

ERRATUM NOTICE
Important: There has been an erratum issued for this article. Read more …
Summary
Este procedimiento se estableció para ser utilizado para el desarrollo de cultivos hepáticos 3D avanzados in vitro, que pueden proporcionar una evaluación más fisiológicamente relevante de los peligros genotóxicos asociados con exposiciones nanomateriales a lo largo de un régimen de dosis aguda o repetida a largo plazo.
Abstract
Debido al rápido desarrollo e implementación de una amplia gama de nanomateriales de ingeniería (ENM), la exposición a ENM es inevitable y el desarrollo de sistemas de prueba in vitro robustos y predictivos es esencial. La toxicología hepática es clave al considerar la exposición a la ENM, ya que el hígado desempeña un papel vital en la homeostasis metabólica y la desintoxicación, además de ser un sitio importante de acumulación de ENM después de la exposición. Sobre la base de esto y la comprensión aceptada de que los modelos de hepatocitos 2D no imitan con precisión las complejidades de las intrincadas interacciones multicelulares y la actividad metabólica observada in vivo, hay un mayor enfoque en el desarrollo de modelos hepáticos 3D fisiológicamente relevantes adaptados para fines in vitro de evaluación de riesgos ENM. De acuerdo con los principios de los 3R para reemplazar, reducir y refinar la experimentación con animales, se ha desarrollado un modelo hepático basado en líneas celulares 3D HepG2, que es un sistema fácil de usar y rentable que puede soportar regímenes de exposición ENM extendidos y repetidos (≤14 días). Estos modelos esferoides (≥500 μm de diámetro) conservan su capacidad proliferativa (es decir, modelos de células divisorias) lo que les permite combinarse con el ensayo de micronucleus 'estándar de oro' para evaluar eficazmente la genotoxicidad in vitro. Su capacidad para informar sobre una gama de puntos finales toxicológicos (por ejemplo, función hepática, (pro)respuesta inflamatoria, citotoxicidad y genotoxicidad) se ha caracterizado utilizando varios ENM en regímenes de exposición agudos (24 h) y a largo plazo (120 h). Este modelo hepático in vitro 3D tiene la capacidad de ser utilizado para evaluar exposiciones enM más realistas, proporcionando así un enfoque in vitro futuro para apoyar mejor la evaluación de riesgos enM de una manera rutinaria y de fácil acceso.
Introduction
Debido al rápido desarrollo e implementación de una amplia gama de nanomateriales de ingeniería (ENM) a través de una plétora de aplicaciones basadas en humanos (por ejemplo, alimentos, cosméticos, ropa, equipos deportivos, electrónica, transporte y medicina), es inevitable que los seres humanos estén expuestos a LAM de forma regular. Con esto, hay mayores preocupaciones de que las características fisioquímicas específicas de tamaño novedosas que consideran estos materiales ventajosos en numerosas aplicaciones podrían causar efectos adversos sobre la salud humana y el medio ambiente de manera concomitante. Actualmente existen muchas actividades internacionales para reflejar activamente exposiciones más fisiológicamente relevantes a estos ENM y evaluar la toxicidad potencial de estos materiales en escenarios agudos, a largo plazo y repetidos de exposición a dosis bajas.
La toxicología hepática es clave a la hora de considerar la exposición a la ENM, ya que es ampliamente conocido que el hígado es un sitio importante de acumulación de ENM después de la exposición1,2. Además, el hígado es el sistema de órganos primarios para el metabolismo y desintoxicación de sustancias que entran en circulación sistémica3. Sobre la base de la comprensión aceptada de que los modelos de hepatocitos 2D no imitan con precisión las complejidades de interacciones multicelulares intrincadas o representan adecuadamente la actividad metabólica observada in vivo, se ha establecido un mayor enfoque en el desarrollo de modelos hepáticos 3D in vitro robustos y fisiológicamente relevantes para tecnologías sustitutivas in vivo4,5. La utilización de tecnologías avanzadas de cultivo 3D mejora la longevidad de los modelos hepáticos in vitro, lo que permite investigar regímenes de exposición repetida a largo plazo. Además, este formato de cultivo avanzado promueve la formación de características fisiológicas y organotípicas mejoradas como biliar canaliculi, procesos de transporte activo y capacidades mejoradas de metabolización de fármacos CYP450, mejorando así la predictividad de los modelos6. Los modelos hepáticos in vitro 3D actuales que consisten en monocultivos (sólo hepatocitos) o co-cultivos (hepatocitos con células no parnquimales) existen en varios formatos, que van desde microtissues o esferoides en placas de adhesión ultrarrápidas, esferoides colgantes, células incrustadas en matrices y/o andamios y plataformas de cultivo celular microfluídico, todos los cuales se consideran modelos in vitro avanzados eficaces para la evaluación de toxicidad hepática6,7. Sin embargo, la mayoría de estos sistemas modelo son de alto mantenimiento, requieren equipos especializados y son caros. Además, estos modelos suelen ser estáticos (es decir, modelos celulares que no cumplen) que impiden su uso en la evaluación de puntos finales de riesgo, como pruebas de genotoxicidad utilizando métodos que cuantifican el daño fijo del ADN. La genotoxicidad es un requisito previo básico en la toxicología regulatoria, y es un componente vital de la evaluación del riesgo de cualquier tóxico8. No hay un solo ensayo que pueda aplicarse para cuantificar todas las formas de daño del ADN que puedan surgir después de la exposición a un agente exógeno. Sin embargo, un componente central de la batería de pruebas de genotoxicidad in vitro es el ensayo de micronucleos, que es una técnica fiable y multifacética que mide el daño cromosómico bruto9. Es una técnica estándar de oro descrita por la Guía de Examen 487 de la OCDE, para evaluar los daños in vitro del ADN y la genotoxicidad y forma parte del requisito de batería de prueba para la evaluación de riesgos reglamentarios10,11.
La línea celular de carcinoma hepatocelular humano, HepG2, se utiliza ampliamente para la detección inicial de evaluación de riesgos, ya que las células están disponibles, relativamente baratas de obtener, fáciles de cultivar y aptos para un cribado de alto rendimiento12,13. Cuando se cultivan en estructuras esféricas 3D, se ha demostrado que recapitulan bien el microambiente hepático y ofrecen un modelo hepático con capacidades proliferativas suficientes para soportar el ensayo micronucleus3. Se estableció un mayor desarrollo de los modelos esferoides HepG2 para mejorar la longevidad y la funcionalidad hepática del modelo con el fin de apoyar la evaluación del riesgo de genotoxicidad a largo plazo, regímenes de exposición repetida (≤14 días). Así, en consonancia con los principios de los 3R para sustituir, reducir y refinar la experimentación con animales, se ha establecido el presente protocolo para proporcionar un modelo hepático in vitro 3D avanzado capaz de evaluar de forma fiable múltiples variables toxicológicas (por ejemplo, funcionalidad hepática, marcadores inflamatorios (pro), citotoxicidad y genotoxicidad) siguiendo exposiciones químicas y ENM agudas, a largo plazo y repetidas de forma rutinaria y de fácil acceso.
Aquí, presentamos un método para establecer una línea celular hepatocito 3D fisiológicamente relevante basada en el sistema modelo in vitro para la evaluación de riesgos de genotoxicidad después de exposiciones agudas o repetidas enM a largo plazo. El protocolo se puede dividir en 6 etapas clave: cultivar células HepG2 criopreservadas; Preparación de esferoides hepG2; Transferencia de esferoide hepG2 de caída colgante a suspensión de agarose; Cosecha de esferoides hepG2; ensayo y puntuación de micronucleus; y análisis de datos.
Protocol
1.Culturing criopreservado células HepG2
NOTA: Las células HepG2, obtenidas de American Type Culture Collection (ATCC) se cultivaron en 1x Dulbecco's Modified Eagle Medium (DMEM) con 4.5g/L D-glucosa y L-glutamina complementada con 10% suero bovino fetal (FBS) y 1% antibiótico de penicilina/estreptomicina.
- Medio de cultivo celular DMEM pre-caliente (incluyendo los suplementos) en un baño de agua de 37 °C durante 30 min.
- Retire un vial de células HepG2 del nitrógeno líquido y descongele en un baño de agua de 37 °C durante 2-3 minutos, mientras arremolina suavemente el vial para permitir el deshielo uniforme de la suspensión celular. Tener cuidado de no sumergir el vial por encima de la O-anillo con el fin de reducir el potencial de contaminación.
- Una vez descongelado, retire el vial del baño de agua y rocíe generosamente con 70% de etanol para descontaminar la superficie exterior del vial antes de colocar debajo de una capucha estéril de cultivo de tejido laminar clase II.
- Pipetear cuidadosamente el contenido del criovial de las células HepG2 en un tubo centrífuga que contiene 9 ml de medio de cultivo celular DMEM pre-calentado (con suplementos).
- Usando un strippette de 10 ml, transfiera 10 ml de la suspensión celular a un matraz de cultivo celular desechable de25 cm 2 e incuba el cultivo durante 3 días (desde la siembra) a un matraz de cultivo celular desechable de 5% CO2 y 37 °C hasta que se alcance ~80% de confluencia antes de someterse a la subcultura en un matraz de cultivo celular desechable más grande de 75 cm2.
- Una vez alcanzada la confluencia del 80%, las células de sub-cultivo en condiciones estériles por trippsinización con solución de trippsina/EDTA del 0,05% se calientan previamente en un baño de agua de 37 °C durante 30 minutos. En ningún momento se debe permitir que las células se sequen.
- A medida que las células forman una monocapa adherente, retire los medios volcando en una olla de desinfectante. A continuación, lave inmediatamente la monocapa para eliminar todos los rastros de los medios existentes enjuagando el matraz dos veces con 3 ml de solución PBS estéril de 1x mantenida a temperatura ambiente. Además, deseche PBS en la olla de desinfectante.
- Una vez eliminado el lavado PBS, añadir 5 ml de solución precalentada 0.05% trypsin-EDTA, asegurando cubrir toda la superficie de las células e incubar células durante 6-8 min a 37 °C y 5% CO2.
- Toque suavemente el matraz para desalojar las células de la parte inferior del matraz y luego agregue 5 ml de medio de cultivo celular DMEM (con suplementos) para neutralizar la enzima trypsin.
- Transfiera la suspensión celular a un tubo centrífuga de 50 ml y pipetee la suspensión celular hacia arriba y hacia abajo a fondo para asegurarse de que las células estén completamente desasociadas.
- Centrífuga la suspensión celular diluida a 230 x g durante 5 min. Deseche el sobrenadante en desinfectante y vuelva a suspender el pellet celular en 25 ml de medio de cultivo celular DMEM (con suplementos).
- Transfiera la suspensión celular a un matraz de cultivo celular desechable de 75 cm2 e incuba a 37 °C y 5% CO2 durante otros 3 días antes de someterse a la preparación de esferoides. Una vez que los HepG2 han tenido tiempo de aclimatarse y una vez más alcanzar ~ 80% confluencia, determinar la concentración celular en preparación para la siembra de esferoides.
2. Preparación de esferoides HepG2
- Repita los pasos de subcultura indicados anteriormente, excepto después de la centrifugación, vuelva a suspender el pellet de celda en 1 ml de medio de cultivo DMEM precalentado en un baño de agua de 37 °C. Suspensión de celda de pipeta arriba y abajo a fondo.
- Puntuación de la viabilidad celular mediante el ensayo trypan blue exclusion (ver OSHA SOP 3.21 Toxinas reproductivas, mutágenos, teratógenos y embriotoxinas – Procedimientos para el manejo y almacenamiento seguros (2019) para orientación de salud y seguridad)14 con una relación de 1:1 de suspensión celular a solución azul trypan prefiltrada del 0,4%.
- Antes del recuento celular, tome 1 ml de solución azul trypan utilizando una jeringa de 1 ml y filtre con una unidad de filtro de 0,45 μm en un tubo estéril de 1 ml.
- Transfiera 10 μL de solución azul trypan filtrada a un tubo de 0,2 ml y agregue 10 μL de suspensión celular. La solución trypan azul filtrada restante se puede almacenar hasta 3 meses a temperatura ambiente para su uso futuro.
- Rocíe bien el hemociclo con un 70% de etanol y séquelo con una toalla de papel estéril antes de asegurar el control de cubierta en la parte superior con vapor de respiración. Deslizar el cubrebosque a través de la superficie humedecida de la respiración induce fuerzas cohesivas mediante la generación de anillos Newton.
- Pipetear suavemente la suspensión de la célula azul trypan hacia arriba y hacia abajo usando una pipeta de 1000 μL (para reducir el estrés pura) antes de añadir 10 μL al hemocítómetro. Asegúrese de que la solución esté dispersa debajo del resbalón de la cubierta y cubra toda la red sin burbujas de aire.

Figura 1: Contar celdas utilizando un hemociclo. Representación diagramamática de un hemocítómetro que resalta de qué cuadrante contar células. Haga clic aquí para ver una versión más grande de esta figura.
- Bajo el microscopio, cuente las células vivas (sin mancha) y muertas (azul manchado) que se encuentran en los cuatro grandes cuadrados de esquina(Figura 1). Excluya las celdas que se encuentren para superponerse o sentarse en el interior dos bordes de los cuadrados de esquina grandes (es decir, en las líneas) en el recuento.
- Mediante el siguiente cálculo, calcule el número medio de células vivas y viables (sin manchar) presentes en la muestra:
Número total de celdas/ml = recuento de células vivas x x 10.000
x 10.000
donde la dilución se refiere a cuántas veces se diluyó la solución de stock en azul trypan (2x en este caso) y # de cuadrados contados se refiere a los cuatro grandes cuadrados de esquina del hemocítómetro contados - Basado en el recuento de celdas HepG2 viable y utilizando la siguiente fórmula:
C1V1=C2V2
donde C1 = la concentración de células viables actualmente,
V1 = el volumen de suspensión celular actualmente,
C2 = se busca la concentración de suspensión celular,
V2 = el volumen de suspensión celular deseado - Prepare una solución de 10 ml de suspensión celular HepG2 con medio de cultivo celular DMEM a una concentración de 2,0 x 105 celdas/ml para lograr 4000 células HepG2 por caída colgante de 20 μL. Mezcle bien la suspensión celular canalizando suavemente hacia arriba y hacia abajo usando una pipeta de 1000 μL para asegurarse de que todas las células estén completamente suspendidas dentro del medio.
- A los pozos de una placa de cultivo celular de 96 pozos, agregue 100 μL de PBS estériles y a temperatura ambiente para evitar que las gotas colgantes se sequen durante la incubación.
- Tome la tapa de una placa estándar de cultivo celular de fondo plano de 96 pozos, invierta y enciba cuidadosamente las gotas de pipeta de 20 μL de la suspensión celular en el centro de cada ranura del pozo de la tapa, como se muestra en la Figura 2. Utilice una pipeta multicanal, pero agregue sólo 2 - 4 gotas a la vez, ya que la siembra múltiple puede afectar la precisión y colocación de las gotas.
- Centrar las gotas dentro de las ranuras de los pozos dispuestos en la tapa; de lo contrario, no colgarán en el centro de los pozos cuando la tapa de la placa se voltee y están en riesgo de caerse en el plato. Voltee suavemente la tapa de la placa de 96 pozos, por lo que las gotas ahora cuelgan y colocan cuidadosamente en la parte superior de la placa de 96 pozos.
- Coloque toda la placa de 96 pozos con tapa suavemente en una incubadora a 37 °C y 5% DE CO2 durante 3 días antes de la transferencia de esferoides sobre la agarose.
NOTA: Se debe tener especial cuidado no sólo al transportar las placas hacia/desde las incubadoras, sino al abrir y cerrar la incubadora en general, ya que el movimiento excesivo puede hacer que las placas se desplacen y los esferoides caigan o se formen incorrectamente.
 x 10.000
x 10.000
Figura 2: Preparación del modelo esferoide in vitro hepG2 3D2. (A) Las células HepG2 sembradas en 20 μL caen sobre la tapa de una placa de 96 pozos. (B) Las células HepG2 post-siembra en el modelo colgante de caída para permitir la formación de esferoides. Haga clic aquí para ver una versión más grande de esta figura.
3. Transferencia de esferoides HepG2 de caída colgante a suspensión de agarose
NOTA: El día 3 después de la siembra en gotas colgantes, los esferoides se transfieren a los pozos de la misma placa de 96 pozos, todos los cuales han sido previamente recubiertos con una fina capa de gel de agarose del 1,5%.
- Preparar geles de agarose y autoclave (es decir, día 2 después de la siembra) antes del día de recubrimiento de la placa (es decir, día 3 después de la siembra).
- Para preparar un gel de agarose del 1,5%, pesar 0,30 g de agarose en una botella de vidrio limpia y luego añadir 20 ml de medio DMEM libre de fenol rojo. Autoclave la agarose para 1 h a 230 °C para esterilización. El recubrimiento de agarose evita que los esferoides HepG2 se adhieran a la base de los pozos y formen una monocapa celular en lugar de retener su estructura esferoide 3D.
- El día 3 post siembra, retire la placa de 96 pozos que contiene los esferoides colgantes hepG2 gota fuera de la incubadora y voltear cuidadosamente la tapa para que los esferoides ya no cuelgan.
- Utilizando una pipeta multicanal, retire y deseche los 100 μL de PBS previamente añadidos a la base de la placa de 96 pozos. Deje que las placas se transmitan durante 2-3 minutos mientras calienta la agarose en preparación para el recubrimiento.
PRECAUCIÓN: Este procedimiento resulta en una agarose líquida muy caliente que si se derrama sobre la piel puede arder y causar lesiones. Además, se debe tener cuidado al manipular la botella de vidrio que contiene la agarose líquida, ya que esto también puede ser muy caliente. - Usando los geles de agarose del 1,5% preparados previamente, calienta la botella de vidrio que contiene el gel de agarose de 20 ml durante 30 s en un microondas al vatio máximo (es decir, 900 W). Para cubrir dos placas de 96 pozos, una botella de 20 ml de gel de agarose pre-preparado del 1,5% debe ser suficiente.
- Una vez derretido, arremolina suavemente la agarose girando la botella de vidrio para eliminar cualquier burbuja y luego añadir 50 μL de agarose en la base de cada pozo.
NOTA: Al agregar la agarose, asegúrese de no inclinar la placa >45° como la agarose se establece rápidamente y no formará una capa plana y nivelada que pueda interrumpir el crecimiento esferoide. Es importante trabajar eficientemente en esta etapa para evitar que la agarose se solidifique antes de que la placa esté completamente recubierta. - Deje que la placa se mantenga durante 2 minutos a temperatura ambiente antes de agregar 100 μL de medio de cultivo celular DMEM pre-calentado (con suplementos) en la parte superior de la capa de agarose sólida en cada pozo.
- Voltea la tapa de la placa de 96 pozos y colócalo de nuevo encima de la placa de 96 pozos para que los esferoides estén colgando una vez más.
- Centrífuga la placa durante 3 minutos a 200 x g con el fin de transferir los esferoides de la gota colgante en los pozos individuales de la placa de 96 pozos. Después de la transferencia, los esferoides HepG2 ahora deben suspenderse en el medio de cultivo celular. Permítales conformarse con 24 h en la incubadora a 37 °C y 5% CO2.
- Exponer esferoides HepG2 de este tamaño a tratamientos químicos o ENM en el día 4 después de la siembra (es decir, 24 h después de la transferencia a placas recubiertas de agarose).
- Para mantener la viabilidad de la célula durante períodos de cultivo extendidos, actualice el medio de cultivo celular cada 3 días. Para ello, aspirar suavemente 50 μL del medio de cultivo celular de la superficie del pozo y reemplazarlo con un nuevo medio de cultivo celular DMEM de 50 μL. No se cuide de eliminar o molestar al esferoide al realizar un cambio medio.
4. Exposición a nanomateriales/químicos
NOTA: El modelo de esferoide hepático HepG2 puede soportar regímenes de exposición basados en ENM y químicos, pero el enfoque principal de este protocolo son las exposiciones ENM. Antes de la exposición, la ENM de ensayo debe dispersarse adecuadamente; esto puede realizarse según lo indicado por el Protocolo de dispersión NanoGenoTox (Acuerdo de concesión Nº 20092101, 2018)15.
- Después de la dispersión según el Protocolo de dispersión NanoGenoTox, diluir la suspensión ENM de la concentración inicial de 2,56 mg/ml a la concentración final deseada en el medio de cultivo celular DMEM precalentado (incluidos los suplementos). Se requiere un volumen total de 5 ml para dosificar una placa de pozo de 96.
- Para exponer el esferoide HepG2 a un producto químico o ENM, utilizando una pipeta de 200 μL, aspirar 50 μL de medio de cultivo celular de la superficie de cada pozo (dejando 50 μL en el pozo para no perturbar los esferoides) y reemplazar con 50 μL de medio que contiene el tóxico de prueba a la dosis requerida.
- Una vez aplicado el material de ensayo, incubar las placas durante el tiempo de exposición deseado a 37 °C y 5% CO2.
- Si se lleva a cabo un régimen de exposición a largo plazo (≥24 h), inmediatamente después de que haya transcurrido el plazo de exposición deseado, coseche los esferoides para el análisis del punto final de micronucleus como se describe a continuación en los pasos 6.1 – 6.4.
- Sin embargo, con regímenes de exposición aguda (por ejemplo, ≤24 h), una vez finalizado el período de exposición, cosecha, piscina y almacenamiento de 50 μL de sobrenadante de cada pozo en la placa de pozo 96 a -80 °C para un análisis bioquímico posterior. Reemplace el medio de cultivo celular por 50 μL de medio fresco que contenga 6 μg/ml de citochalasina B y deje incubar durante 1 – 1,5 ciclos celulares (es decir, 24 - 26 h para HepG2) en preparación para la cosecha de ensayos micronucleus de bloque de citoquinas.
NOTA: Para los regímenes de exposición aguda (≤24 h), se puede aplicar el ensayo de micronucleus de bloque de citoquinesis con citochalasina B, pero para regímenes de exposición a largo plazo (≥24 h), la versión mononuclear (sin citochalasina B) del ensayo debe utilizarse como se describe a continuación en la Figura 4.
5. Cosecha de esferoides HepG2
NOTA: Después de los tratamientos químicos o de exposición a LAM, se puede cosechar tejido medio o esferoide de cultivo celular para el análisis de múltiples endpoints. Dependiendo del análisis de punto final, los esferoides se pueden cosechar individualmente (por ejemplo, para el análisis de imágenes) o agrupados (por ejemplo, para el ensayo de micronucleus de bloques de citoquinesis).
- Retire la placa de 96 pozos de la incubadora.
- Usando una pipeta de 200 μL, aspirar los 100 μL del medio de cultivo celular, incluido el tejido esferoide de cada pozo y recogerlo en un tubo estéril de centrífuga de 15 ml. Cuídate para evitar el contacto con la agarose.
- Una vez recogido, centrífuga la suspensión esferoide a 230 x g durante 5 minutos. Retire el sobrenadante y guárdelo a -80 °C para un análisis posterior de la variable (por ejemplo, pruebas de función hepática).
- Vuelva a suspender el pellet de esferoides en 1 ml de PBS estéril y a temperatura ambiente (1x).
- Una vez lavado, centrífuga la suspensión esferoide de nuevo a 230 x g durante 3 minutos. Deseche el sobrenadante, vuelva a suspender en 500 μL de solución 0.05% trypsin-EDTA e incubar durante 6-8 min a 37 °C y 5% CO2.
- Después de la incubación, canalizar suavemente las células trippsinizadas hacia arriba y hacia abajo para desasociar completamente y volver a suspender las células HepG2 antes de neutralizar con 1 ml de medio de cultivo celular DMEM.
- Centrífuga la suspensión celular diluida a 230 x g durante 5 min. Deseche el sobrenadante en desinfectante y vuelva a suspender el pellet celular en 2 ml de PBS a temperatura ambiente (1x).
- Centrífuga la suspensión de la celda a 230 x g durante 5 minutos. Deseche el sobrenadante en desinfectante y luego vuelva a suspender el pellet celular una vez más en 2 ml de PBS frío (1x). Asegúrese de que las células estén bien dispersas para evitar que grupos de células oscurezcan el campo de visión cuando se montan en diapositivas de microscopio.
6. Ensayo y puntuación de micronucleus
Para el método manual del ensayo de micronucleus, se requiere una citocentrífuga para producir un citodot (una región definida y concentrada de las células) en el centro de la diapositiva del microscopio. Este proceso admite una puntuación más eficiente de la diapositiva, ya que permite al anotador localizar fácilmente las celdas de interés, en lugar de evaluar toda una diapositiva donde las celdas se pueden propagar ampliamente.
- Sumerja las diapositivas de microscopio esmerilado (tres por dosis) en un 70% de etanol seguido de ddH2O y deje secar al aire durante 5 minutos.
- Coloque las diapositivas de microscopio preparadas en el embudo de cubeta como se muestra en la Figura 3A,donde la diapositiva de vidrio (iii) se coloca en el soporte metálico (iv) con una tarjeta de filtro (ii) y embudo de cubeta (i) asegurado en la parte superior.
- Organice embudos de cubeta en la citocentrífuga con el embudo hacia arriba, para que se puedan añadir directamente 100 μL de suspensión celular en cada uno de ellos.
- Citospin durante 5 min a 500 x g para asegurar que las células se distribuyan uniformemente en la superficie de la diapositiva.

Figura 3: Configuración de citospin para preparar células tratadas en diapositivas de microscopio. (A) Muestra los componentes individuales, (i) embudo de cubeta, (ii) tarjeta de filtro, (iii) diapositiva de microscopio de vidrio y (iv) soporte de metal necesario para citospin células HepG2 en diapositivas de microscopio. (B) El embudo de cubeta final configurado. (C) La correcta colocación del embudo de cubeta dentro de la citocentrífuga. Haga clic aquí para ver una versión más grande de esta figura.
- Deje las diapositivas secas al aire antes de la fijación en frío helado, 90% metanol durante 10 minutos.
- Una vez fijos, deje las diapositivas para secar al aire durante la noche a temperatura ambiente antes de almacenarlas a -20 °C durante un período de hasta 6 meses.
- Cuando sea necesario, retire las diapositivas de microscopio pre-preparadas del congelador -20 °C y deje que se caliente a temperatura ambiente antes de realizar la tinción de Giemsa.
PRECAUCIÓN: De conformidad con el Reglamento (CE) Nº 1272/2008 [CLP], la solución de tinción de Giemsa es un líquido altamente inflamable que puede ser tóxico si se ingiere y causa daños en el contacto con los ojos, la piel o si se inhala. Consulte la hoja de SDS asociada para obtener información detallada sobre almacenamiento, manipulación y salud y seguridad sobre este producto químico antes de su uso. - Mientras las diapositivas se descongelan, prepare una solución de tinción Giemsa del 20% (25 ml necesarios para manchar ~30 diapositivas) diluida en tampón de fosfatasa (pH 6.8). Mezcle bien girando suavemente la solución antes de filtrar con papel de filtro plegado colocado en un embudo.
- Con una pipeta Pasteur, añadir de 3 a 5 gotas de solución giemsa filtrada al citodot en cada tobogán y dejar durante 8 – 10 minutos.
- El lavado se desliza en dos lavados sucesivos de tampón de fosfatasa antes de enjuagar brevemente bajo agua fría para eliminar cualquier exceso de mancha sobrante. Deje repos deslizantes al aire seco.
- Una vez seco, en una campana de humo, sumerja los toboganes manchados en xileno durante 10 s antes de añadir una gota de medio de montaje al centro del citodot y colocar un cubrebotas de vidrio en la parte superior.
- Deje que las diapositivas del microscopio en la campana del humo durante la noche se sequen antes de la puntuación manual; se pueden almacenar indefinidamente a temperatura ambiente.
7. Análisis de datos
- Como se describe en las Directrices de examen 487 (2014)11de la OCDE , para evaluar y cuantificar el daño del ADN inducido como resultado de la exposición a un ENM o agente químico, utilizar un microscopio ligero (objetivo de 100x con aceite de inmersión) 2000 células mononucleadas o 1000 binucleadas por réplica biológica para puntuar para la presencia de micronuclei, como se muestra en la Figura 4.

Figura 4: Micronucleus ensayo de puntuación árbol de decisión. Árbol de decisión esquemático para resaltar la necesidad de diferentes procedimientos de evaluación de puntuación y citotoxicidad al utilizar el ensayo de micronucleus con modelos 3D siguiendo regímenes de exposición agudos o a largo plazo. Las exposiciones agudas (≤24 h) permiten el uso del ensayo de micronucleus bloqueado por citoquinesis, mientras que las exposiciones a largo plazo (≥24 h) requieren la versión mononuclear del ensayo; ambos se describen en la Guía de Examen 487 de la OCDE. Haga clic aquí para ver una versión más grande de esta figura.
- Sobre la base de la proporción de micronuclei presentes por número de células mononucleadas o binucleadas puntuadas, calcular un porcentaje del valor de genotoxicidad.
- Con el fin de evaluar el daño de ADN observado no es como resultado de los desechos celulares causados por una alta proporción de células apoptóticas, tomar una medida de citotoxicidad junto. En este caso, dependiendo de la presencia de citochalasina B, utilice el cálculo CPBI o RVCC (como se describe en la Figura 4). La genotoxicidad sólo debe evaluarse en muestras en las que la citotoxicidad sea inferior al 55% ± 5% tal como se define en la Guía de examen 48711de la OCDE.
Representative Results
La idoneidad de este modelo esferoide hepático 3D basado en líneas celulares para el cultivo a largo plazo y la evaluación de riesgos genotóxicos se evaluó mediante la realización de caracterización basal para determinar la viabilidad y la funcionalidad hepática del modelo durante la duración de 14 días en el cultivo, así como su aplicabilidad para el ensayo de micronucleus.
Caracterización basal del modelo esferoide hepg2 hepático 3D
Antes de cualquier evaluación toxicológica in vitro, es importante comprobar que los esferoides 3D HepG2 se han formado correctamente antes de realizar la transferencia de agarose o tratamiento químico/ENM. Los esferoides hepG2 producidos utilizando el método de caída colgante suelen tardar de 2 a 3 días después de la siembra (4000 células/esferoides) en formar esferoides compactos en forma esférica con un diámetro promedio de 495,52 μm W x 482,69 μm H como se muestra en la Figura 5A-5C. Los esferoides hepG2 que se han formado correctamente y son aceptables para ser utilizados para la evaluación toxicológica in vitro deben tener una estructura compacta, en forma esférica con una superficie lisa y sin proyecciones visuales. La Figura 5 proporciona ejemplos de buena calidad (Figura 5D-F) y una mala calidad (Figura 5G-I) esferoides. Este último debe descartarse. Por lo general, el 90-95% de los esferoides formados por placa se formarán correctamente y serán viables para una mayor experimentación.

Figura 5: Imágenes de microscopía ligera que muestran la morfología natural de los esferoides HepG2 formados a través del método de caída colgante. (A-C) mostrar día 2 y (D-I) Día 4 Esferoides hepG2 después de la siembra. (D-F) son ejemplos de esferoides HepG2 de buena calidad, mientras que (G-I) muestra esferoides mal formados. Todas las imágenes fueron tomadas con un objetivo X20 usando un microscopio. La barra de escala representa 20 μm. Haga clic aquí para ver una versión más grande de esta figura.
Para confirmar aún más la viabilidad esferoide hepg2, se puede realizar un ensayo básico colorimétrico bromocresol verde albúmina (BCG) o ensayo Urea para evaluar su funcionalidad similar al hígado. La funcionalidad similar al hígado se evaluó de acuerdo con la viabilidad utilizando el ensayo trypan blue exclusion durante un período de cultivo de 14 días para determinar la longevidad del modelo de esferoides hepáticos y establecer si podría apoyar la evaluación de riesgos a largo plazo o repetida basada en ENM/química (Figura 6). La concentración de albúmina se mantuvo constante durante el período de cultivo. La producción de Urea muestra un aumento en la concentración de urea producida por esferoide durante una semana en el cultivo antes de llegar a una meseta para el día 7. Es importante tener en cuenta que los niveles de albúmina y urea producidos en los esferoides 3D HepG2 son sustancialmente más altos que los observados en la misma línea celular cultivada en un formato 2D. De hecho, los cultivos 2D de células HepG2, pico de albúmina y niveles de urea fueron 0,001 mg/ml y 0,010 ng/μL respectivamente. Además, en trabajos anteriores publicados por Shah et al. utilizando un sistema esferoide HepG2 casi idéntico, los autores destacan una notable mejora en la actividad metabólica (CYP1A1 y CYP1A2) en los sistemas de modelos in vitro HepG2 3D en comparación con las células HepG2 cultivadas en 2D5.

Figura 6: Datos de caracterización basal de 14 días para esferoides hepáticos HepG2. Después de la transferencia de caída colgante, (A) destaca la viabilidad del modelo esferoide HepG2 durante un período de 14 días, mientras que (B) y (C) resaltan la funcionalidad de albúmina y urea similar al hígado respectivamente. Datos medios ± SEM presentados, n = 4. Haga clic aquí para ver una versión más grande de esta figura.
Con el inevitable desarrollo de un núcleo necrótico, una limitación conocida de los cultivos esferoides hepáticos 3D, la viabilidad de este modelo basado en HepG2 tuvo que establecerse para demostrar que era capaz de sostener regímenes de exposición a largo plazo (5-10 días) manteniendo la capacidad proliferativa necesaria para soportar el ensayo micronucleus5. De hecho, este modelo de esferoide hepático 3D se ha demostrado que conserva >70% viabilidad durante 10 días en el cultivo. Basándose en esto y en conjunto con la funcionalidad hepática sostenida observada durante el período de cultivo ≥14 días, este modelo de esferoide hepático 3D puede así apoyar regímenes de exposición en ENM repetidos a largo plazo de hasta 10 días de duración (es decir, antes de que la viabilidad de los esferoides caiga por debajo del 70%). Como referencia, se aconseja que los niveles de albúmina para esferoides HepG2 sembrados a 4000 células/esferoides deben ser ≥20.0 ng/μL, mientras que la producción de urea debe ser ≥0.25 ng/μL antes de realizar una evaluación toxicológica in vitro con este modelo.
Evaluación de genotoxicidad de nanomateriales modificados
Para la evaluación de la genotoxicidad, el ensayo de micronucleus se utilizó para determinar la presencia de micronuclei después de exposiciones agudas (24 h) y a largo plazo (120 h) ENM. Aflatoxina B1 es un conocido carcinógeno hepático16,17 y es un control positivo recomendado para el ensayo de micronucleus. Los experimentos de optimización han demostrado que 0,1 μM de Alfatoxin B1 induce una respuesta genotóxica positiva significativa (≥2,0 veces mayor) en los esferoides hepológicos 3D HepG2 y, por lo tanto, se utiliza en cada ensayo de micronucleus realizado con este modelo. Para garantizar la validez de los resultados del ensayo de micronucleus utilizando el modelo esferoide HepG2, la frecuencia de micronucleus de fondo para las células HepG2 utilizadas en este modelo in vitro 3D debe estar dentro de un rango de 0.6% – 1.2%. Como resultado, Alfatoxin B1 debe inducir una respuesta genotóxica de al menos dos veces mayor que la observada con el control negativo; por lo tanto, 0,1 μM de Alfatoxina B1 debe inducir una frecuencia de micronuclei entre el 1,5% y el 3,0%. Utilizando estos parámetros de control, la genotoxicidad in vitro asociada a ENM se puede evaluar de forma fiable. Sobre la base de la Orientación 487 de la OCDE sobre ensayos, es importante tener en cuenta que al probar un ENM o un producto químico, las concentraciones seleccionadas no deben inducir más del 55 % ± 5% de citotoxicidad (indicado por una reducción de los valores de CPBI o RVCC en relación con el control negativo)11. La Figura 7 ilustra los datos generados cuando la Aflatoxina B1 y dos ENM (dióxido de titanio (TiO2)y astilla (Ag)) fueron evaluados después de exposiciones agudas y a largo plazo en los esferoides HepG2, y se analizó el potencial genotóxico posterior utilizando el ensayo de micronucleus. Ambas ENM evaluadas se probaron a una dosis no citotóxica y baja de 5,00 μg/ml sobre una exposición aguda (24 h) y un régimen de exposición a largo plazo (120 h). Se puede observar una tendencia similar de genotoxicidad tanto en tiO2 como en ag enms, por lo que la respuesta de genotoxicidad elevada que resultó después de la exposición de 24 h no fue evidente después de una exposición a largo plazo de 5 días. Esto fue a pesar de la genotoxicidad sostenida inducida por el control positivo de la Aflatoxina B1 en ambos momentos.

Figura 7: Evaluación de la genotoxicidad después de la exposición TiO2 y Ag ENM en esferoides hepG2. Evaluación de la genotoxicidad (frecuencia de micronucleus)utilizando la exposición aguda (24 horas) yb)a largo plazo (120 horas) a 5,00 μg/ml de TiO2 y Ag ENM. El control negativo es sólo un medio, mientras que el control positivo es de 0,1 μM de aflatoxina B1. Datos medios (n=2) presentados ± SD. Significado indicado en relación con el control negativo: * = p≤ 0,05. Haga clic aquí para ver una versión más grande de esta figura.
Discussion
Las aplicaciones para modelos hepáticos 3D varían considerablemente dependiendo de la variable bioquímica particular o la vía de resultado adversa que se dirige. Cada modelo tiene sus beneficios y limitaciones, desde la variación interdonor en los modelos primarios de hepatocitocito humano (PHH) hasta la reducción de la actividad del citocromo p450 en modelos basados en líneas celulares, pero todos son valiosos por derecho propio6,12,18,19. Al evaluar la genotoxicidad hay limitaciones en la compatibilidad de los modelos con los puntos finales aprobados por la reglamentación, como el ensayo de micronucleos in vitro, ya que se requiere proliferación activa. Esto es necesario, ya que la evaluación de la genotoxicidad requiere la cuantificación de los daños fijos del ADN para ser evaluados después de la división celular cuando hay oportunidad de reparación del ADN para corregir lesiones transitorias. Desafortunadamente, los esferoides a base de hepatocitos altamente diferenciados (es decir, HepaRG) o microtissues PHH, que se consideran que exhiben las características hepáticas más fisiológicamente relevantes forman modelos estáticos (no proliferativos)12,19,20. Como resultado, el modelo esferoide 3D HepG2 presentado aquí proporciona un modelo adecuado y alternativo capaz de soportar pruebas de genotoxicidad. Los esferoides basados en líneas celulares HepG2 tienen suficientes células divisorias activas en la superficie externa de los esferoides manteniendo características básicas similares al hígado, como la producción de albúmina y urea y alguna actividad cyp4505,12,19. Principalmente este modelo hepático in vitro se ha desarrollado para complementar el ensayo de micronucleus, ya que este es uno de los dos ensayos in vitro recomendados en la batería para pruebas de genotoxicidad8,10,11,21. Sin embargo, el modelo se puede aplicar fácilmente a las tecnologías de análisis de secuenciación de ADN y expresión génica (ARN), mientras que tiene el potencial de adaptarse y utilizarse aún más para otras variables de daño de ADN, como el ensayo del cometa. No obstante, es importante considerar el papel que desempeña la interferencia en el MEDE en algunos análisis de puntos finales. Por ejemplo, los análisis basados en citometría de flujo pueden no ser adecuados para la evaluación de la genotoxicidad en OCA específicamente debido a la interferencia de partículas22.
Un factor limitante de los modelos esferoides que se someten activamente a la división celular es su tamaño. La optimización de la densidad de siembra es crítica, ya que debe haber suficientes células que permitan que el modelo siga proliferando; pero no demasiado alto un número de células, lo que resulta en el esferoide se vuelve demasiado compacto, lo que conduce a un aumento del núcleo necrótico. Se cree que la causa de esta necrosis es la difusión restringida de oxígeno y nutrientes, ya que se cree que el límite de esta difusión es de aproximadamente 100 – 150 μm de tejido23,24. Sin embargo, esto depende del tipo de celda, el número de celda, las interacciones de andamios y las condiciones de cultivo25. Desde entonces, se ha demostrado que aproximadamente 700 μm de diámetro es el límite para evitar la aparición prematura de necrosis en el centro de esferoides C3A, la siembra de 4000 células HepG2 por esferoide asegura que el diámetro del modelo en el momento de la exposición sea ≤500 μm26. Además, Shah et al. establecieron que las células HepG2 sembradas por encima de 5000 células por esferoide presentaban una reducción del 25% en la viabilidad después de 7 días de cultivo, que podrían pertenecer al diámetro medio de 680 μm y una disponibilidad limitada de nutrientes en una caída colgante de 20 μL5. Para superar esto, el modelo ideado en el presente protocolo se somete a un paso crítico donde la gota colgante se transfiere a pozos recubiertos de agarose después de la formación inicial del esferoide. Esto garantiza un mayor volumen de medio de cultivo está presente para mantener el número cada vez mayor de células dentro de los esferoides. Como resultado, el modelo esferoide HepG2 sigue siendo más del 70% viable después de 10 días en cultivo y se puede utilizar para la evaluación de riesgos a largo plazo invitro.
Mientras que el modelo esferoide HepG2 puede soportar regímenes de exposición tanto agudos como a largo plazo, el medio de cultivo celular refrescante durante períodos de cultivo prolongado está restringido para este modelo, ya que no se recomienda la sustitución completa del medio debido a la pérdida potencial de los esferoides. Se presume que con las exposiciones en el ENM, la tendencia a las dispersiones homogéneas de ENM a aglomerar y sedimentos es alta. Sin embargo, es notable que la velocidad a la que un sedimento ENM puede variar dependiendo de los parámetros de partículas (por ejemplo, tamaño, forma y densidad) y se puede determinar teóricamente utilizando el modelo de sedimentación in vitro, difusión y dosimetría (ISDD), o sus derivados recientes, a menudo referidos cuando con respecto a la exposición ENM (suspensión) se acerca a27,28. Con esto es mente, se supone que si sólo el 50% del medio de cultivo celular se elimina cuidadosamente de la superficie del cultivo celular, la interrupción y posterior eliminación de la dosis de ENM debe ser en teoría mínima. Sin embargo, con el movimiento browniano en juego, esto puede no ser estrictamente el caso y se deben seguir trabajando en la deposición y sedimentación de cada ENM en particular que se va a probar para garantizar que la dosimetría correcta se conserve a lo largo de los regímenes de exposición a largo plazo27. Principalmente, se trata de una limitación potencial a tener en cuenta al realizar regímenes de dosificación repetidos, ya que esto podría ser crítico para la concentración final y acumulada. Por otra parte, las exposiciones basadas en productos químicos, si bien no están exentos de sus propias limitaciones a tener en cuenta, ofrecen un enfoque más simplista en el sentido de que las sustancias químicas tienden a permanecer en solución y, por lo tanto, una sustitución directa de la concentración química original, además de la concentración recién añadida, garantiza que cualquier producto químico perdido durante la actualización de los medios de comunicación se sustituye en consecuencia29. Las aplicaciones futuras incluirían la evaluación de la idoneidad del modelo de regímenes de exposición repetidos durante períodos de cultivo a largo plazo, ya que las estrategias de dosificación repetidas son cruciales para evaluar la capacidad de un sistema de órganos en particular para mejorar o superar los efectos adversos, si los hubiera, inducidos por la bioacumulación de una sustancia xenóbiótica.
En conclusión, este modelo hepático in vitro 3D tiene la capacidad de ser utilizado para evaluar una gama de escenarios de exposición realistas, proporcionando así un enfoque in vitro futuro para apoyar mejor tanto la evaluación de riesgos enemis como químicos de una manera rutinaria y de fácil acceso.
Disclosures
Los autores no tienen nada que revelar.
Acknowledgments
Los autores quieren reconocer que esta investigación ha recibido financiación del programa de investigación e innovación Horizonte 2020 de la Unión Europea para el proyecto PATROLS, en virtud del acuerdo de subvención Nº 760813
Materials
| Name | Company | Catalog Number | Comments |
| Aflotoxin B1 | Sigma Aldrich, UK | A6636-5MG | |
| Agarose | Sigma Aldrich, UK | A9539-50G | |
| Autoclave Tape | |||
| BCG Albumin Assay | Sigma Aldrich, UK | MAK124 | |
| Bovine Serum Albumin Powder | Sigma Aldrich, UK | A9418 | |
| Cell Freezing Aid | Thermo Fisher Scientific, UK | 5100-0001 - Mr Frosty | |
| Centrifuge | Eppendorf | 5810 R | |
| Cytochalasin B | Merck, UK | 250233 | |
| Cytology Metal Clips | |||
| Cytospin 4 Centrifuge | ThermoFisher Scientific, UK | CM00730202 | |
| DMEM with 4.5g/L D-Glucose, L-Glutamine | GIBCO, Paisley, UK | 41965-039 | |
| DMEM, phenol-red free with 4.5g/L D-Glucose, L-Glutamine with Hepes | GIBCO, Paisley, UK | 21063-029 | |
| DPX Mounting Medium | FisherScientific, UK | D/5330/05 | |
| Ethanol | FisherScientific, UK | 10048291 | |
| FBS | GIBCO, Paisley, UK | 10270-106 | |
| Filter Cards for Shandon Cytospin | FisherScientific, UK | 15995742 | |
| Frosted Glass Slides | ThermoFisher Scientific, UK | ||
| Giemsa's Stain Improved R66 Solution, Gurr | VWR Chemicals, UK | MFCD00081642 | |
| Glass Coverslips (24 x 60) | Deckglaser, VWR | ECN631-1575 | |
| Haemocytometer and Coverslip | |||
| Immersion Oil for Microscope | Zeiss, UK | 518F, ISO8034 | |
| Laminar Class II Tissue Culture Hood | Scanlaf Mars | ||
| Light Microscope | Zeiss, UK | Axiovert 40C | |
| Liquid Nitrogen | |||
| Methanol | FisherScientific, UK | 10284580 | |
| Microwave | |||
| Non-Filtered, Sterile 200µl and 1000µl Pipette tips | Greiner-Bio-One, UK | ||
| NuncMicroWell 96-Well Microplates | ThermoFisher Scientific, Denmark | 167008 | |
| P1000 and P200 micropipettes | |||
| P300 and P50 multi-channel pipettes | |||
| PBS pH 7.4 1X, MgCl2 and CaCl2 Free | GIBCO, Paisley, UK | 14190-094 | |
| Pen/Strep | GIBCO, Paisley, UK | 15140-122, Penicillin/Strepmyocin 100X or 10,000U/ml | |
| Phosphatase Buffer Tablets | GIBCO, Paisley, UK | 10582-013 | |
| Pipette Boy | |||
| Simport Scientific CytoSep Funnels for Shandon Cytospin 4 Centrifuges | FisherScientific, UK | 11690581 | |
| Sonifier SFX 550 240V CE 1/2" - Probe | Branson, USA | 101-063-971R | |
| T-25 and T-75 Tissue Culture Flask | Greiner-Bio-One, UK | T-25 (690175) and T-75 (660175) | |
| Trypan Blue Solution | Sigma Aldrich, UK | T8154-100mL | |
| Urea Assay Kit | Sigma Aldrich, UK | MAK006 | |
| Virkon Disinfectant | DuPont, UK | Rely+On Virkon | |
| Water Bath (37?C) | Grant JBNova 18 | ||
| Weighing Balance | |||
| Xylene | FisherScientific, UK | 10588070 | |
| 0.05% Trypsin-EDTA | GIBCO, Paisley, UK | 5300-054 | |
| 0.2mL and 1.0mL Eppendorf Tubes | Greiner-Bio-One, UK | ||
| 0.45µm Filter Unit | Millex HA, MF-Millipore, UK | SLHA033SS | |
| 1.0mL Syringe | BD Plastipak, FisherScientific, UK | 300185 | |
| 20mL LS Scintillation Glass Vials, 22-400 Foil Lined PP Caps | DWK Life Sciences GmbH, Germany | WHEA986581 | |
| 37?C and 5% CO2 ISO Class 5 Hepa Filter Incubator | NUAIRE DHD Autoflow | ||
| 3mL Pasteur Pipette | Greiner-Bio-One, UK | ||
| 50mL Conical Falcon Tubes | Greiner-Bio-One, UK | ||
| 50mL or 100mL Glass Bottles | |||
| 50mL Skirted Falcon Tubes | Greiner-Bio-One, UK | ||
| 5mL, 10mL and 25mL Pipettes | Greiner-Bio-One, UK | ||
| 9.4cm Square, Petri Dish | Greiner-Bio-One, UK | 688161 |
References
- Geiser, M., Kreyling, W. G. Deposition and biokinetics of inhaled nanoparticles. Particle and Fibre Toxicology. 7, 2 (2010).
- Modrzynska, J. Toxicological effects of nanoparticle deposition in the liver. Kgs. Lyngby, Denmark: Technical University of Denmark. , (2018).
- Elje, E., et al. The comet assay applied to HepG2 liver spheroids. Mutation Research/Genetic Toxicology and Environmental Mutagenesis. 845, 403033 (2019).
- Breslin, S., O'Driscoll, L. Three-dimensional cell culture: the missing link in drug discovery. Drug Discovery Today. 18, 240-249 (2013).
- Shah, U. -K., et al. A three-dimensional in vitro HepG2 cells liver spheroid model for genotoxicity studies. Mutation Research/Genetic Toxicology and Environmental Mutagenesis. 825, 51-58 (2018).
- Lauschke, V. M., Hendriks, D. F. G., Bell, C. C., Andersson, T. B., Ingelman-Sundberg, M. Novel 3D Culture Systems for Studies of Human Liver Function and Assessments of the Hepatotoxicity of Drugs and Drug Candidates. Chemical Research in Toxicology. 29, 1936-1955 (2016).
- van Grunsven, L. A. 3D in vitro models of liver fibrosis. Advanced Drug Delivery Reviews. 121, 133-146 (2017).
- Corvi, R., Madia, F. In vitro genotoxicity testing - can the performance be enhanced. Food and Chemical Toxicology. 106, 600-608 (2017).
- Doak, S. H., Manshian, B., Jenkins, G. J. S., Singh, N. In vitro genotoxicity testing strategy for nanomaterials and the adaptation of current OECD guidelines. Mutation Research/Genetic Toxicology and Environmental Mutagenesis. 745, 104-111 (2012).
- Fenech, M. Cytokinesis-block micronucleus cytome assay. Nature Protocols. 2, 1084-1104 (2007).
- OECD. OECD Guidelines. Test 489: In vivo Mammalian Alkaline Comet Assay. , (2016).
- Gerets, H. H. J., et al. Characterization of primary human hepatocytes, HepG2 cells, and HepaRG cells at the mRNA level and CYP activity in response to inducers and their predictivity for the detection of human hepatotoxins. Cell Biology and Toxicology. 28, 69-87 (2012).
- Sison-Young, R. L., et al. A multicenter assessment of single-cell models aligned to standard measures of cell health for prediction of acute hepatotoxicity. Archives of Toxicology. 91, 1385-1400 (2017).
- European Guidelines 2019. European Agency for Safety and Health at Work. , Available from: https://osha.europa.eu/en/safety-and-health-legislation/european-guidelines (2019).
- Jensen, K. A. The NANOGENOTOX Dispersion Protocol for NANoREG. European Union Grant Agreement n° 2009. 21, 01 (2014).
- Marchese, S., et al. Aflatoxin B1 and M1: Biological Properties and Their Involvement in Cancer Development. Toxins. 10, 214 (2018).
- Rushing, B. R., Selim, M. I. Aflatoxin B1: A review on metabolism, toxicity, occurrence in food, occupational exposure, and detoxification methods. Food and Chemical Toxicology. 124, 81-100 (2019).
- Kermanizadeh, A., Brown, D. M., Moritz, W., Stone, V. The importance of inter-individual Kupffer cell variability in the governance of hepatic toxicity in a 3D primary human liver microtissue model. Scientific Reports. 9, 7295 (2019).
- Berger, B., et al. Comparison of Liver Cell Models Using the Basel Phenotyping Cocktail. Frontiers in Pharmacology. 7, 443 (2016).
- Ramaiahgari, S. C., et al. A 3D in vitro model of differentiated HepG2 cell spheroids with improved liver-like properties for repeated dose high-throughput toxicity studies. Archives of Toxicology. , (2014).
- Li, Y., et al. Factors affecting the in vitro micronucleus assay for evaluation of nanomaterials. Mutagenesis. 32 (1), 151-159 (2016).
- Kirkland, D., Reeve, L., Gatehouse, D., Vanparys, P. A core in vitro genotoxicity battery comprising the Ames test plus the in vitro micronucleus test is sufficient to detect rodent carcinogens and in vivo genotoxins. Mutation Research/Genetic Toxicology and Environmental Mutagenesis. 721 (1), 27-73 (2011).
- Curcio, E., et al. Mass transfer and metabolic reactions in hepatocyte spheroids cultured in rotating wall gas-permeable membrane system. Biomaterials. 28, 5487-5497 (2007).
- Glicklis, R., Merchuk, J. C., Cohen, S. Modeling mass transfer in hepatocyte spheroids via cell viability, spheroid size, and hepatocellular functions. Biotechnology and Bioengineering. 86, 672-680 (2004).
- Asthana, A., Kisaalita, W. S. Microtissue size and hypoxia in HTS with 3D cultures. Drug Discovery Today. 17, 810-817 (2012).
- Gaskell, H., et al. Characterization of a functional C3A liver spheroid model. Toxicology Research. 5, 1053-1065 (2016).
- Cho, E. C., Zhang, Q., Xia, Y. The effect of sedimentation and diffusion on cellular uptake of gold nanoparticles. Nature Nanotechnology. 6, 385-391 (2011).
- Hinderliter, P. M., et al. ISDD: A computational model of particle sedimentation, diffusion and target cell dosimetry for in vitro toxicity studies. Particle and Fiber Toxicology. 7, 36 (2010).
- Kramer, N. I., di Consiglio, E., Blaauboer, B. J., Testai, E. Biokinetics in repeated-dosing in vitro drug toxicity studies. Toxicology in vitro. 30, 217-224 (2015).
Tags
Bioingeniería Número 160 Modelos hepáticos in vitro Nanomateriales Evaluación de Riesgos Exposición a Largo Plazo Nano(geno)toxicología Daño en el ADNErratum
Formal Correction: Erratum: Advanced 3D Liver Models for In vitro Genotoxicity Testing Following Long-Term Nanomaterial Exposure
Posted by JoVE Editors on 01/26/2021.
Citeable Link.
An erratum was issued for: Advanced 3D Liver Models for In vitro Genotoxicity Testing Following Long-Term Nanomaterial Exposure. The Representative Results section was updated.
Figure 6 in the Representative Results section was updated from:
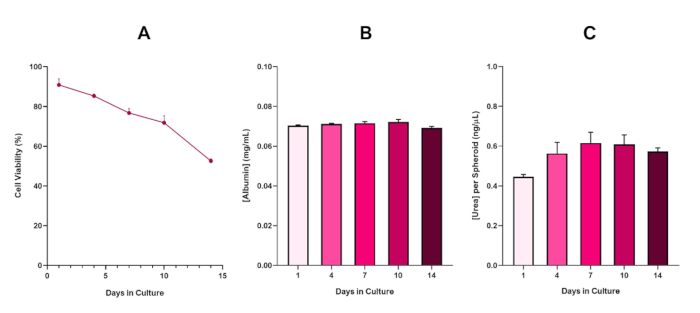
to:
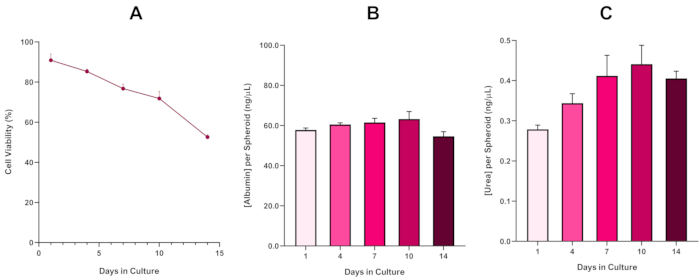
The fourth paragraph in the Representative Results section was updated from:
With the inevitable development of a necrotic core, a known limitation of 3D liver spheroid cultures, the viability of this HepG2 based model had to be established to demonstrate it was able to sustain long-term (5-10 day) exposure regimes whilst maintaining the proliferative capability required to support the micronucleus assay5. Indeed, this 3D liver spheroid model has been shown to retain >70% viability over 10 days in culture. Based on this and in conjunction with the sustained liver-like functionality observed over the ≥14 day culture period, this 3D liver spheroid model can thus support long-term, repeated ENM exposure regimes up to 10 days long (i.e., before viability of the spheroids drop below 70%). For reference, it is advised that albumin levels for HepG2 spheroids seeded at 4000 cells/spheroid should be ≥0.06 mg/mL whilst urea production should be ≥0.4 ng/µL before conducting an in vitro toxicological assessment with this model.
to:
With the inevitable development of a necrotic core, a known limitation of 3D liver spheroid cultures, the viability of this HepG2 based model had to be established to demonstrate it was able to sustain long-term (5-10 day) exposure regimes whilst maintaining the proliferative capability required to support the micronucleus assay5. Indeed, this 3D liver spheroid model has been shown to retain >70% viability over 10 days in culture. Based on this and in conjunction with the sustained liver-like functionality observed over the ≥14 day culture period, this 3D liver spheroid model can thus support long-term, repeated ENM exposure regimes up to 10 days long (i.e., before viability of the spheroids drop below 70%). For reference, it is advised that albumin levels for HepG2 spheroids seeded at 4000 cells/spheroid should be ≥50.0 ng/μL whilst urea production should be ≥0.25 ng/µL before conducting an in vitro toxicological assessment with this model.
