller för X-ray analyser rapporteras sällan.
Reaktivt, används olika N -oxid derivat för att aktivera tillräckligt lutidine ringen, antingen en nukleofil eller elektrofil sätt. Nukleofil eller elektrofil faktorn påverkas av substituenterna. Med pyridin ringen är elektron-återkalla grupper, är den viktigaste faktorn nukleofil karakteristiska1. Gratis N-oxid föreningar är sällan isolerade som lämplig kristaller för Röntga analys på grund av den delocalized laddningen i aromatiska ringen. Den utläggning faktorn är dock kritiska till att stabilisera den negativa tätheten av syre15.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. reaktion
- Placera i ett dragskåp en öppnad runda 100 mL-mätkolv med 0,5 mol (29,8 mL) isättiksyra och tillsätt 0.051 mol (5.82 mL) 3,5-dimethylpyridine och 5 mL H2O2 (35%). Hålla den blandning reaktionen under konstant magnetisk omrörning, vid en inre temperatur på 80 ° C i 5 h.
- Efter reaktionstiden, svalka kolven till 24 ° C med is (gör inte exponera ättiksyra gaser på isen), och Anslut den till en hög vakuum destillationsenheten för 90-120 min att ta bort överflödig ättiksyra.
Varning: Använd inte hett material. Vänta tills glas når en hanterbar temperatur. Detta kommer också att undvika ångor in toppen av destillationsenheten. - Tillsätt destillerat vatten (10 mL) två gånger för att säkerställa avlägsnande av alla spår av ättiksyra och koncentrera blandningen mycket som möjligt.
2. valens justering och utvinning
- Lös i bi-destillerade vatten isolerade trögflytande och transparent produkten och använda en potentiometer för justering av pH till 10 med ren solid Na2CO3.
- Placera försiktigt lösningen i en 250 mL separation tratt och extrahera den 5 gånger med 250 mL CHCl3 att förbättra avkastningen. Återställa det organiska lagret och torka den över solid Na2så4 för 30 min maximala, som innehåller produkten. Om nödvändigt, extrahera vattenfasen med önskad mängd CHCl3.
Försiktighet: CHCl3 kan orsaka dåsighet och yrsel; hanterar med omsorg och släpper ett dragskåp. - Ta bort lösningsmedlet under reducerat tryck med en hög vakuum destillation enhet, fram till bildandet av ett mycket hygroskopiskt klart beige kristallint pulver (70%).
3. kristallisation
- Lös 4,3 g kristallint pulver i 50 mL kall hög utförs vätskekromatografi (HPLC) grad dietyleter. Vakuum filtrera lösningen att ta bort spår av solid utgångsmaterial eller även damm. Häll ett glas petriskål filtratet, lämnar den att sakta avdunsta vid 4 ° C i ett laboratorium kylskåp.
- Säkerställa att efter två dagar, klart färglösa kristaller erhålls. Mät sedan smältpunkten, som bör vara i spänna av 310-311 K.
4. analys av 3,5-Lutidine N -oxid Dehydrate
- Ta bort de kristaller som bildas, prismatiska form och färglös, genom dekantering från kolvens väggar för ytterligare Röntga analys. Om inte omedelbart används, hålla kristallerna i dietyleter att undvika crystal återfuktning.
- Lös upp 0,010 g 3,5-lutidine N -oxid torkar i 0,4 mL CDCl3 att utföra NMR H1 och C13 analys att bevisa effektiviteten i förfarandet.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Protokollet är i huvudsak en förlängning av Ochiais teknik1. Dock tillämpas lägre temperatur och kortare tid. Den här enkla metoden kan användas för att få en mångsidig ligand, som är en substituerad pyridin N-oxid derivat. För att bekräfta bildandet av 1, föredras NMR 1H och 13C analys att testa effektiviteten av förfarandet.
Den kemiska förskjutningen visar bildandet av 1. Signalen på 2.28 ppm (delar per miljon) motsvarar de sex motsvarande hydrogens av två metylgrupperna i 3 och 5 positioner, som uppfattar magnetfältet i mindre andel än permanenten magnetiska. Det finns två uppsättningar av septuplets: en hör till protonen i c position på 7,9, vilket dubblar storleken på andra signalen på 6,9 som tillhör protonen i position a. figur 1 visar de kemiska förskjutningarna provocerade av närvaron av den syre atom bonde d att kväveatomen av pyridin ringen. Syreatomen är electro-återkalla och närmare väteatomerna att syreatomen (c och a) Visa förskjutning till en högre frekvens än för de metyl hydrogens (b).
Samma process ritas för NMR 13C spectrum, figur 2, där signalerna för de närmare kolatomer till syreatomen (c och en) visar frekvens separation mellan deras signaler Δc = 1 300 Hz och Δen = 200 Hz. De metyl kolatomer visar än en gång, inte någon förändring. Det IR-spektrumet kan användas för att se framgången med metoden samt.
ORTEP diagrammet, figur 4, visar förekomsten av två molekyler vatten kring asymmetrisk molekylen. Dessa molekyler tros stabilisera den N-O bond. I liknande fall, det har beskrivits för pyridin N-oxid och relaterade aromatiska kväveoxider. Det finns en betydande stabilisera π-typ O→N back-donation, återspeglas i ett beräknat bond beställer högre än 1 och ett antal elektron ensamstående par O Atomen lägre än 36.
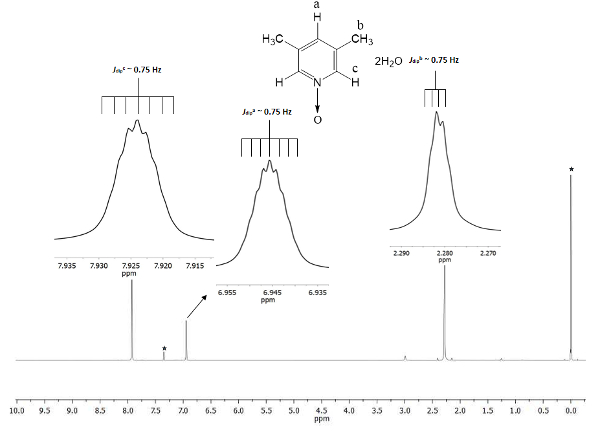
Figur 1 . TMS refererade CDCl3 500 MHz NMR 1H spektrum av 1. De integrationer och kemiska förskjutningarna av tre signaler håller med tre olika typer av väteatomer i lutine N-oxid. Klicka här för att se en större version av denna siffra.
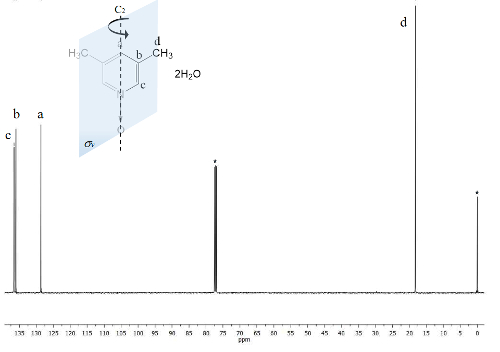
Figur 2 . TMS refererade CDCl3 100 MHz NMR 13C spektrum av 1. Tre signaler observeras för de fem aromatiska kolväten och en för de två metylgrupperna. Klicka här för att se en större version av denna siffra.
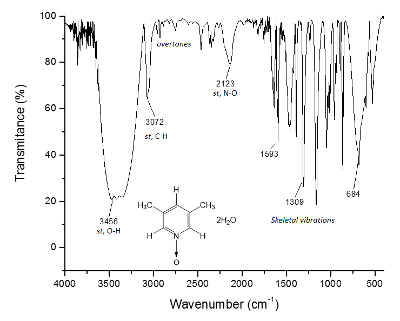
Figur 3 . IR-spektrum 1. O-H obligationer, framför 3 300 cm-1, ansvarar för Supramolekylära strukturerabildandet och crystal bildandet. Klicka här för att se en större version av denna siffra.

Figur 4 . ORTEP diagram över 1 där två molekyler av H2O former överbrygga Väteförbindelser med lutidine's syre, köra sina väteatomer mot syreatomen. Denna siffra har ändrats från Merino García o.a. 12 Klicka här för att se en större version av denna siffra.

Figur 5 . Bilder av lämplig röntgendiffraktion kristaller av 1 i dietyleter (överst) och på open air (nederst). En av dessa kristaller var bekräftat i en röntgendiffraktometer och visade en diffraktion väg av röntgen, som förtalats och förfinats av speciella computational program i en molekylär och kristallin struktur24,25, 26,27,28. Klicka här för att se en större version av denna siffra.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Det protokoll som presenteras här är en konventionell metod att länka en syreatom till kväveatomen av den 3,5-lutidine som en metod för funktionalisering av substrat. Denna teknik är också väl etablerad avkastning röntgen lämplig uttorkad kristaller (figur 5, bilder tagna med en Sony DSC-HX300 Cyber-shot-kamera). Så långt vi är berörda, har inte många rapporter beskrivs produktionen av sådana kristaller16. Många föreningar växa perfekt kristaller för Röntga analys när de är kelaterat av olika metaller17,18,19,20. När det kristallint pulvret bildas, är det viktigt att extrahera den från deras moderlut använder en Kitasato kolv och en Buchner tratt. Använda gummislangar, Kitasato kolven är ansluten till en vakuum linje och ovanpå det Buchner tratten placeras med ett filterpapper. När vakuumet har varit påslagen, fuktas pappersfiltret med en liten mängd vätska som kristalliserade produkten. Detta förhindrar att det kristallint pulvret sipprar in Buchner tratten av vakuum effekt. Efter att pappersfiltret, skakas lösningen med kristallint pulver för att säkerställa att allt kristallint pulver är filtrerat, och ingen kvar i botten av kolven. Lösningen hälls snabbt över Buchner tratten. Den kristalliniskt pulver erhållet är kvar i ca 10 min på filterpapperet, och sedan dammsuga är avstängd och kristallint pulver lossnat från papperet och lagras i en ogenomskinlig glasflaska, märkt med dess kod och förvaras vid 4 ° C tills vidare analys. Filtermediat vätskan hälls i ett glas petriskål, lämnar det till långsamt avdunstning vid 4 ° C att förbättra bildandet av adekvat kristaller för Röntga analys.
Det är viktigt att notera att detta protokoll använder lösningsmedel och material som är lätt att få och allmänhet finns på någon forskningslaboratorium. Den pH-justeringen genom tillsats av Na2CO3 och den konsekventa magnetisk omrörningen är kritiska till avkastningen av den slutliga produkten. Det är dock viktigt att vara extra försiktig uppmärksam i alla steg i processen, särskilt i utvinning scenen där inga spår av utgångsmaterial måste vara närvarande för att ha råd med bildandet av kristallint pulver och därefter kristaller. Således kan denna extraktion/reningssteg övervakas av antingen NMR eller IR-spektroskopi för att säkerställa kvaliteten på produkten.
För att säkerställa reproducerbarheten av detta protokoll, är NMR ett utmärkt verktyg. Även fina detaljer är synliga i spektrumet. Alla signaler visas som indragen i figur 1. Dessa inläggningar skildrar tydligt en split, nämligen multiplicity, av alla signaler. Till exempel protoner b (Jdipb ~ 0,75 Hz) visar fyra toppar på krönet av signalen, med en separation bland dem (Δtopp-topp) illamående konstant av ~ 0,0075 ppm. 0,0075 ppm kan omvandlas till energi med hjälp av följande ekvation21
 Ekvation 1
Ekvation 1

Omformningen rekommenderas eftersom de signaler som utspelas kommer från tvåpolig rumsliga interaktionen mellan tre hydrogens atomkärnor av metylgruppen, och även om de är längre än de 4 enkelbindningar med protoner c och a, de kan uppfatta deras tvåpolig magnetiska momentum interaktioner22. Dessutom ger gratis sigma limning rotation i metylgruppen super hyperfine proton-proton samspelet ska vara synlig i multiplicityen av signalen. Septuplets av protons en och c på 6,9 och 7,9 ppm, respektive härrör från samma tvåpolig naturfenomen. I dessa fall, protoner en och c kan skilja protonerna i metylgruppen samma rotation dynamiska. Senast, som förväntat, beräknade Jdip för a, b och c har knappt samma värde ~ 0,75 Hz. Dessa kvantiteter av interaktioner bekräfta den väte kärnor rumsliga ordningen i hela den magnetisk anisotropin.
Däremot, gör 1 C2v symmetri motsvarande kolatomer23. 13C spektrumet, figur 2, visar de typiska signalen för metylgrupper bifogas aromatiska ringar, kolatomer d vid 18 ppm. Dessutom en signal på 129 ppm är synliga på denna region på grund av mindre elektronegativa elementet påverkas kol en. Vid höga frekvenser presenteras signalen för mer utsatta kolatomer atomkärnor till det magnetiska fältet på 137 ppm22.
Den presenterade metoden är mycket användbar för syntesen av pyridin N-oxider, som ger bra avkastning, i rimlig tid med mjuk reaktionsbetingelser och billiga och lätt tillgängliga reagens, som inte kräver ytterligare katalysatorer. Dessa villkor kan användas för vetenskapliga och pedagogiska gemenskapen för att få ett brett spektrum av pyridines N-oxider som prekursorer för andra molekyler av intresse. Den lämpliga metoden ger möjlighet att förvärva grundläggande experimentella och konceptuella verktyg i utbildningslaboratorier för studenter, bevisar en framgångsrik syntes av föreningar och lycka att se bildandet av kristaller. Det är dock viktigt att understryka att, som någon kemisk reaktion, det är nödvändigt att vidta alla försiktighetsåtgärder eftersom generellt de reagens som används är farliga.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Alla författarna förklarar någon intressekonflikt.
Acknowledgments
Detta arbete har stötts av Vicerrectoría de Investigación y Estudios de Posgrado från BUAP, spridning av vetenskap och projekt nr. REOY-NAT14, 15, 16-G. ELIAS-NAT17. RMG tack CONACyT (Mexiko) för stipendium 417887.
Materials
| Name | Company | Catalog Number | Comments |
| 3,5-lutidine | Sigma-Aldrich | L4206-500ML | |
| Glacial acetic acid | Fermont | 3015 | |
| Hidrogen peroxide (35%) | Sigma-Aldrich | 349887-500ML | |
| Na2CO3 anhydrous | Productos Químicos Monterrey | 1792 | |
| Na2SO4 anhydrous | Alfa reactivos | 25051-C | |
| CHCl3 | Fermont | 6205 | |
| Ethyl eter | Mercury Chemist | QME0309 | |
| Distilled water | Comercializadora Química Poblana | not-existent |
References
- Ochiai, E. Recent Japanese work on the chemistry of pyridine 1-oxide and related compounds. J. Org. Chem. 18 (5), 534-551 (1953).
- Solomons, T. W. G. Organic Chemistry 2nd Edition. , John Wiley & Sons. 1110 (1976).
- Albini, A., Pietra, S. Heterocyclic N-Oxides. , CRC Press. ISBN: 0849345529 328 (1991).
- Koukal, P., Ulc, J., Necas, D., Kotora, Heterocyclic N.-Oxides. Topics in Heterocyclic Chemistry. 53, 29-58 (2017).
- Wen-Man, Z., Jian-Jun, D., Xu, J., Jun, X., Huan-Jian, X. Visible-Light-Induced C2 alkylation of pyridine N.-oxides. J. Org. Chem. 82 (4), 2059-2066 (2017).
- Merino García, M. R., Ríos-Merino, F. J., Bernès, S., Reyes-Ortega, Y. Crystal structure of 3,5-dimethylpyridine N-oxide dihydrate. Acta Cryst. 72 (12), 1687-1690 (2016).
- Sarma, R., Karmakar, A., Baruah, J. B. N-Oxides in Metal-Containing Multicomponent Molecular Complexes. Inorg. Chem. 47 (3), 763-765 (2008).
- Youssif, S. Recent trends in the chemistry of pyridine N-oxides. ARKIVOC. 2001, 242-268 (2001).
- Chucholowski, A. W., Uhlendorf, S. Base catalyzed rearrangement of 5-cyanomethyl-2-isoxazolines; novel pathway for the formation of 2-aminopyridine N-oxides. Tetrahedron Lett. 31 (14), 1949-1952 (1990).
- Thellend, A., Battioni, P., Sanderson, W., Mansuy, D. Oxidation of N-Heterocycles by H2O2 Catalyzed by a Mn-Porphyrin: An Easy Access to N-Oxides Under Mild Conditions. Synthesis. 1997 (12), 1387-1388 (1997).
- Copéret, C., Adolfson, H., Tinh-Alfredo, V. K. h, Yudin, A. K., Sharpless, K. B. A simple and Efficient Method for the Preparation of Pyridine N-Oxides. J. Org. Chem. 63 (5), 1740-1741 (1998).
- Ferrer, M., Sánchez-Baeza, F., Messeguer, A. On the preparation of amine N-oxides by using dioxiranes. Tetrahedron. 53 (46), 15877-15888 (1997).
- Adam, W., Briviba, K., Duschek, F., Golsch, D., Kiefer, W., Sies, H. Formation of singlet oxygen in the deoxygenation of heteroarene N-oxides by dimethyldioxirane. J. Chem. Soc. Chem. Commun. 1995 (18), 1831-1832 (1995).
- Murray, R. W., Singh, M. A Facile One-Step Synthesis of C-Arylnitrones Using Dimethyldioxirane. J.Org.Chem. 55 (9), 2954-2957 (1990).
- Kim, S. W., Um, T., Shin, S. Brønsted acid-catalyzed α-halogenation of ynamides from halogenated solvents and pyridine-N-oxides. Chem. Commun. 53 (18), 2733-2736 (2017).
- Campeau, L., Rousseaux, R., Fagnou, K. A solution to the 2-pyridyl organometallic cross-coupling problem: regioselective catalytic direct arylation of pyridine N-oxides. J. Am. Chem. Soc. 127 (51), 18020-18021 (2005).
- Gang, L., et al. Metal-free methylation of a pyridine N-oxide C-H bond by using peroxides. Org. Biomol. Chem. 13 (46), 11184-11188 (2015).
- May, D., Nyman,, Hampden-Smith, M. J., Duesler, E. N. Synthesis, characterization, and reactivity of group 12 metal thiocarboxylates M(SOCR)2Lut2[M) Cd, Zn; R ) CH3, C(CH3)3; Lut ) 3,5-Dimethylpyridine (Lutidine)]. Inorg. Chem. 36 (10), 2218-2224 (1997).
- Cho, S. H., Hwang, S. J., Chang, S. Palladium-Catalyzed C-H Functionalization of Pyridine N-Oxides: Highly Selective Alkenylation and Direct Arylation with Unactivated Arenes. J. Am. Chem. Soc. 130 (29), 9254-9256 (2008).
- Ide, Y., et al. Spin-crossover between high-spin (S = 5/2) and low-spin (S = 1/2) states in six-coordinate iron(III) porphyrin complexes having two pyridine-N. oxide derivatives. Dalton Trans. 46 (1), 242-249 (2017).
- Drago, R. S. Physical Methods in Chemistry. , Saunders College Publishing USA. 750 (1977).
- Cervantes-Mejía, V., et al. Branched Polyamines Functionalized with Proposed Reaction Pathways Based on 1H-NMR, Atomic Absorption and IR Spectroscopies. American Journal of Analytical Chemistry. 5 (16), 1090-1101 (2014).
- Huheey, J. E., Keiter, E. A., Keiter, R. L. Inorganic Chemistry: Principles of Structure and Reactivity, 4th Edition. , Oxford University Press. Mexico. ISBN: 9706131620 1023 (1997).
- Rigaku, CrysAlisPRO. , (2013).
- Sheldrick, G. M. SHELXT - Integrated space-group and crystal-structure determination. Acta Cryst. 71 (1), 3-8 (2015).
- Sheldrick, G. M. Crystal structure refinement with SHELXL. Acta Cryst. 71 (1), 3-8 (2015).
- Sheldrick, G. M. A short history of SHELX. Acta Cryst. 64 (1), 112-122 (2008).
- Macrae, C. F., et al. Mercury CSD 2.0 - new features for the visualization and investigation of crystal structures. J. Appl. Cryst. 41 (2), 466-470 (2008).
- ChemBioDraw Ultra 13. , PerkinElmer. (2013).
