

ERRATUM NOTICE
Important: There has been an erratum issued for this article. Read more …
Summary
Detta förfarande fastställdes för att användas för att utveckla avancerade 3D lever kulturer in vitro, som kan ge en mer fysiologiskt relevant bedömning av de genotoxiska faror som är associerade med nanomaterial exponeringar över både en akut eller långsiktig, upprepad dos regimer.
Abstract
På grund av den snabba utvecklingen och implementeringen av ett brett spektrum av konstruerade nanomaterial (ENM) är exponering för ENM oundviklig och utvecklingen av robusta, prediktiva in vitro-testsystem är avgörande. Levertoxikologi är nyckeln vid bedömning av ENM-exponering, eftersom levern tjänar en viktig roll i metabolisk homeostas och avgiftning samt är en viktig plats för ENM-ackumulering efter exponering. Baserat på detta och den accepterade förståelsen att 2D hepatocyt modeller inte exakt efterlikna komplexiteten i invecklade multi-cellular interaktioner och metabolisk aktivitet observeras in vivo, det finns ett större fokus på utvecklingen av fysiologiskt relevanta 3D levermodeller skräddarsydda för ENM farobedömning ändamål in vitro. I enlighet med principerna i 3R för att ersätta, minska och förfina djurförsök har en 3D HepG2 cellinjebaserad levermodell utvecklats, vilket är ett användarvänligt, kostnadseffektivt system som kan stödja både utökade och upprepade ENM-exponeringssystem (≤14 dagar). Dessa sfäroidmodeller (≥500 μm i diameter) behåller sin proliferativa kapacitet (dvs. delningscellmodeller) så att de kan kopplas ihop med mikrokärnanalysen "guldstandard" för att effektivt bedöma genotoxitoxiciteten in vitro. Deras förmåga att rapportera om en rad toxikologiska effektmått (t.ex. leverfunktion, (pro-)inflammatoriskt svar, cytotoxicitet och genotoxicity) har kännetecknats av flera ENMs över både akuta (24 h) och långsiktiga (120 h) exponeringssystem. Denna 3D-in vitro-levermodell har kapacitet att användas för att utvärdera mer realistiska ENM-exponeringar, vilket ger en framtida in vitro-strategi för att bättre stödja ENM-farobedömning på ett rutinmässigt och lättillgängligt sätt.
Introduction
På grund av den snabba utvecklingen och implementeringen av ett brett spektrum av konstruerade nanomaterial (ENM) i en mängd mänskliga tillämpningar (t.ex. livsmedel, kosmetika, kläder, sportutrustning, elektronik, transport och medicin) är det oundvikligt att människor regelbundet kommer att utsättas för ENM. Med detta finns det en ökad oro för att de nya, storleksspecifika fysiokemiska egenskaperna som anser att dessa material är fördelaktiga i många tillämpningar kan orsaka negativa effekter på människors hälsa och miljön samtidigt. För närvarande finns det många internationella aktiviteter för att aktivt återspegla mer fysiologiskt relevanta exponeringar för dessa ENM och bedöma dessa materials potentiella toxicitet över akuta, långsiktiga och upprepade lågdosexponeringsscenarier.
Levertoxikologi är nyckeln när man överväger ENM-exponering, eftersom det är allmänt känt att levern är en viktig plats för ENM-ackumulering efterexponering 1,2. Dessutom är levern det primära organsystemet för metabolism och avgiftning av ämnen som kommer in i systemisk cirkulation3. Baserat på den accepterade förståelsen att 2D hepatocyt modeller inte korrekt efterliknar komplexiteten i invecklade multicellulära interaktioner eller på lämpligt sätt representerar metabolisk aktivitet observeras in vivo, har ett större fokus på att utveckla robusta och fysiologiskt relevanta in vitro 3D levermodeller för in vivo ersättningsteknik fastställts4,5. Genom att använda avancerad 3D-kulturteknik förbättras livslängden för in vitro-levermodeller som möjliggör långsiktiga, upprepade exponeringssystem som ska undersökas. Dessutom främjar detta avancerade kulturformat bildandet av förbättrade fysiologiska, organotypiska egenskaper som gallkanaliculi, aktiva transportprocesser och förbättrad CYP450 läkemedelsmetaboliserande kapacitet, vilket förbättrar förutsägbarheten hos modellerna6. Nuvarande 3D in vitro levermodeller bestående av monokulturer (endast hepatocyter) eller samkulturer (hepatocyter med nonparenchymala celler) finns i flera format, allt från mikrotissues eller sfäroider i ultralåga vidhäftningsplattor, hängande droppe sfäroider, celler inbäddade i matriser och/eller byggnadsställningar och mikrofluidiska cellodlingsplattformar, som alla anses vara effektiva avancerade in vitro-modeller för bedömning avlevertoxicitet 6,7. Majoriteten av dessa modellsystem är dock högt underhåll, kräver specialiserad utrustning och är dyra. Dessutom är dessa modeller ofta statiska (dvs. icke-dela upp cellmodeller) som förhindrar deras användning vid bedömning av faroslutpunkter, såsom genotoxiitetstestning med hjälp av metoder som kvantifierar fasta DNA-skador. Genotoxicity är en grundläggande förutsättning inom regulatorisk toxikologi, och det är en viktig komponent i riskbedömningen av alla toxiskamedel 8. Det finns ingen enda analys som kan tillämpas för att kvantifiera alla former av DNA-skador som kan uppstå efter exponering för ett exogent medel. En kärnkomponent i in vitro-genotoxicitytestbatteriet är dock mikrokärnaanalysen, som är en tillförlitlig och mångfacetterad teknik som mäter bruttokromosomskador9. Det är en guldstandardteknik som beskrivs i OECD:s testriktlinje 487, för bedömning av DNA-skador och genotoxitoxicitet in vitro och ingår i testbatterikravet för regulatorisk farobedömning10,11.
Den mänskliga hepatocellulära carcinom cellinjen, HepG2, används i stor utsträckning för inledande farobedömning screening eftersom cellerna är lättillgängliga, relativt billiga att källa, enkla att odla och mottagliga för hög genomströmning screening12,13. När de odlas i 3D-sfäriska strukturer har de visat sig rekapitulera levermikromiljön väl och erbjuda en levermodell med tillräcklig proliferativ förmåga att stödja mikrokärnans analys3. Vidareutveckling av HepG2-sfäroidmodellerna fastställdes för att förbättra modellens livslängd och leverliknande funktionalitet för att stödja bedömning av genotoxicity-faror över långsiktiga, upprepade exponeringssystem (≤14 dagar). I enlighet med principerna i de 3R som ska ersätta, minska och förfina djurförsök har detta protokoll upprättats för att tillhandahålla en avancerad 3D-in vitro-levermodell som på ett tillförlitligt sätt kan utvärdera flera toxikologiska effektmått (t.ex. leverfunktionalitet, (pro-)inflammatoriska markörer, cytotoxicitet och genotoxicity) efter akuta, långsiktiga och upprepade kemiska och ENM-exponeringar på ett rutinmässigt och lättillgängligt sätt.
Här presenterar vi en metod för att fastställa ett fysiologiskt relevant 3D hepatocyt cellinje baserat in vitro modellsystem för genotoxicity farobedömning efter akut eller långsiktiga, upprepade ENM exponeringar. Protokollet kan delas upp i 6 viktiga steg: odling av kryopreserverade HepG2-celler; HepG2 sfäroid förberedelse; HepG2 sfäroid överföring från hängande droppe till agarose suspension; HepG2 sfäroid skörd; Mikrokärnaanalys och poängsättning. och dataanalys.
Protocol
1.Odling av kryopreserverade HepG2-celler
OBS: HepG2 celler, erhållna från American Type Culture Collection (ATCC) odlades i 1x Dulbeccos Modified Eagle Medium (DMEM) med 4,5 g/L D-glukos och L-glutamin kompletterat med 10% fostervint serum (FBS) och 1% penicillin/streptomycin antibiotikum.
- Förvärmt DMEM-cellkulturmedium (inklusive kosttillskott) i ett 37 °C vattenbad i 30 min.
- Ta bort en flaska HepG2-celler från flytande kväve och tina i ett 37 °C vattenbad i 2-3 minuter, samtidigt som injektionsflaskan försiktigt virvlas bort för att möjliggöra enhetlig upptining av cellupphängningen. Var försiktig så att injektionsflaskan inte sänks ned ovanför O-ringen för att minska risken för kontaminering.
- När injektionsflaskan har tinats upp, ta bort injektionsflaskan från vattenbadet och spraya generöst med 70% etanol för att sanera injektionsflaskans yttre yta innan du placerar under en steril laminär vävnadskulturhuv av klass II.
- Pipett innehållet i kryovialen av HepG2-celler i ett centrifugrör som innehåller 9 ml förvärmt DMEM-cellkulturmedium (med kosttillskott).
- Använd en 10 ml strippette, överför 10 ml av cellfjädringen till en 25 cm2 engångscellkulturkolv och inkuberar kulturen i 3 dagar (från sådd) vid 5% CO2 och 37 °C tills ~80% konfluens uppnås innan den genomgår underkultur till en större 75 cm2 engångscellskulturkolv.
- När 80% confluency uppnås, subkulturceller under sterila förhållanden genom trypsinisering med 0,05% trypsin/ EDTA-lösning förvärmd i ett 37 °C vattenbad i 30 min. Cellerna får inte vid något tillfälle torka ut.
- När celler bildar ett vidhäftande monoskikt, ta bort mediet genom att tippa in i en desinfektionsavfallspott. Tvätta sedan omedelbart monoskiktet för att avlägsna alla spår av befintligt medium genom att skölja kolven två gånger med 3 ml steril 1x PBS-lösning som hålls vid rumstemperatur. Kassera också PBS i desinfektionsavfallsgrytan.
- När PBS-tvätten har tagits bort, tillsätt 5 ml förvärmd 0,05% trypsin-EDTA-lösning, se till att täcka hela cellytan och inkubera celler i 6-8 min vid 37 °C och 5% CO2.
- Knacka försiktigt på kolven för att lossa cellerna från kolvens botten och tillsätt sedan 5 ml DMEM-cellkulturmedium (med kosttillskott) för att neutralisera trypsinenzymet.
- Överför cellfjädringen till ett 50 ml centrifugeringsrör och pipett cellupphängningen upp och ner noggrant för att säkerställa att cellerna är helt disassocierade.
- Centrifugera den utspädda cellupphängningen vid 230 x g i 5 min. Kassera supernatanten i desinfektionsmedel och suspendera cellpellets i 25mL DMEM-cellkulturmedium (med kosttillskott).
- Överför cellupphängningen till en 75 cm2 engångscellskulturkolv och inkubera vid 37 °C och 5 % CO2 i ytterligare 3 dagar innan den genomgår sfäroidpreparat. När HepG2s har haft tid att acklimatisera sig och återigen nå ~ 80% confluency, bestämma cellkoncentrationen som förberedelse för sfäroid sådd.
2. HepG2 sfäroid förberedelse
- Upprepa de steg för underkulturen som anges ovan, utom efter centrifugation, suspendera cellpelleten igen i 1 ml DMEM-odlingsmedium som förvärmts i ett vattenbad på 37 °C. Pipettcellsupphängning upp och ner noggrant.
- Poängcellens livskraft med trypan blue exclusion assay (se OSHA SOP 3.21 Reproduktiva toxiner, mutagener, teratogener och embryotoxiner – Procedurer för säker hantering och lagring (2019) för hälso- och säkerhetsvägledning)14 med ett 1:1-förhållande mellan cellfjädring och förfiltrerad 0,4% trypanblå lösning.
- Innan cellräkningen, ta 1 ml trypanblå lösning med en 1 ml spruta och filtrera med en filterenhet på 0,45 μm i ett sterilt 1 ml-rör.
- Överför 10 μL filtrerad trypanblå lösning till ett 0,2 ml-rör och tillsätt 10 μL cellfjädring. Återstående filtrerad trypanblå lösning kan lagras upp till 3 månader vid rumstemperatur för framtida användning.
- Spraya haemocytometern noggrant med 70% etanol och torka torrt med en steril pappershandduk innan du säkrar täcket på toppen med andningsånga. Att skjuta täcket över den fuktade ytan inducerar sammanhållna krafter genom att generera Newton-ringar.
- Pipetten trypanblå cellens fjädring upp och ner med en 1000 μL-pipett (för att minska ren stress) innan du lägger till 10 μL till haemocytometern. Se till att lösningen är spridd under täckspingen och täcker hela gallret utan luftbubblor.

Figur 1: Räkna celler med hjälp av en haemocytometer. Diagrammatisk representation av en haemocytometer som belyser vilken kvadrant man ska räkna celler från. Klicka här om du vill visa en större version av den här figuren.
- Under mikroskopet, räkna de levande (osedda) och döda (färgade blå) cellerna som finns i de fyra stora hörnrutorna (figur 1). Exkludera celler som befunnits överlappa eller sitta på de inre två kanterna på de stora hörnrutorna (dvs. på linjerna) i räkningen.
- Beräkna det genomsnittliga antalet levande, livskraftiga celler (obeklivade) som finns i provet med hjälp av följande beräkning:
Totalt antal celler/ml = Antal levande celler x x 10 000
x 10 000
där utspädning avser hur många gånger stamlösningen späddes ut i trypanblått (2x i detta fall) och # antal räknade rutor avser de fyra stora hörnrutorna på den haemocytometer som räknas - Baserat på det livskraftiga antalet HepG2-celler och med hjälp av följande formel:
C1V1=C2V2
där C1 = koncentrationen av livskraftiga celler för närvarande.
V1 = volymen av cellfjädring för närvarande,
C2 = koncentrationen av cellsuspension önskas,
V2 = volymen av cellfjädring önskas - Förbered en 10 ml stamlösning av HepG2-cellupphängning med DMEM-cellkulturmedium vid en koncentration av 2,0 x 105 celler/ml för att uppnå 4000 HepG2-celler per 20 μL hängande droppe. Blanda cellfjädringen noggrant genom att försiktigt leda upp och ner med en 1000 μL-pipett för att säkerställa att alla celler är helt upphängda i mediet.
- Till brunnarna på en 96-brunns cellkulturplatta, tillsätt 100 μL steril, rumstemperatur PBS för att förhindra att de hängande dropparna torkar ut under inkubation.
- Ta locket på en vanlig platt botten 96-brunns cellkulturplatta, vänd den och försiktigt pipett 20 μL droppar cellfjädring i mitten av varje brunnsspår på locket, som visas i figur 2. Använd en flerkanalspipett men tillsätt endast 2 - 4 droppar samtidigt eftersom flera sådd kan påverka droppens noggrannhet och placering.
- Centrera dropparna i spåren på brunnarna som läggs ut på locket; Annars kommer de inte att hänga i mitten av brunnarna när locket på plattan vänds och riskerar att falla av i plattan. Vänd försiktigt locket på 96-brunnsplattan, så dropparna hänger nu och placera försiktigt ovanpå 96-brunnsplattan.
- Placera hela 96 brunnsplattan försiktigt med lock i en inkubator vid 37 °C och 5 % CO2 i 3 dagar före sfäroidöverföring till agarosa.
OBS: Extra försiktighet måste vidtas inte bara vid transport av plattorna till/från inkubatorerna, utan när inkubatorn öppnas och stängs i allmänhet eftersom överdriven rörelse kan leda till att plattorna skiftar och sfäroiderna antingen faller eller bildas felaktigt.
 x 10 000
x 10 000
Bild 2: 3D HepG2 in vitro-sfäroidmodellberedning. (A) HepG2-cellerna som sås i 20 μL droppar på locket på en 96-brunnsplatta. B)HepG2-cellerna efter sådd i hängande droppmodell för att möjliggöra sfäroidbildning. Klicka här om du vill visa en större version av den här figuren.
3. HepG2 sfäroid överföring från hängande droppe till agarose suspension
OBS: På dag 3 efter sådd i hängande droppar överförs sfäroiderna till brunnarna på samma 96-brunnsplatta som alla tidigare har belagts med ett fint lager av 1,5% agarose gel.
- Förbered agarosageler och autoklav (dvs. dag 2 efter sådd) före dagen för plåtbeläggning (dvs. dag 3 efter sådd).
- För att förbereda en 1,5% agarose gel, väg 0,30 g agarose i en ren glasflaska och tillsätt sedan 20 ml fenolrött DMEM-medium. Autoklav agarosen i 1 timme vid 230 °C för sterilisering. Agarose beläggningen förhindrar HepG2 sfäroider från att följa basen av brunnar och bilda en cellulär monolayer istället för att behålla sin 3D sfäroid struktur.
- På dag 3 efter sådd, ta bort 96-brunnsplattan som innehåller HepG2 hängande droppe sfäroider ur inkubatorn och vänd försiktigt locket så att sfäroiderna inte längre hänger.
- Använd en flerkanalspipett och ta bort de 100 μL PBS som tidigare lagts till basen på 96-brunnsplattan. Låt plattorna luftas i 2-3 minuter medan du värmer agarosen som förberedelse för beläggning.
VARNING: Detta förfarande resulterar i mycket varm, flytande agarose som, om den spills på huden, kan brinna och orsaka skada. Dessutom måste försiktighet vidtas vid hantering av glasflaskan som innehåller den flytande agarosen eftersom även denna kan vara mycket varm. - Använd de 1,5% agarose geler som tidigare förberetts, värm glasflaskan som innehåller 20 mL agarose gel för 30 s i en mikrovågsugn vid maximal watt (dvs. 900 W). För att täcka två 96-brunnsplattor bör en 20 ml flaska förberedda 1,5% agarose gel vara tillräcklig.
- När den har smält, snurra försiktigt agarosen genom att rotera glasflaskan för att ta bort eventuella bubblor och tillsätt sedan 50 μL agarose i basen av varje brunn.
OBS: När du lägger till agarose, se till att inte vinkla plattan >45° eftersom agarosen sätter snabbt och inte bildar ett platt, jämnt lager som kan störa sfäroidtillväxten. Det är viktigt att arbeta effektivt i detta skede för att förhindra att agarosen stelnar innan plattan är helt belagd. - Låt plattan stå i 2 minuter vid rumstemperatur innan du lägger till 100 μL förvärmt DMEM-cellkulturmedium (med kosttillskott) ovanpå det fasta agarosaskiktet i varje brunn.
- Vänd på locket på 96-brunnsplattan och placera tillbaka ovanpå 96-brunnsplattan så att sfäroiderna nu hänger igen.
- Centrifugera plattan i 3 min vid 200 x g för att överföra sfäroiderna från den hängande droppen till de enskilda brunnarna på 96-brunnsplattan. Efter överföringen bör HepG2-sfäroiderna nu suspenderas i cellkulturmediet. Låt dem nöja sig med 24 timmar i inkubatorn vid 37 °C och 5 % CO2.
- Exponera HepG2-sfäroider av denna storlek för antingen kemiska eller ENM-behandlingar på dag 4 efter sådd (dvs. 24 timmar efter överföring till agarosabelagda plattor).
- För att bibehålla cellens livskraft under längre odlingsperioder, uppdatera cellkulturmediet var tredje dag. För att göra detta, aspirera försiktigt 50 μL av cellkulturmediet från brunnens yta och ersätt med en frisk 50 μL DMEM-cellkulturmedium. Var försiktig så att du inte tar bort eller stör sfäroiden när du utför en medelstor förändring.
4. Nanomaterial/kemisk exponering
OBS: HepG2 lever sfäroid modell kan stödja både ENM och kemiskt baserade exponering regimer, men det primära fokuset för detta protokoll är ENM exponeringar. Före exponeringen skall testet ENM vara lämpligt spridda. Detta kan utföras enligt nanogenotoxspridningsprotokollet (bidragsavtal nr 20092101, 2018)15.
- Efter spridning enligt NanoGenoTox Dispersion Protocol, späd enm suspension från startkoncentrationen av 2,56 mg/mL till den slutliga önskade koncentrationen i förvärmda DMEM cell kultur medium (inklusive kosttillskott). En total volym på 5 ml krävs för att dosera en 96 brunnsplatta.
- För att utsätta HepG2-sfäroiden för antingen en kemikalie eller ENM, med hjälp av en 200 μL-pipett, aspirera 50 μL cellkulturmedium från ytan av varje brunn (lämna 50 μL i brunnen för att inte störa sfäroiderna) och ersätta med 50 μL medium som innehåller testtoxiska medel vid önskad dos.
- När testmaterialet har applicerats, inkubera plattorna under önskad exponeringstid vid 37 °C och 5 % CO2.
- Om ett långsiktigt (≥24 h) exponeringssystem utförs, skörda sfäroiderna för mikrokärnors endpointanalys omedelbart efter det att den önskade exponeringstiden har gått ut, enligt beskrivningen nedan i steg 6.1– 6.4.
- Med akuta exponeringssystem (t.ex. ≤24 h) skördar, poolar och lagrar 50 μL supernatant från varje brunn i 96 brunnsplattan vid -80 °C för ytterligare biokemisk analys senare. Ersätt cellkulturmediet med 50 μL färskt medium som innehåller 6 μg/ml Cytochalasin B och låt inkubera i 1 – 1,5 cellcykler (dvs. 24 – 26 h för HepG2) som förberedelse för cytokinesblockets mikronukleustestskörd.
OBS: För akuta (≤24 h) exponeringsregimer kan cytokinesblockets mikrokärnaanalys med Cytochalasin B tillämpas, men för långsiktiga (≥24 h) exponeringsregimer måste den mononukleära versionen (utan Cytochalasin B) av analysen användas enligt beskrivningen nedan i figur 4.
5. HepG2 sfäroid skörd
OBS: Efter antingen kemiska eller ENM-exponeringsbehandlingar kan både cellkulturell medium eller sfäroidvävnad skördas för analys av flera effektmått. Beroende på slutpunktsanalysen kan sfäroider antingen skördas individuellt (t.ex. för bildanalys) eller poolas tillsammans (t.ex. för cytokinesblock micronucleus-analys).
- Ta bort 96-brunnsplattan från inkubatorn.
- Använd en 200 μL-pipett, aspirera 100 μL cellkulturmedium inklusive sfäroidvävnaden från varje brunn och samla i ett sterilt 15 ml centrifugerör. Var noga med att undvika kontakt med agarosen.
- När centrifugera sfäroidupphängningen vid 230 x g i 5 min. Ta bort supernaten och förvara vid -80 °C för ytterligare endpointanalys (t.ex. leverfunktionstester) senare.
- Suspendera pelleten av sfäroider i 1 ml steril, rumstemperatur PBS (1x).
- När den har tvättats, centrifugera sfäroidupphängningen igen vid 230 x g i 3 min. Kassera supernatanten, suspendera igen i 500 μL 0,05% trypsin-EDTA-lösning och inkubera i 6-8 min vid 37 °C och 5% CO2.
- Efter inkubation, försiktigt pipettera de trypsiniserade cellerna upp och ner för att helt avassociera och suspendera HepG2-cellerna innan de neutraliseras med 1 ml DMEM-cellkulturmedium.
- Centrifugera den utspädda cellupphängningen vid 230 x g i 5 min. Kassera supernatanten i desinfektionsmedel och suspendera cellpellet i 2mL rumstemperatur PBS (1x).
- Centrifugera cellupphängningen vid 230 x g i 5 min. Kassera supernatmedlet i desinfektionsmedel och suspendera sedan cellpelleten igen i 2 ml kall PBS (1x). Se till att cellerna är väl spridda för att förhindra att klumpar av celler skymtar synfältet när de monteras på mikroskopbilder.
6. Mikrokärnaanalys och poängsättning
För den manuella metoden för mikrokärnaanalysen krävs en cytocentrifuge för att producera en cytodot (en definierad, koncentrerad cellregion) i mitten av mikroskopbilden. Denna process stöder effektivare poängsättning av bilden eftersom det gör det möjligt för målskytten att enkelt hitta de celler av intresse, i motsats till att utvärdera en hel bild där cellerna kan spridas i stor utsträckning.
- Doppa frostade mikroskopr (tre per dos) i 70% etanol följt av ddH2O och låt lufttorka i 5 min.
- Placera förberedda mikroskopglas i cuvettetratt enligt figur 3A, där glasglaset (iii) placeras i metallstödet (iv) med ett filterkort (ii) och cuvettetratt (i) fastsatt ovanpå.
- Ordna cuvettetrattar i cytocentrifugen med tratten uppåt, så att 100 μL cellfjädring kan tillsättas direkt i var och en.
- Cytospin i 5 min vid 500 x g för att säkerställa att cellerna fördelas jämnt på bildens yta.

Figur 3: Cytospininställning för att förbereda behandlade celler på mikroskopbilder. (A) Visar de enskilda komponenterna, (i) cuvette tratt, (ii) filterkort, iii) glasmikroskopsutschbana och (iv) metallstöd som krävs för att cytospina HepG2-celler på mikroskopbilder. B)Den sista cuvettetratten. C)Korrekt placering av cuvettetratten i cytocentrifugen. Klicka här om du vill visa en större version av den här figuren.
- Låt rutschkanorna lufttorka innan fixeringen i iskall, 90% metanol i 10 min.
- När bilderna är fasta, låt rutschkanorna lufttorka över natten vid rumstemperatur innan de förvaras vid -20 °C i upp till 6 månader.
- Vid behov, ta bort de förberedda mikroskoprutschbanorna från -20 °C frys och låt värma till rumstemperatur innan giemsafärgning.
VARNING: Enligt förordning (EG) nr 1272/2008 [CLP] är Giemsa färglösning en mycket brandfarlig vätska som kan vara giftig vid förtäring och orsaka skador vid kontakt med ögon, hud eller vid inandning. Se tillhörande SDS-blad för detaljerad lagring, hantering och hälso- och säkerhetsråd om denna kemikalie före användning. - Medan diabilderna tinar, förbered en 20% Giemsa färgningslösning (25 ml krävs för att färga ~ 30 bilder) utspädd i fosfatasbuffert (pH 6,8). Blanda noggrant genom att försiktigt virvla runt lösningen innan du filtrerar med vikt filterpapper placerat i en tratt.
- Använd en Pasteur-pipett, tillsätt 3 – 5 droppar filtrerad Giemsa-lösning till cytodoten på varje bild och låt den vara i 8 – 10 min.
- Tvätta diabilder i två på varandra följande fosfatasbuffert tvättar innan du kort sköljer under kallt vatten för att ta bort eventuella överflödiga fläckar kvar. Låt rutschkanorna lufttorka.
- När de är torra, i en rökhuv, doppa målade diabilder i xylen i 10 s innan du lägger till en droppe monteringsmedium i mitten av cytodoten och en plats ett glaslock ovanpå.
- Låt mikroskopet glida i rökhuven över natten för att torka innan manuell poängsättning; De kan förvaras på obestämd tid i rumstemperatur.
7. Dataanalys
- Enligt OECD:s testriktlinjer 487 (2014)11, för att bedöma och kvantifiera DNA-skador som framkallats till följd av exponering för ett enm- eller kemiskt agens, använd ett lätt mikroskop (100x mål med nedsänkningsolja) 2000 mononukleerade eller 1000 binukleära celler per biologisk replikat för att poäng för förekomst av mikrokärnor, enligt figur 4.

Figur 4: Micronucleus assay poäng bedömning beslut träd. Schematiskt beslutsträd för att belysa behovet av olika bedömningsförfaranden för poängsättning och cytotoxicitet vid användning av mikrokärnansanalysen med 3D-modeller efter akuta eller långsiktiga exponeringssystem. Akuta (≤24 h) exponeringar tillåter användning av cytokinesen blockerad mikrokärnaanalys, medan långsiktiga (≥24 h) exponeringar kräver den mononukleära versionen av analysen. båda beskrivs i OECD:s testriktlinje 487. Klicka här om du vill visa en större version av den här figuren.
- Beräkna en procentandel genotoxicityvärde baserat på andelen mikrokärnor per antal mononukleära eller binucleated celler.
- För att bedöma de observerade DNA-skadorna är inte ett resultat av cellskräp som orsakas av en hög andel apoptotiska celler, ta ett mått av cytotoxicitet vid sidan av. I detta fall, beroende på förekomsten av Cytochalasin B, använd antingen CPBI eller RVCC beräkning (enligt beskrivningen i figur 4). Genotoxicity får endast utvärderas i prover där cytotoxiciteten är mindre än 55 % ± 5 % enligt definitionen i OECD:s testriktlinje 48711.
Representative Results
Lämpligheten av denna cellinjebaserade 3D lever sfäroid modell för långsiktiga kultur och genotoxic farobedömning utvärderades genom att genomföra baslinjen karakterisering för att bestämma livskraft och lever-liknande funktionaliteten hos modellen under varaktigheten av 14 dagar i kultur samt dess tillämplighet för micronucleus analys.
Baslinje karakterisering av 3D HepG2 Lever sfäroid modell
Före någon toxikologisk bedömning in vitro är det viktigt att kontrollera att 3D HepG2-sfäroiderna har bildats ordentligt innan agarosöverföringen eller kemisk/ENM-behandling utförs. HepG2-sfäroider som produceras med hjälp av metoden för hängande droppe tar vanligtvis 2 - 3 dagar efter sådd (4000 celler/sfäroid) för att bilda kompakta, sfäriskt formade sfäroider med en genomsnittlig diameter på 495,52 μm W x 482,69 μm H enligt figur 5A-5C. HepG2-sfäroider som har bildats korrekt och som är godtagbara för att användas för toxikologisk bedömning in vitro måste ha en kompakt, sfärisk formad struktur med en slät yta och inga visuella projektioner. Figur 5 ger exempel på god kvalitet (figur 5D-F) och en sfäroid av dålig kvalitet(figur 5G-I). Varav den senare bör kasseras. Vanligtvis kommer 90-95% av sfäroider som bildas per platta att bildas korrekt och vara livskraftiga för ytterligare experiment.

Figur 5: Lätta mikroskopibilder som visar den naturliga morfologin hos HepG2-sfäroiderna som bildas via hängande droppmetod. (A-C) visa dag 2 och (D-I) Dag 4 HepG2 lever sfäroider efter sådd. (D-F) är exempel på hepg2-sfäroider av god kvalitet medan (G-I) visar dåligt formade sfäroider. Alla bilder togs på ett X20-mål med hjälp av ett mikroskop. Skalstrecket representerar 20 μm. Klicka här för att se en större version av denna siffra.
För att ytterligare bekräfta HepG2 sfäroid livskraft, en grundläggande kolorimetrisk Bromocresol Green Albumin (BCG) Analys eller Urea Assay kan utföras för att bedöma deras leverliknande funktionalitet. Leverliknande funktionalitet bedömdes i linje med livskraften med trypan blue exclusion assay under en 14-dagars kulturperiod för att bestämma livslängden för leversfäroidmodellen och fastställa om den kunde stödja långsiktig eller upprepad ENM/ kemisk baserad farobedömning (figur 6). Albuminkoncentrationen förblev konsekvent under kulturperiodens varaktighet. Urea produktion visar en ökning av koncentrationen av urea produceras per sfäroid över en vecka i kultur innan du når en platå dag 7. Det är viktigt att notera att nivåerna av albumin och urea som produceras i 3D HepG2-sfäroiderna är betydligt högre än de som observerats i samma cellinje odlad i ett 2D-format. Faktum är att 2D-kulturer av HepG2-celler, toppalbumin och urea nivåer var 0,001 mg/mL respektive 0,010 ng/μL. Dessutom, i tidigare arbete publicerat av Shah et al. med hjälp av ett nästan identiskt HepG2 sfäroidsystem, belyser författarna en anmärkningsvärd förbättring av metabolisk aktivitet (CYP1A1 och CYP1A2) i 3D HepG2 in vitro-modellsystem jämfört med de 2D-odlade HepG2-cellerna5.

Figur 6: 14-dagars baslinjekarakteriseringsdata för Leversfäroider från HepG2. Efter överföring från hängande droppe belyser (A) livskraften hos HepG2-sfäroidmodellen under en 14-dagarsperiod medan (B) och (C) belyser den leverliknande albumin- respektive ureafunktionen. Genomsnittliga data ± SEM presenteras, n = 4. Klicka här om du vill visa en större version av den här figuren.
Med den oundvikliga utvecklingen av en nekrotisk kärna, en känd begränsning av 3D lever sfäroid kulturer, var livskraften hos denna HepG2-baserade modell tvungen att fastställas för att visa att den kunde upprätthålla långsiktiga (5-10 dagar) exponering regimer samtidigt upprätthålla den proliferativa förmåga som krävs för att stödja mikrokärnan analys5. Faktum är att denna 3D lever sfäroid modell har visat sig behålla >70% livskraft över 10 dagar i kultur. Baserat på detta och i samband med den ihållande leverliknande funktionalitet som observerats under ≥14-dagarsperioden kan denna 3D leversfäroidmodell därmed stödja långsiktiga, upprepade ENM-exponeringssystem upp till 10 dagar långa (dvs. innan sfäroidens livskraft sjunker under 70%). Som referens rekommenderas att albuminnivåerna för HepG2-sfäroider som sås vid 4000 celler/sfäroid bör vara ≥20,0 ng/μL medan karbamidproduktionen bör vara ≥0,25 ng/μL innan en toxikologisk bedömning in vitro med denna modell görs.
Genotoxicity bedömning av konstruerade nanomaterial
För genotoxicity bedömning användes mikronucleus analysen för att bestämma förekomsten av mikrokärnor efter både akut (24 h) och långsiktiga (120 h) ENM exponeringar. Aflatoxin B1 är ett känt leverkarcinogen16,17 och är en rekommenderad positiv kontroll för mikrokärnans analys. Optimeringsexperiment har visat att 0,1 μM Alfatoxin B1 inducerar ett signifikant positivt (≥2,0 gånger ökning) genotoxiskt svar i 3D HepG2 leversfäroider och används därför i varje mikrokärnaanalys som utförs med denna modell. För att säkerställa giltigheten av mikrokärnans analysresultat med hepG2-sfäroidmodellen bör bakgrundsmikrokärnafrekvensen för HepG2-celler som används i denna 3D-in vitro-modell ligga inom ett intervall på 0,6% - 1,2%. Som ett resultat bör Alfatoxin B1 inducera ett genotoxiskt svar som är minst tvåfaldigt högre än det som ses med den negativa kontrollen. Således bör 0,1 μM Alfatoxin B1 inducera en mikrokärnor frekvens mellan 1,5% – 3,0%. Med hjälp av dessa kontrollparametrar kan ENM-associerad genotoxiitet in vitro sedan bedömas på ett tillförlitligt sätt. På grundval av OECD:s testriktlinje 487 är det viktigt att notera att de valda koncentrationerna vid testning av en ENM eller kemikalie inte bör inducera mer än 55 % ± 5 % cytotoxicitet (indikeras av en minskning av CPBI- eller RVCC-värdena i förhållande till den negativa kontrollen)11. Figur 7 illustrerar de data som genererades när Aflatoxin B1 och två ENMs (titandioxid (TiO2)och sliver (Ag)) utvärderades efter både akuta och långsiktiga exponeringar i HepG2 sfäroider, och efterföljande genotoxic potential analyserades med hjälp av micronucleus analys. Båda de bedömda enm-transaktionsstrukturerna testades med en icke-cytotoxisk låg dos på 5, 00 μg/ml över en akut (24 timmar) exponering och ett långsiktigt (120 timmar) exponeringssystem. En liknande trend för genotoxicity i både TiO2 och Ag ENMs kan observeras, varigenom det förhöjda genotoxicitysvaret som resulterade efter 24 h exponering inte var uppenbart efter en långvarig 5 dagars exponering. Detta trots ihållande genotoxicity framkallas av Aflatoxin B1 positiva kontroll vid båda tidpunkterna.

Figur 7: Genotoxicity bedömning efter TiO2 och Ag ENM exponering på HepG2 lever sfäroider. Bedömning av genotoxicity (mikrokärnafrekvens) med hjälp av mikrokärnans analysstation (A) akut (24 timmar) och (B) långvarig (120 timmars) exponering för 5, 00 μg/ml TiO2 och Ag ENM. Negativ kontroll är endast ett medium, medan den positiva kontrollen är 0,1 μM aflatoxin B1. Medeldata (n=2) ± SD. Signifikans som anges i förhållande till den negativa kontrollen: * = p≤ 0,05. Klicka här om du vill visa en större version av den här figuren.
Discussion
Tillämpningar för 3D lever modeller varierar avsevärt beroende på den specifika biokemiska endpoint eller negativa resultatet utbildningsavsnittet som riktas. Varje modell har sina fördelar och begränsningar, från interdonorvariation i primära human hepatocytmodeller (PHH) till minskad cytokrom p450-aktivitet i cellinjebaserade modeller, men alla är värdefulla i sin egenrätt 6,12,18,19. Vid bedömning av genotoxicity finns det begränsningar i modellernas kompatibilitet med regulatoriska godkända effektmått såsom in vitro mikronukleusanalys, eftersom aktiv spridning krävs. Detta är nödvändigt, eftersom genotoxicity bedömning kräver kvantifiering av fasta DNA-skador att bedömas post cell uppdelning när det finns möjlighet för DNA reparation att korrigera övergående skador. Tyvärr bildar mycket differentierade hepatocyter (dvs. HepaRG) baserade sfäroider eller PHH-mikrotissues, som anses uppvisa de mest fysiologiskt relevanta leverliknande egenskaperna statiska (icke-proliferativa) modeller12,19,20. Som ett resultat ger 3D HepG2-sfäroidmodellen som presenteras här en lämplig, alternativ modell som kan stödja genotoxicitytestning. HepG2 cellinjebaserade sfäroider har tillräckligt aktivt dela celler på den yttre ytan av sfäroider samtidigt upprätthålla grundläggande lever-liknande egenskaper, såsom albumin och urea produktion och vissa CYP450 aktivitet5,12,19. Huvudsakligen har denna in vitro levermodell utvecklats för att komplettera mikrokärnans analys, eftersom detta är en av de två in vitro-analyser som rekommenderas i batteriet för genotoxicitytestning8,10,11,21. Modellen kan dock enkelt tillämpas på DNA-sekvenseringsanalys och genuttrycksteknik (RNA), medan den har potential att anpassas ytterligare och användas för andra DNA-skador, såsom kometanalysen. Det är dock viktigt att överväga vilken roll ENM-inblandning spelar i vissa slutpunktsanalyser. Flödescytometribaserade analyser kanske till exempel inte är lämpliga för ENM-genotoxisbedömning specifikt på grund av partikelinterferens22.
En begränsande faktor för sfäroidmodeller som aktivt genomgår celldelning är deras storlek. Optimering av såddtäthet är avgörande eftersom det måste finnas tillräckligt med celler som gör att modellen kan fortsätta att föröka sig; men inte för högt cellnummer, vilket resulterar i att sfäroiden blir alltför kompakt, vilket leder till en ökad nekrotisk kärna. Orsaken till denna nekros tros vara begränsad syre- och näringsspridning, eftersom gränsen för denna diffusion tros vara cirka 100 - 150 μmvävnad 23,24. Detta beror dock på celltyp, cellnummer, ställningsinteraktioner och odlingsförhållanden25. Sedan dess har det visat sig att cirka 700 μm diameter är gränsen för att undvika för tidig ansenlig nekros i mitten av C3A-sfäroider, sådd 4000 HepG2-celler per sfäroid säkerställer att modellens diameter vid exponeringstiden är ≤500 μm26. Dessutom fastställde Shah et al. att HepG2-celler som såddes över 5000 celler per sfäroid uppvisade en 25% minskning av livskraften efter 7 dagar i kultur, vilket kan hänföra sig till den genomsnittliga diametern på 680 μm och begränsad tillgänglighet av näringsämnen i en 20 μL hängandedroppe 5. För att övervinna detta genomgår modellen som utformats i det nuvarande protokollet ett kritiskt steg där den hängande droppen överförs till agarosebelagda brunnar efter inledande bildandet av sfäroiden. Detta säkerställer att en större volym odlingsmedium finns för att upprätthålla det ständigt växande antalet celler inom sfäroiderna. Som ett resultat förblir HepG2-sfäroidmodellen över 70% livskraftig efter 10 dagar i kultur och kan användas för långsiktig farobedömning in vitro.
Medan HepG2-sfäroidmodellen kan stödja både akuta och långsiktiga exponeringssystem, är uppfriskande cellkulturmedel under längre odlingsperioder begränsat för denna modell eftersom fullständig ersättning av mediet inte rekommenderas på grund av den potentiella förlusten av sfäroiderna. Det antas att tendensen till homogena ENM-dispersioner till agglomerat och sediment är hög vid ENM-exponeringar. Det är dock anmärkningsvärt att den hastighet med vilken ett ENM-sediment kan variera beroende på partikelparametrarna (t.ex. storlek, form och densitet) och kan bestämmas teoretiskt med hjälp av in vitro-sedimenterings-, diffusions- och dosimetrimodellen (ISDD) eller dess senaste derivat, som ofta nämns när det gäller ENM-exponering (suspension)närmar sig 27,28. Med detta är sinnet antas det att om endast 50% av cellkulturmediet försiktigt avlägsnas från cellkulturens yta, bör störningen och efterföljande avlägsnande av ENM-dosen i teorin vara minimal. Med brownsk rörelse i spel är det dock inte fallet och ytterligare arbete med nedfall och sedimentering av varje enskild enm som ska testas bör vidtas för att säkerställa att korrekt dosimetri bibehålls i de långsiktiga exponeringssystemen27. I första hand är detta en potentiell begränsning att överväga när man utför upprepade doseringssystem eftersom detta kan vara avgörande för den slutliga, ackumulerade koncentrationen. Kemikaliebaserade exponeringar å andra sidan erbjuder, även om de inte är utan sina egna begränsningar att beakta, ett mer förenklat tillvägagångssätt genom att kemiska ämnen tenderar att förbli i lösning och därmed en direkt ersättning av den ursprungliga kemiska koncentrationen utöver den nyligen tillagda koncentrationen säkerställer att alla kemikalier som går förlorade under förfriskningar i medierna ersättsi enlighet med detta 29. Framtida tillämpningar skulle omfatta utvärdering av modellens lämplighet för upprepade exponeringssystem under långsiktiga odlingsperioder, eftersom upprepade dosstrategier är av avgörande betydelse för att bedöma ett visst organsystems förmåga att lindra eller övervinna eventuella negativa effekter som orsakas av bioackumulering av ett xenobiotiskt ämne.
Sammanfattningsvis har denna 3D-in vitro-levermodell kapacitet att användas för att utvärdera en rad realistiska exponeringsscenarier och därigenom ge en framtida in vitro-metod för att bättre stödja både ENM och kemisk farobedömning på ett rutinmässigt och lättillgängligt sätt.
Disclosures
Författarna har inget att avslöja.
Acknowledgments
Författarna vill erkänna att denna forskning har fått finansiering från EU:s forsknings- och innovationsprogram Horisont 2020 för PATROLS-projektet, enligt bidragsavtal nr 760813.
Materials
| Name | Company | Catalog Number | Comments |
| Aflotoxin B1 | Sigma Aldrich, UK | A6636-5MG | |
| Agarose | Sigma Aldrich, UK | A9539-50G | |
| Autoclave Tape | |||
| BCG Albumin Assay | Sigma Aldrich, UK | MAK124 | |
| Bovine Serum Albumin Powder | Sigma Aldrich, UK | A9418 | |
| Cell Freezing Aid | Thermo Fisher Scientific, UK | 5100-0001 - Mr Frosty | |
| Centrifuge | Eppendorf | 5810 R | |
| Cytochalasin B | Merck, UK | 250233 | |
| Cytology Metal Clips | |||
| Cytospin 4 Centrifuge | ThermoFisher Scientific, UK | CM00730202 | |
| DMEM with 4.5g/L D-Glucose, L-Glutamine | GIBCO, Paisley, UK | 41965-039 | |
| DMEM, phenol-red free with 4.5g/L D-Glucose, L-Glutamine with Hepes | GIBCO, Paisley, UK | 21063-029 | |
| DPX Mounting Medium | FisherScientific, UK | D/5330/05 | |
| Ethanol | FisherScientific, UK | 10048291 | |
| FBS | GIBCO, Paisley, UK | 10270-106 | |
| Filter Cards for Shandon Cytospin | FisherScientific, UK | 15995742 | |
| Frosted Glass Slides | ThermoFisher Scientific, UK | ||
| Giemsa's Stain Improved R66 Solution, Gurr | VWR Chemicals, UK | MFCD00081642 | |
| Glass Coverslips (24 x 60) | Deckglaser, VWR | ECN631-1575 | |
| Haemocytometer and Coverslip | |||
| Immersion Oil for Microscope | Zeiss, UK | 518F, ISO8034 | |
| Laminar Class II Tissue Culture Hood | Scanlaf Mars | ||
| Light Microscope | Zeiss, UK | Axiovert 40C | |
| Liquid Nitrogen | |||
| Methanol | FisherScientific, UK | 10284580 | |
| Microwave | |||
| Non-Filtered, Sterile 200µl and 1000µl Pipette tips | Greiner-Bio-One, UK | ||
| NuncMicroWell 96-Well Microplates | ThermoFisher Scientific, Denmark | 167008 | |
| P1000 and P200 micropipettes | |||
| P300 and P50 multi-channel pipettes | |||
| PBS pH 7.4 1X, MgCl2 and CaCl2 Free | GIBCO, Paisley, UK | 14190-094 | |
| Pen/Strep | GIBCO, Paisley, UK | 15140-122, Penicillin/Strepmyocin 100X or 10,000U/ml | |
| Phosphatase Buffer Tablets | GIBCO, Paisley, UK | 10582-013 | |
| Pipette Boy | |||
| Simport Scientific CytoSep Funnels for Shandon Cytospin 4 Centrifuges | FisherScientific, UK | 11690581 | |
| Sonifier SFX 550 240V CE 1/2" - Probe | Branson, USA | 101-063-971R | |
| T-25 and T-75 Tissue Culture Flask | Greiner-Bio-One, UK | T-25 (690175) and T-75 (660175) | |
| Trypan Blue Solution | Sigma Aldrich, UK | T8154-100mL | |
| Urea Assay Kit | Sigma Aldrich, UK | MAK006 | |
| Virkon Disinfectant | DuPont, UK | Rely+On Virkon | |
| Water Bath (37?C) | Grant JBNova 18 | ||
| Weighing Balance | |||
| Xylene | FisherScientific, UK | 10588070 | |
| 0.05% Trypsin-EDTA | GIBCO, Paisley, UK | 5300-054 | |
| 0.2mL and 1.0mL Eppendorf Tubes | Greiner-Bio-One, UK | ||
| 0.45µm Filter Unit | Millex HA, MF-Millipore, UK | SLHA033SS | |
| 1.0mL Syringe | BD Plastipak, FisherScientific, UK | 300185 | |
| 20mL LS Scintillation Glass Vials, 22-400 Foil Lined PP Caps | DWK Life Sciences GmbH, Germany | WHEA986581 | |
| 37?C and 5% CO2 ISO Class 5 Hepa Filter Incubator | NUAIRE DHD Autoflow | ||
| 3mL Pasteur Pipette | Greiner-Bio-One, UK | ||
| 50mL Conical Falcon Tubes | Greiner-Bio-One, UK | ||
| 50mL or 100mL Glass Bottles | |||
| 50mL Skirted Falcon Tubes | Greiner-Bio-One, UK | ||
| 5mL, 10mL and 25mL Pipettes | Greiner-Bio-One, UK | ||
| 9.4cm Square, Petri Dish | Greiner-Bio-One, UK | 688161 |
References
- Geiser, M., Kreyling, W. G. Deposition and biokinetics of inhaled nanoparticles. Particle and Fibre Toxicology. 7, 2 (2010).
- Modrzynska, J. Toxicological effects of nanoparticle deposition in the liver. Kgs. Lyngby, Denmark: Technical University of Denmark. , (2018).
- Elje, E., et al. The comet assay applied to HepG2 liver spheroids. Mutation Research/Genetic Toxicology and Environmental Mutagenesis. 845, 403033 (2019).
- Breslin, S., O'Driscoll, L. Three-dimensional cell culture: the missing link in drug discovery. Drug Discovery Today. 18, 240-249 (2013).
- Shah, U. -K., et al. A three-dimensional in vitro HepG2 cells liver spheroid model for genotoxicity studies. Mutation Research/Genetic Toxicology and Environmental Mutagenesis. 825, 51-58 (2018).
- Lauschke, V. M., Hendriks, D. F. G., Bell, C. C., Andersson, T. B., Ingelman-Sundberg, M. Novel 3D Culture Systems for Studies of Human Liver Function and Assessments of the Hepatotoxicity of Drugs and Drug Candidates. Chemical Research in Toxicology. 29, 1936-1955 (2016).
- van Grunsven, L. A. 3D in vitro models of liver fibrosis. Advanced Drug Delivery Reviews. 121, 133-146 (2017).
- Corvi, R., Madia, F. In vitro genotoxicity testing - can the performance be enhanced. Food and Chemical Toxicology. 106, 600-608 (2017).
- Doak, S. H., Manshian, B., Jenkins, G. J. S., Singh, N. In vitro genotoxicity testing strategy for nanomaterials and the adaptation of current OECD guidelines. Mutation Research/Genetic Toxicology and Environmental Mutagenesis. 745, 104-111 (2012).
- Fenech, M. Cytokinesis-block micronucleus cytome assay. Nature Protocols. 2, 1084-1104 (2007).
- OECD. OECD Guidelines. Test 489: In vivo Mammalian Alkaline Comet Assay. , (2016).
- Gerets, H. H. J., et al. Characterization of primary human hepatocytes, HepG2 cells, and HepaRG cells at the mRNA level and CYP activity in response to inducers and their predictivity for the detection of human hepatotoxins. Cell Biology and Toxicology. 28, 69-87 (2012).
- Sison-Young, R. L., et al. A multicenter assessment of single-cell models aligned to standard measures of cell health for prediction of acute hepatotoxicity. Archives of Toxicology. 91, 1385-1400 (2017).
- European Guidelines 2019. European Agency for Safety and Health at Work. , Available from: https://osha.europa.eu/en/safety-and-health-legislation/european-guidelines (2019).
- Jensen, K. A. The NANOGENOTOX Dispersion Protocol for NANoREG. European Union Grant Agreement n° 2009. 21, 01 (2014).
- Marchese, S., et al. Aflatoxin B1 and M1: Biological Properties and Their Involvement in Cancer Development. Toxins. 10, 214 (2018).
- Rushing, B. R., Selim, M. I. Aflatoxin B1: A review on metabolism, toxicity, occurrence in food, occupational exposure, and detoxification methods. Food and Chemical Toxicology. 124, 81-100 (2019).
- Kermanizadeh, A., Brown, D. M., Moritz, W., Stone, V. The importance of inter-individual Kupffer cell variability in the governance of hepatic toxicity in a 3D primary human liver microtissue model. Scientific Reports. 9, 7295 (2019).
- Berger, B., et al. Comparison of Liver Cell Models Using the Basel Phenotyping Cocktail. Frontiers in Pharmacology. 7, 443 (2016).
- Ramaiahgari, S. C., et al. A 3D in vitro model of differentiated HepG2 cell spheroids with improved liver-like properties for repeated dose high-throughput toxicity studies. Archives of Toxicology. , (2014).
- Li, Y., et al. Factors affecting the in vitro micronucleus assay for evaluation of nanomaterials. Mutagenesis. 32 (1), 151-159 (2016).
- Kirkland, D., Reeve, L., Gatehouse, D., Vanparys, P. A core in vitro genotoxicity battery comprising the Ames test plus the in vitro micronucleus test is sufficient to detect rodent carcinogens and in vivo genotoxins. Mutation Research/Genetic Toxicology and Environmental Mutagenesis. 721 (1), 27-73 (2011).
- Curcio, E., et al. Mass transfer and metabolic reactions in hepatocyte spheroids cultured in rotating wall gas-permeable membrane system. Biomaterials. 28, 5487-5497 (2007).
- Glicklis, R., Merchuk, J. C., Cohen, S. Modeling mass transfer in hepatocyte spheroids via cell viability, spheroid size, and hepatocellular functions. Biotechnology and Bioengineering. 86, 672-680 (2004).
- Asthana, A., Kisaalita, W. S. Microtissue size and hypoxia in HTS with 3D cultures. Drug Discovery Today. 17, 810-817 (2012).
- Gaskell, H., et al. Characterization of a functional C3A liver spheroid model. Toxicology Research. 5, 1053-1065 (2016).
- Cho, E. C., Zhang, Q., Xia, Y. The effect of sedimentation and diffusion on cellular uptake of gold nanoparticles. Nature Nanotechnology. 6, 385-391 (2011).
- Hinderliter, P. M., et al. ISDD: A computational model of particle sedimentation, diffusion and target cell dosimetry for in vitro toxicity studies. Particle and Fiber Toxicology. 7, 36 (2010).
- Kramer, N. I., di Consiglio, E., Blaauboer, B. J., Testai, E. Biokinetics in repeated-dosing in vitro drug toxicity studies. Toxicology in vitro. 30, 217-224 (2015).
Tags
Bioengineering utgåva 160 In Vitro Levermodeller Nanomaterial Farobedömning Långtidsexponering Nano(geno)toxikologi DNA-skadorErratum
Formal Correction: Erratum: Advanced 3D Liver Models for In vitro Genotoxicity Testing Following Long-Term Nanomaterial Exposure
Posted by JoVE Editors on 01/26/2021.
Citeable Link.
An erratum was issued for: Advanced 3D Liver Models for In vitro Genotoxicity Testing Following Long-Term Nanomaterial Exposure. The Representative Results section was updated.
Figure 6 in the Representative Results section was updated from:
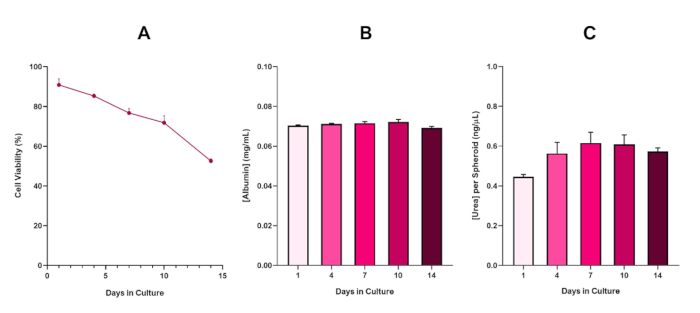
to:
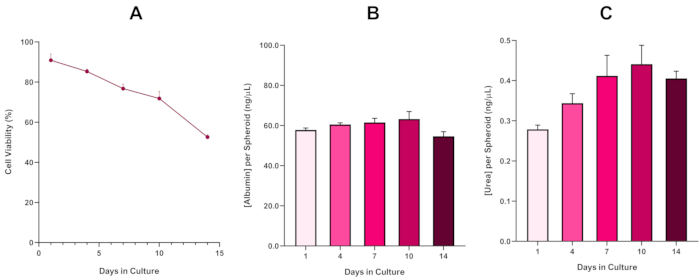
The fourth paragraph in the Representative Results section was updated from:
With the inevitable development of a necrotic core, a known limitation of 3D liver spheroid cultures, the viability of this HepG2 based model had to be established to demonstrate it was able to sustain long-term (5-10 day) exposure regimes whilst maintaining the proliferative capability required to support the micronucleus assay5. Indeed, this 3D liver spheroid model has been shown to retain >70% viability over 10 days in culture. Based on this and in conjunction with the sustained liver-like functionality observed over the ≥14 day culture period, this 3D liver spheroid model can thus support long-term, repeated ENM exposure regimes up to 10 days long (i.e., before viability of the spheroids drop below 70%). For reference, it is advised that albumin levels for HepG2 spheroids seeded at 4000 cells/spheroid should be ≥0.06 mg/mL whilst urea production should be ≥0.4 ng/µL before conducting an in vitro toxicological assessment with this model.
to:
With the inevitable development of a necrotic core, a known limitation of 3D liver spheroid cultures, the viability of this HepG2 based model had to be established to demonstrate it was able to sustain long-term (5-10 day) exposure regimes whilst maintaining the proliferative capability required to support the micronucleus assay5. Indeed, this 3D liver spheroid model has been shown to retain >70% viability over 10 days in culture. Based on this and in conjunction with the sustained liver-like functionality observed over the ≥14 day culture period, this 3D liver spheroid model can thus support long-term, repeated ENM exposure regimes up to 10 days long (i.e., before viability of the spheroids drop below 70%). For reference, it is advised that albumin levels for HepG2 spheroids seeded at 4000 cells/spheroid should be ≥50.0 ng/μL whilst urea production should be ≥0.25 ng/µL before conducting an in vitro toxicological assessment with this model.
