

ERRATUM NOTICE
Important: There has been an erratum issued for this article. Read more …
Summary
Denne procedure blev indført for at udvikle avancerede 3D-leverkulturer in vitro, som kan give en mere fysiologisk relevant vurdering af de genotoksiske farer, der er forbundet med nanomaterialeeksponering over både en akut eller langvarig, gentagen dosis regimer.
Abstract
På grund af den hurtige udvikling og implementering af en bred vifte af manipulerede nanomaterialer (ENM) er eksponering for ENM uundgåelig, og udviklingen af robuste, prædiktive in vitro-testsystemer er afgørende. Levertoksikologi er nøglen, når man overvejer ENM eksponering, som leveren tjener en afgørende rolle i metaboliske homøostase og afgiftning samt at være et vigtigt sted for ENM akkumulering post eksponering. Baseret på dette og den accepterede forståelse af, at 2D-hepatocytmodeller ikke nøjagtigt efterligner kompleksiteten af indviklede multicellulære interaktioner og metabolisk aktivitet observeret in vivo, er der større fokus på udviklingen af fysiologisk relevante 3D-levermodeller skræddersyet til ENM-farevurderingsformål in vitro. I overensstemmelse med principperne i 3R'erne til at erstatte, reducere og forfine dyreforsøg er der udviklet en 3D HepG2 cellelinjebaseret levermodel, som er et brugervenligt og omkostningseffektivt system, der kan understøtte både udvidede og gentagne ENM-eksponeringsordninger (≤14 dage). Disse sfæroide modeller (≥ 500 μm i diameter) bevarer deres proliferative kapacitet (dvs. dividere cellemodeller), så de kan kombineres med mikrokerneanalysen »guldstandard« for effektivt at vurdere genotoksicitet in vitro. Deres evne til at rapportere om en række toksikologiske endpoints (f.eks. leverfunktion, (pro)inflammatorisk respons, cytotoksicitet og genotoksicitet) er blevet karakteriseret ved hjælp af flere ENMs på tværs af både akutte (24 h) og langsigtede (120 h) eksponeringsregimer. Denne 3D-in vitro-levermodel har kapacitet til at blive anvendt til at evaluere mere realistiske ENM-eksponeringer, hvilket giver en fremtidig in vitro-tilgang til bedre at understøtte ENM-farevurdering på en rutinemæssig og lettilgængelig måde.
Introduction
På grund af den hurtige udvikling og implementering af en bred vifte af manipulerede nanomaterialer (ENM) på tværs af en overflod af menneskebaserede applikationer (f.eks. fødevarer, kosmetik, tøj, sportsudstyr, elektronik, transport og medicin) er det uundgåeligt, at mennesker regelmæssigt vil blive udsat for ENM. Med dette er der øget bekymring for, at de nye, størrelsesspecifikke fysiokemiske egenskaber, der anser disse materialer for fordelagtige i mange applikationer, kan forårsage negative virkninger på menneskers sundhed og miljøet samtidig. I øjeblikket er der mange internationale aktiviteter på plads for aktivt at afspejle mere fysiologisk relevante eksponeringer for disse ENM og vurdere disse materialers potentielle toksicitet i scenarier med akut, langvarig og gentagen lavdosiseksponering.
Levertoksikologi er nøglen, når man overvejer ENM-eksponering, da det er almindeligt kendt, at leveren er et vigtigt sted for ENM-akkumulering efter eksponering1,2. Desuden er leveren det primære organsystem til metabolisme og afgiftning af stoffer, der kommer i systemiskcirkulation 3. Baseret på den accepterede forståelse af , at 2D hepatocytmodeller ikke nøjagtigt efterligner kompleksiteten af indviklede flercellede interaktioner eller passende repræsenterer metabolisk aktivitet observeret in vivo , er der etableret et større fokus på at udvikle robuste og fysiologisk relevante in vitro 3D-levermodeller for in vivo-erstatningsteknologier4,5. Brug af avancerede 3D-kulturteknologier forbedrer levetiden af in vitro-levermodeller, hvilket giver mulighed for langsigtede, gentagne eksponeringsordninger, der skal undersøges. Derudover fremmer dette avancerede kulturformat dannelsen af forbedrede fysiologiske, organotypiske træk som galdekanaliculi, aktive transportprocesser og forbedrede CYP450-lægemiddelmetaboliseringsfunktioner, hvilket forbedrer forudsigeligheden af modellerne6. Nuværende 3D-in vitro-levermodeller bestående af monokulturer (kun hepatocytter) eller samkulturer (hepatocytter med ikke-parenchymale celler) findes i flere formater, lige fra mikrotissuer eller sfæroider i ultralow vedhæftningsplader, hængende dråbesprædroider, celler indlejret i matricer og/eller stilladser og mikrofluidiske cellekulturplatforme, som alle anses for effektive avancerede in vitro-modeller til vurdering af levertoksicitet6,7. Men de fleste af disse modelsystemer er høj vedligeholdelse, kræver specialiseret udstyr og er dyre. Desuden er disse modeller ofte statiske (dvs. ikke-opdelte cellemodeller), der forhindrer deres anvendelse i vurderingen af fareslutpunkter, såsom genotoksicitetstest ved hjælp af metoder, der kvantificerer fast DNA-skade. Genotoksicitet er en central forudsætning for lovgivningsmæssig toksikologi, og det er en vigtig del af risikovurderingen af ethvert toksiksmiddel8. Der er ingen enkelt analyse, der kan anvendes til at kvantificere alle former for DNA-skader, der kan opstå efter eksponering for et eksogent middel. En kernekomponent i in vitro-genotoksicitetstestbatteriet er imidlertid mikrokerneanalysen, som er en pålidelig og mangefacetteret teknik, der måler bruttokromosomskader9. Det er en guldstandardteknik, der er beskrevet i OECD's testretningslinje 487 til vurdering af in vitro-DNA-skader og genotoksicitet og er en del af kravet om testbatterier til lovgivningsmæssig farevurdering10,11.
Den menneskelige hepatocellulære karcinomcellelinje, HepG2, anvendes bredt til indledende risikovurderingsscreening, da cellerne er let tilgængelige, relativt billige at købe, enkle at kultur og modtagelige for høj gennemløbsscreening12,13. Når de dyrkes i 3D-sfæriske strukturer, har de vist sig at opsummere levermikromiljøet godt og tilbyde en levermodel med tilstrækkelige proliferative evner til at understøtte mikrokerneanalysen3. Yderligere udvikling af HepG2-sfæroidmodellerne blev etableret for at forbedre modellens levetid og leverlignende funktionalitet for at understøtte vurdering af genotoksicitetsfare over langsigtede, gentagne eksponeringsordninger (≤14 dage). I overensstemmelse med principperne i 3R'erne til erstatning, reduktion og forfinelse af dyreforsøg er denne protokol således blevet udarbejdet for at tilvejebringe en avanceret 3D-in vitro-levermodel, der er i stand til pålideligt at evaluere flere toksikologiske slutpunkter (f.eks. leverfunktionalitet, (pro-)inflammatoriske markører, cytotoksicitet og genotoksicitet) efter akut, langvarig og gentagen kemisk og ENM-eksponering på en rutinemæssig og let tilgængelig måde.
Her præsenterer vi en metode til at etablere et fysiologisk relevant 3D-hepatocytcellelinjebaseret in vitro-modelsystem til vurdering af genotoksicitetsfare efter akutte eller langsigtede, gentagne ENM-eksponeringer. Protokollen kan opdeles i 6 centrale faser: dyrkning cryopreserved HepG2 celler; HepG2 sfæroid forberedelse; HepG2 sfæroid overførsel fra hængende dråbe til agarose suspension; HepG2 sfærisk høst; mikrokerneanalyse og -scoring og dataanalyse.
Protocol
1.Culturing cryopreserved HepG2 celler
BEMÆRK: HepG2 celler, opnået fra American Type Culture Collection (ATCC) blev dyrket i 1x Dulbecco's Modified Eagle Medium (DMEM) med 4,5 g / L D-glukose og L-glutamin suppleret med 10% føtal kvæg serum (FBS) og 1% penicillin / streptomycin antibiotikum.
- Forvarmet DMEM-cellekulturmedium (herunder kosttilskud) i et 37 °C vandbad i 30 min.
- Et hætteglas HepG2-celler fjernes fra flydende nitrogen og tøes op i et 37 °C vandbad i 2-3 minutter, mens hætteglasset hvirvles forsigtigt, så cellens affjedring kan optøs ensartet. Pas på ikke at nedsænke hætteglasset over O-ringen for at reducere risikoen for forurening.
- Når det er optøet, skal du fjerne hætteglasset fra vandbadet og sprøjte generøst med 70% ethanol for at dekontaminere hættens ydre overflade, før du placerer under en steril, klasse II laminar vævskulturhætte.
- Pipetter forsigtigt indholdet af kryoformet af HepG2-celler i et centrifugerør, der indeholder 9 mL forvarmet DMEM-cellekulturmedium (med kosttilskud).
- Ved hjælp af en 10 mL strippette overføres 10 mL af celleophænget til en 25 cm2 engangscellekulturkolbe og inkuberes kulturen i 3 dage (fra såning) ved 5% CO2 og 37 °C, indtil ~ 80% sammenløb nås, før den undergår subkultur til en større 75 cm2 engangscellekulturkolbe.
- Når 80% sammenløb er nået, sub-dyrkning celler under sterile forhold ved trypsinization med 0,05% trypsin / EDTA løsning forvarmet i en 37 ° C vandbad i 30 min. Cellerne må på intet tidspunkt tørre ud.
- Når celler danner et klæbende monolag, skal du fjerne mediet ved at tippe ind i en desinfektionsmiddelaffaldspotte. Derefter vaskes straks monolayer for at fjerne alle spor af eksisterende medier ved at skylle kolben to gange med 3 ml steril 1x PBS-opløsning, der opbevares ved stuetemperatur. Kassér også PBS i desinfektionsmiddelaffaldspotten.
- Når PBS-vask er fjernet, tilsættes 5 ml forvarmet 0,05% trypsin-EDTA-opløsning, hvilket sikrer, at hele overfladen af cellerne og inkuber cellerne i 6-8 minutter ved 37 °C og 5% CO2.
- Bank forsigtigt på kolben for at løsne cellerne fra bunden af kolben og tilsæt derefter 5 mL DMEM cellekulturmedium (med kosttilskud) for at neutralisere trypsinenzymet.
- Celleaffjedringen overføres til et 50 mL centrifugerør, og celleaffjedringen pipetters grundigt op og ned for at sikre, at cellerne er helt adskilte.
- Centrifuge den fortyndede celle suspension på 230 x g i 5 min. Det supernatant kasseres i desinfektionsmiddel, og cellepillen suspenderes igen i 25 mL DMEM-cellekulturmedie (med kosttilskud).
- Celleaffjedringen overføres til en 75 cm2 engangscellekulturkolbe, og der inkuberes ved 37 °C og 5% CO2 i yderligere 3 dage, før den gennemgår kugleformet præparat. Når HepG2s har haft tid til at akklimatisere og endnu en gang nå ~ 80% sammenløb, bestemme cellekoncentrationen som forberedelse til sfæroid såning.
2. HepG2 sfæroid forberedelse
- Gentag de ovenfor anførte trin for subkultur, undtagen efter centrifugering, suspenderer cellepillen igen i 1 mL DMEM-dyrkningsmedie, der er forvarmet i et 37 °C vandbad. Pipette celle suspension op og ned grundigt.
- Score celle levedygtighed ved hjælp af Trypan Blue Exclusion Assay (se OSHA SOP 3.21 Reproduktive toksiner, mutagener, teratogener og embryotoksiner – Procedurer for sikker håndtering og opbevaring (2019) for sundheds- og sikkerhedsvejledning)14 med et 1:1-forhold mellem celleaffjedring og forfiltreret 0,4% Trypan blå opløsning.
- Før celletællingen tages 1 ml Trypan blå opløsning med en 1 ml sprøjte, og der filtreres med en filterenhed på 0,45 μm i et sterilt, 1 ml rør.
- 10 μL filtreret, Trypanblå opløsning overføres til et 0,2 ml rør, og der tilsættes 10 μL celleaffjedring. Resterende filtreret Trypan blå opløsning kan opbevares op til 3 måneder ved stuetemperatur til fremtidig brug.
- Sprøjt hæmocytometeret grundigt med 70% ethanol og tør det af med et sterilt køkkenrulle, før du sikrer dækslet på toppen ved hjælp af åndedrætsdampe. Glidende coverlip over ånde fugtet overflade inducerer sammenhængende kræfter ved at generere Newton ringe.
- Rør forsigtigt Trypan blå celleophæng op og ned ved hjælp af en 1000 μL pipette (for at reducere ren og skær stress), før der tilsættes 10 μL til hæmofometeret. Sørg for, at opløsningen er spredt under dækslet slip og dækker hele nettet uden luftbobler.

Figur 1: Tælle celler ved hjælp af et hæmocytometer. Diagrammatisk repræsentation af et hæmocytometer, der fremhæver, hvilken kvadrant der skal tælles celler fra. Klik her for at se en større version af dette tal.
- Under mikroskopet tælles de levende (uindfattede) og døde (farvede blå) celler, der findes i de fire store hjørnekanter(figur 1). Udelad celler, der findes til overlapning eller sidde på de indre to kanter af de store hjørne firkanter (dvs. på linjerne) i optællingen.
- Ved hjælp af følgende beregning beregnes det gennemsnitlige antal levende, levedygtige celler (ikke indgroet), der findes i stikprøven:
Samlet antal celler/mL = Antal levende celler x x 10.000
x x 10.000
hvor fortynding refererer til, hvor mange gange bestanden opløsningen blev fortyndet i Trypan blå (2x i dette tilfælde) og # af optalte firkanter refererer til de fire store hjørne firkanter af hæmocytometeret tælles - Baseret på det levedygtige HepG2-celleantal og ved hjælp af følgende formel:
C1V1=C2V2
hvor C1 = koncentrationen af levedygtige celler i øjeblikket
V1 = mængden af celleaffjedring i øjeblikket
C2 = koncentrationen af celleaffjedring ønskede,
V2 = mængden af celleaffjedring ønskede - Forbered en 10 ml lageropløsning af HepG2 celleaffjedring med DMEM cellekultur medium i en koncentration på 2,0 x 105 celler/mL for at opnå 4000 HepG2 celler pr. 20 μL hængende dråbe. Celleaffjedringen blandes grundigt ved forsigtigt at pipere op og ned ved hjælp af en 1000 μL pipette for at sikre, at alle celler er helt ophængt i mediet.
- Til brøndene på en 96-brønds cellekulturplade tilsættes 100 μL steril, stuetemperatur PBS for at forhindre, at de hængende dråber tørrer ud under inkubation.
- Tag låget på en flad standardcellekulturplade med 96 brønde, vend den om og pipetter forsigtigt 20 μL dråber af celleophænget ind i midten af hver brøndrille af låget, som vist i figur 2. Brug en multi-kanals pipette, men tilføj kun 2 - 4 dråber på én gang, da flere såning kan påvirke nøjagtigheden og placeringen af dråberne.
- Centrer dråberne i rillerne af brøndene lagt ud på låget; ellers hænger de ikke i midten af brøndene, når pladens låg vendes og risikerer at falde af i pladen. Vend forsigtigt låget på 96-brøndspladen, så dråberne nu hænger og placeres forsigtigt oven på 96-brøndspladen.
- Placer hele 96 brøndpladen med låg forsigtigt i en inkubator ved 37 °C og 5% CO2 i 3 dage før kugleformet overførsel på agarose.
BEMÆRK: Der skal udvises ekstra forsigtighed, ikke kun ved transport af pladerne til/fra inkubatorerne, men ved åbning og lukning af inkubatoren generelt, da overdreven bevægelse kan få pladerne til at skifte, og kugleformerne enten falder eller dannes forkert.
 x x 10.000
x x 10.000
Figur 2: 3D HepG2 in vitro sfæroid modelpræparat. (b) HepG2-cellerne efter såning i den hængende dråbemodel for at muliggøre sfæroiddannelse. Klik her for at se en større version af dette tal.
3. HepG2 sfæroid overførsel fra hængende dråbe til agarose suspension
BEMÆRK: På dag 3 post såning i hængende dråber, er de sfæroider overføres til brøndene i samme 96-godt plade, som alle tidligere har været belagt med et fint lag på 1,5% agarose gel.
- Forbered agarose geler og autoklave (dvs. dag 2 efter såning) før dagen for pladebelægning (dvs. dag 3 efter såning).
- For at forberede en 1,5% agarose gel, vejer 0,30 g agarose i en ren, glasflaske og tilsæt derefter 20 mL phenol-rødfri DMEM medium. Agarosen skal autoklavers i 1 time ved 230 °C med henblik på sterilisering. Agarosebelægningen forhindrer HepG2-sfæroiderne i at klæbe til bunden af brønde og danne et cellulært monolayer i stedet for at bevare deres 3D-sfæroidstruktur.
- På dag 3 post såning, fjerne 96-brønd plade, der indeholder HepG2 hængende dråbe sfæroider ud af inkubatoren og forsigtigt vende låget, så spheroids ikke længere hænger.
- Ved hjælp af en multikanalpipette fjernes og kasseres de 100 μL PBS, der tidligere er tilsat bunden af 96-brøndspladen. Lad pladerne lufttørre i 2-3 minutter, mens agarose opvarmes som forberedelse til belægning.
ADVARSEL: Denne procedure resulterer i meget varm, flydende agarose, som, hvis den spildes på huden, kan brænde og forårsage skade. Desuden skal man være forsigtig, når du håndterer glasflasken, der indeholder flydende agarose, da dette også kan være meget varmt. - Brug de 1,5% agarose geler, der tidligere er tilberedt, opvarmes glasflasken indeholdende 20 mL agarosegel til 30 s i en mikrobølgeovn ved den maksimale watt (dvs. 900 W). For at belægge to 96-brønds plader skal en 20 mL flaske forforberedt 1,5% agarosegel være tilstrækkelig.
- Når den er smeltet, skal du forsigtigt hvirvle agarose ved at dreje glasflasken for at fjerne eventuelle bobler og derefter tilføje 50 μL agarose i bunden af hver brønd.
BEMÆRK: Når du tilføjer agarose, skal du sørge for ikke at vinkle pladen >45°, da agarose indstilles hurtigt og ikke danner et fladt, niveaulag, der kan forstyrre kugleformet vækst. Det er vigtigt at arbejde effektivt på dette tidspunkt for at forhindre agarose i at størkne, før pladen er helt belagt. - Lad pladen stå i 2 minutter ved stuetemperatur, før der tilsættes 100 μL forvarmet DMEM cellekulturmedium (med kosttilskud) oven på det faste agaroselag i hver brønd.
- Vend låget på 96-brøndspladen og læg tilbage på toppen af 96-brøndspladen, så sfæroiderne nu hænger igen.
- Centrifuge pladen i 3 min ved 200 x g for at overføre spheroids fra den hængende dråbe til de enkelte brønde af 96-brønds pladen. Efter overførslen bør HepG2-sfæroiderne nu suspenderes i cellekulturmediet. Lad dem afregne i 24 timer i inkubatoren ved 37 °C og 5% CO2.
- Udsæt HepG2-sfæroider af denne størrelse til enten kemiske eller ENM-behandlinger på dag 4 efter såning (dvs. 24 timer efter overførsel til agarosebelagte plader).
- For at opretholde celle levedygtighed over længere kultur perioder, opdatere cellekultur medium hver 3 dage. For at gøre dette skal du forsigtigt suge 50 μL af cellekulturmediet fra brøndens overflade og erstatte med et frisk 50 μL DMEM-cellekulturmedium. Pas på ikke at fjerne eller forstyrre kugleformet, når du udfører en medium ændring.
4. Nanomateriale/kemisk eksponering
BEMÆRK: HepG2-leverspærremodellen kan understøtte både ENM- og kemikaliebaserede eksponeringsregimer, men det primære fokus for denne protokol er ENM-eksponeringer. Før eksponering skal testen ENM være tilstrækkeligt spredt. Dette kan udføres som anvist af NanoGenoTox-spredningsprotokollen (tilskudsaftale nr. 20092101, 2018)15.
- Efter spredning i henhold til NanoGenoTox Dispersion-protokollen fortyndes ENM-suspensionen fra startkoncentrationen på 2,56 mg/mL til den endelige ønskede koncentration i forvarmet DMEM-cellekulturmedium (herunder kosttilskud). Der kræves et samlet volumen på 5 mL for at dosere en 96 brøndplade.
- For at udsætte HepG2-kugleformet for enten et kemikalie eller en ENM ved hjælp af en 200 μL pipette, indsugningsmediet 50 μL cellekulturmedie fra overfladen af hver brønd (hvilket efterlader 50 μL i brønden for ikke at forstyrre spheroiderne) og erstattes med 50 μL medium, der indeholder testtoksiksivet ved den krævede dosis.
- Når prøvematerialet er påført, inkuberes pladerne i den ønskede eksponeringstid ved 37 °C og 5% CO2.
- Hvis der gennemføres et langvarigt (≥24 h) eksponeringssystem, skal du straks efter udløbet af den ønskede eksponeringsfrist høste spheroiderne til mikrokerneslutpunktanalyse som beskrevet nedenfor i trin 6.1 – 6.4.
- Med ordninger for akut eksponering (f.eks. ≤24 h), når eksponeringsperioden er afsluttet, høstes, samles og opbevares 50 μL supernatant fra hver brønd i 96-brøndpladen ved -80 °C til yderligere biokemisk analyse senere. Cellekulturmediet erstattes med 50 μL friskt medium, der indeholder 6 μg/mL Cytochalasin B, og der skal inkuberes i 1 – 1,5 cellecyklusser (dvs. 24 – 26 timer for HepG2) som forberedelse til cytokinesisblokkens mikronukleus assayhøst.
BEMÆRK: For akut (≤24 h) eksponeringsregimer kan cytokineseblokmikrokerneanalysen med Cytochalasin B anvendes, men for langvarige (≥24 h) eksponeringsregimer skal den mononukleare version (uden Cytochalasin B) af analysen anvendes som beskrevet nedenfor i figur 4.
5. HepG2 sfæroid høst
BEMÆRK: Efter enten kemiske eller ENM eksponering behandlinger, både cellekultur medium eller sfæroid væv kan høstes til flere endpoint analyse. Afhængigt af slutpunktsanalysen kan sfæroider enten høstes individuelt (f.eks. til billedanalyse) eller samles (f.eks. til cytokinesisblokmikrokerneanalyse).
- Fjern 96-brøndspladen fra inkubatoren.
- Ved hjælp af en 200 μL pipette suges 100 μL cellekulturmedie, herunder sfæroidvævet fra hver brønd, og der opsamles i et sterilt centrifugerør på 15 mL. Pas på at undgå kontakt med agarose.
- Når den er indsamlet, centrifuges kugleformet suspension ved 230 x g i 5 min. Supernatanten fjernes og opbevares ved -80 °C til yderligere slutpunktsanalyse (f.eks. leverfunktionstest) senere.
- Pelletlen af sfæroider igen suspenderes i 1 mL steril, stuetemperatur PBS (1x).
- Når den er vasket, centrifuges kugleformet igen ved 230 x g i 3 min. Supernatanten kasseres igen, suspenderes igen i 500 μL af 0,05% trypsin-EDTA-opløsning og inkuberes i 6-8 min ved 37 °C og 5% CO2.
- Efter inkubation, forsigtigt pipette de trypsiniserede celler op og ned til fuldt ud at adskille og re-suspendere HepG2 celler før neutralisere med 1 mL DMEM cellekultur medium.
- Centrifuge den fortyndede celle suspension på 230 x g i 5 min. Supernatanten kasseres i desinfektionsmiddel, og cellepillen suspenderes igen i 2mL af rumtemperaturen PBS (1x).
- Centrifuge celleaffjedringen ved 230 x g i 5 min. Supernatanten kasseres i desinfektionsmiddel, og cellepillen suspenderes igen i 2 mL kold PBS (1x). Sørg for, at cellerne er godt spredt for at forhindre klumper af celler tilsløre synsfeltet, når monteret på mikroskop dias.
6. Micronucleus assay og scoring
Til den manuelle metode til mikrokerneanalysen kræves der en cytocentrifuge for at producere en cytodot (et defineret, koncentreret celleområde) midt på mikroskopdiaset. Denne proces understøtter en mere effektiv scoring af diaset, da det gør det muligt for målscoreren nemt at finde de celler af interesse, i modsætning til at evaluere et helt dias, hvor cellerne kan spredes bredt.
- Dip matteret mikroskop dias (tre per dosis) i 70% ethanol efterfulgt af ddH2O og lad lufttørre i 5 min.
- Placer forberedt mikroskop glider ind i cuvette tragt som vist i figur 3A, hvor glasset dias (iii) er placeret i metal støtte (iv) med et filter kort (ii) og cuvette tragt (i) sikret på toppen.
- Arranger cuvette tragte i cytocentrifuge med tragten vender op, så 100 μL celle suspension kan tilføjes direkte i hver enkelt.
- Cytospin i 5 min ved 500 x g for at sikre, at cellerne fordeles jævnt på overfladen af diaset.

Figur 3: Cytospinopsætning til klargøring af behandlede celler på mikroskopdias. (A) Viser de enkelte komponenter, (i) cuvettetragt, (ii) filterkort, (iii) glasmikroskopdias og (iv) metalstøtte, der kræves for at cytospin HepG2-celler på mikroskopdias. (B) Den endelige cuvette tragt oprettet. (C) Korrekt placering af cuvettetragten i cytocentrifuge. Klik her for at se en større version af dette tal.
- Lad rutsjebanerne lufttørre før fiksering i iskold, 90% methanol i 10 minutter.
- Når de er fastgjort, skal rutsjebanerne lufttørre natten over ved stuetemperatur, før de opbevares ved -20 °C i op til 6 måneder.
- Når det er nødvendigt, skal du fjerne de forberedte mikroskopdias fra fryseren på -20 °C og gøre det muligt at varme til stuetemperatur, før giemsa-farvningen foretages.
ADVARSEL: I henhold til forordning (EF) nr. Se det tilhørende SDS-ark for detaljeret opbevaring, håndtering og sundheds- og sikkerhedsrådgivning om dette kemikalie inden brug. - Mens dias er optøning, forberede en 20% Giemsa farvning løsning (25 ml kræves for at plette ~ 30 dias) fortyndet i fosfatase buffer (pH 6,8). Bland grundigt ved forsigtigt at hvirvle opløsningen, før du filtrerer ved hjælp af foldet filterpapir placeret i en tragt.
- Ved hjælp af en Pasteur pipette tilsættes 3 – 5 dråber filtreret Giemsa-opløsning til cytodot på hvert dias og efterlades i 8 - 10 minutter.
- Vask rutsjebaner i to på hinanden følgende fosfatase buffer vasker før kortvarigt skylning under koldt vand for at fjerne overskydende plet rester. Lad rutsjebanerne lufttørre.
- Når tør, i en røghætte, dip farvede dias i xylen i 10 s, før du tilføjer en dråbe montering medium til midten af cytodot og et sted et glas coverslip på toppen.
- Lad mikroskopet glide i røghætten natten over for at tørre før manuel scoring; de kan opbevares på ubestemt tid ved stuetemperatur.
7. Dataanalyse
- Som beskrevet i OECD's retningslinjerne for test 487 (2014)11skal du bruge et let mikroskop (100x mål med nedsænkningsolie) 2000 monokerner eller 1000 binucleerede celler pr. biologisk replikat til at score for forekomst af mikronuklei som vist i figur 4.

Figur 4: Micronucleus assay scoring beslutning træ. Skematisk beslutning træ for at fremhæve nødvendigheden af forskellige scoring og cytotoksicitet vurderingsprocedurer, når du bruger micronucleus assay med 3D-modeller efter akut eller langvarig eksponering regimer. Akutte (≤24 h) eksponeringer tillader brug af cytokinese blokeret mikrokerne assay, mens langsigtede (≥24 h) engagementer kræver den mononukleare version af analysen; begge er beskrevet i OECD's testretningslinje 487. Klik her for at se en større version af dette tal.
- Baseret på andelen af mikrokerner, der findes pr. antal mononukleerede eller binucleerede celler, beregnes en procentdel af genotoksicitetsværdien.
- For at vurdere den observerede DNA-skade er ikke som følge af cellerester forårsaget af en høj andel af apoptotiske celler, tage et mål for cytotoksicitet sammen. I dette tilfælde skal du afhængigt af tilstedeværelsen af Cytochalasin B anvende enten CPBI- eller RVCC-beregning (som beskrevet i figur 4). Genotoksicitet må kun vurderes i prøver, hvor cytotoksiciteten er mindre end 55 % ± 5 % som defineret i OECD's testretningslinje 48711.
Representative Results
Egnetheden af denne celle-line baseret 3D lever sfæroid model for langsigtet kultur og genotoksisk fare vurdering blev evalueret ved at gennemføre baseline karakterisering at bestemme levedygtigheden og lever-lignende funktionalitet af modellen i løbet af 14 dage i kultur samt dens anvendelighed for micronucleus assay.
Baseline Karakterisering af 3D HepG2 Lever Sfæroid Model
Forud for enhver in vitro toksikologisk vurdering er det vigtigt at kontrollere, at 3D HepG2-sfæroiderne er dannet korrekt, før de udfører agaroseoverførslen eller kemisk /ENM-behandling. HepG2-sfæroider, der produceres ved hjælp af den hængende dråbemetode, tager normalt 2 - 3 dage efter såning (4000 celler / sfæroid) for at danne kompakte, sfæriske formede sfæroider med en gennemsnitlig diameter på 495,52 μm W x 482,69 μm H som vist i figur 5A-5C. HepG2-sfæroider, der er dannet korrekt og kan accepteres anvendt til in vitro-toksikologisk vurdering, skal have en kompakt, sfærisk formet struktur med en glat overflade og ingen visuelle fremskrivninger. Figur 5 giver eksempler på god kvalitet (figur 5D-F) og spadseroider af dårlig kvalitet ( figur5G-I). Sidstnævnte bør kasseres. Typisk vil 90-95% af sfæroider dannet pr plade dannes korrekt og være levedygtige for yderligere eksperimenter.

Figur 5: Lysmikroskopibilleder, der viser hepG2-sfæroidernes naturlige morfologi dannet ved hjælp af den hængende dråbemetode. (A-C) viser dag 2 og (D-I) Dag 4 HepG2 lever sfæroider efter såning. (D-F) er eksempler på hepG2-sfæroider af god kvalitet, mens (G-I) viser dårligt dannede sfæroider. Alle billeder blev taget på et X20 mål ved hjælp af et mikroskop. Skalalinjen repræsenterer 20 μm. Klik her for at se en større version af dette tal.
For yderligere at bekræfte HepG2 sfæroid levedygtighed, en grundlæggende colorimetric Bromocresol Green Albumin (BCG) Assay eller Urea Assay kan udføres for at vurdere deres lever-lignende funktionalitet. Leverlignende funktionalitet blev vurderet i overensstemmelse med levedygtigheden ved hjælp af Trypan Blue Exclusion Assay over en 14-dages kulturperiode for at bestemme levetiden af leverspærremodellen og fastslå, om den kunne understøtte langsigtet eller gentagen ENM/kemisk baseret farevurdering (Figur 6). Albuminkoncentrationen forblev konsistent i kulturperiodens varighed. Urinstofproduktionen viser en stigning i koncentrationen af urinstof produceret pr. kugleformet over en uge i kultur, før den når et plateau dag 7. Det er vigtigt at bemærke, at niveauet af albumin og urinstof produceret i 3D HepG2-sfæroiderne er væsentligt højere end det, der observeres i samme cellelinje dyrket i et 2D-format. Faktisk var 2D-kulturer af HepG2-celler, peak albumin og urinstofniveauer henholdsvis 0,001 mg / mL og 0,010 ng / μL. Desuden, i tidligere arbejde udgivet af Shah et al. ved hjælp af en næsten identisk HepG2 sfæroid system, forfatterne fremhæve en bemærkelsesværdig forbedring i metabolisk aktivitet (CYP1A1 og CYP1A2) i 3D HepG2 in vitro model systemer i forhold til 2D dyrkede HepG2 celler5.

Figur 6: 14-dages basiskarakteriseringsdata for HepG2-leverspærreoider. Efter overførsel fra hængende dråbe fremhæver (A) levedygtigheden af HepG2-kugleformet model over en 14-dages periode, mens (B) og (C) fremhæver henholdsvis leverlignende albumin- og urinstoffunktionalitet. Gennemsnitsdata ± præsenteret for SEM, n = 4. Klik her for at se en større version af dette tal.
Med den uundgåelige udvikling af en nekrotisk kerne, en kendt begrænsning af 3D-lever sfæroid kulturer, levedygtigheden af denne HepG2 baseret model skulle etableres for at påvise, at det var i stand til at opretholde langsigtede (5-10 dage) eksponering regimer og samtidig opretholde den proliferative kapacitet, der kræves for at støtte mikrokerne assay5. Faktisk har denne 3D-lever sfæroid model vist sig at bevare 70% levedygtighed over 10 dage i kultur. Baseret på dette og i forbindelse med den vedvarende leverlignende funktionalitet observeret i løbet af ≥14 dages kulturperiode kan denne 3D-leverspærremodel således understøtte langsigtede, gentagne ENM-eksponeringsordninger op til 10 dage lange (dvs. før sfæroidernes levedygtighed falder til under 70%). Til reference anbefales det, at albuminniveauerne for HepG2-sfæroider, der er seedet ved 4000 celler/sfæroide, skal ≥20,0 ng/μL, mens urinstofproduktionen skal ≥0,25 ng/μL, før der foretages en in vitro-toksikologisk vurdering med denne model.
Genotoksicitet Vurdering af manipulerede nanomaterialer
Ved vurdering af genotoksicitet blev mikrokerneanalysen anvendt til at bestemme tilstedeværelsen af mikrokerner efter både akutte (24 h) og langsigtede (120 h) ENM-eksponeringer. Aflatoksin B1 er en kendt lever kræftfremkaldende16,17 og er en anbefalet positiv kontrol for micronucleus assay. Optimeringseksperimenter har vist, at 0,1 μM Alfatoxin B1 fremkalder en betydelig positiv (≥2,0 fold stigning) genotoksisk respons i 3D HepG2 leverspærre og dermed anvendes i hver mikrokerneanalyse udført med denne model. For at sikre gyldigheden af mikrokerneanalyseresultaterne ved hjælp af HepG2-kugleformen skal baggrundsmikrokernefrekvensen for HepG2-celler, der anvendes i denne 3D-in vitro-model, ligge inden for et interval på 0,6% – 1,2%. Som følge heraf bør Alfatoxin B1 fremkalde en genotoksisk reaktion, der er mindst dobbelt højere end den, der ses med den negative kontrol; Således bør 0,1 μM Alfatoxin B1 fremkalde en mikrokernefrekvens mellem 1,5% – 3,0%. Ved hjælp af disse kontrolparametre kan enm-associeret genotoksicitet in vitro derefter vurderes pålideligt. På grundlag af OECD's testretningslinje 487 er det vigtigt at bemærke, at de udvalgte koncentrationer ved test af en ENM eller et kemikalie ikke må fremkalde mere end 55 % ± 5 % cytotoksicitet (angivet ved en reduktion i CPBI- eller RVCC-værdierne i forhold til den negative kontrol)11. Figur 7 illustrerer de data, der genereres, når aflatoksin B1 og to ENMs (titandioxid (TiO2) og splint (Ag)) blev evalueret efter både akutte og langsigtede eksponeringer i HepG2-sfæroiderne, og efterfølgende genotoksisk potentiale blev analyseret ved hjælp af mikrokerneanalysen. Begge vurderede ENM'er blev testet ved en ikke-cytotoksisk, lav dosis på 5,00 μg/mL over en akut (24 h) eksponering og langvarig (120 h) eksponeringsordning. En lignende tendens til genotoksicitet på tværs af både TiO2 og Ag ENMs kan observeres, hvorved den forhøjede genotoksicitetsrespons, der resulterede efter 24 timers eksponering, ikke var tydelig efter en langvarig 5-dages eksponering. Dette var på trods af vedvarende genotoksicitet forårsaget af aflatoksin B1 positiv kontrol på begge tidspunkter.

Figur 7: Genotoksicitetsvurdering efter TiO2- og Ag ENM-eksponering på HepG2-leversprædroider. Vurdering af genotoksicitet (mikrokernefrekvens) ved hjælp af mikrokerneanalyseposten (A) akut (24 timer) og (B) langvarig (120 timer) eksponering for 5,00 μg/mL TiO2 og Ag ENM. Negativ kontrol er kun et medie, mens den positive kontrol er 0,1 μM af aflatoksin B1. Gennemsnitlige data (n=2), der præsenteres ± SD. Betydning angivet i forhold til den negative kontrol: * = p≤ 0,05. Klik her for at se en større version af dette tal.
Discussion
Ansøgninger om 3D-levermodeller varierer betydeligt afhængigt af det særlige biokemiske slutpunkt eller den negative resultatvej, der målrettes mod. Hver model har sine fordele og begrænsninger, fra interdonor variation i primære menneskelige hepatocyt (PHH) modeller til reduceret cytochrom p450 aktivitet i celle-line baserede modeller, men alle er værdifulde i deres egen ret6,12,18,19. Ved vurdering af genotoksicitet er der begrænsninger i modellernes kompatibilitet med regulatoriske godkendte slutpunkter såsom in vitro-mikrokerneanalysen, da aktiv spredning er påkrævet. Dette er nødvendigt, da genotoksicitetsvurdering kræver kvantificering af fast DNA-skade, der skal vurderes efter celledeling, når der er mulighed for DNA-reparation for at korrigere forbigående læsioner. Desværre, meget differentieret hepatocyt (dvs. HepaRG) baseret sfæroider eller PHH mikrotissues, som anses for at udvise de mest fysiologisk relevante lever-lignende egenskaber form statisk (ikke-proliferative) modeller12,19,20. Som et resultat giver 3D HepG2-kugleformet model præsenteret her en passende, alternativ model, der er i stand til at understøtte genotoksicitetstest. HepG2 celle-line baseret sfæroider har tilstrækkelig aktivt dividere celler på ydersiden af spheroids samtidig opretholde grundlæggende lever-lignende egenskaber, såsom albumin og urinstof produktion og nogle CYP450 aktivitet5,12,19. Hovedsagelig denne in vitro lever model er blevet udviklet til at supplere micronucleus assay, da dette er en af de to in vitro assays anbefales i batteriet til genotoksicitet test8,10,11,21. Modellen kan dog let anvendes til DNA-sekventeringsanalyse og RNA-teknologier (Gene Expression), mens den har potentialet til at blive yderligere tilpasset og udnyttet til andre DNA-skadesslutpunkter, såsom kometanalysen. Ikke desto mindre er det vigtigt at overveje den rolle, som ENM-interferens spiller i nogle slutpunktsanalyser. F.eks. er flowcytometribaserede analyser muligvis ikke egnede til ENM-genotoksicitetsvurdering specifikt på grund af partikelinterferens22.
En begrænsende faktor for sfæroide modeller, der aktivt gennemgår celledeling, er deres størrelse. Optimering af såningstætheden er kritisk, da der skal være nok celler, der gør det muligt for modellen at fortsætte med at formere sig; men ikke for højt et cellenummer, hvilket resulterer i, at kugleformet bliver alt for kompakt, hvilket fører til en øget nekrotisk kerne. Årsagen til denne nekrose menes at være begrænset ilt og næringsstof diffusion, som grænsen for denne diffusion menes at være ca 100 – 150 μm væv23,24. Dette afhænger dog af celletype, cellenummer, stilladsinteraktioner og kulturforhold25. Siden har det vist sig, at ca. 700 μm diameter er grænsen for at undgå for tidlig nekrose i midten af C3A-sfæroider, såning 4000 HepG2-celler pr. kugleform sikrer, at modellens diameter på eksponeringstidspunktet er ≤500 μm26. Desuden fastslog Shah et al., at HepG2-celler, der var seedet over 5000 celler pr. kugleformet, udviste en 25% reduktion i levedygtigheden efter 7 dage i kultur, hvilket kunne vedrøre den gennemsnitlige diameter på 680 μm og begrænset tilgængelighed af næringsstoffer i en 20 μL hængende dråbe5. For at overvinde dette gennemgår modellen, der er udtænkt i denne protokol, et kritisk trin, hvor den hængende dråbe overføres til agarosebelagte brønde efter den første dannelse af kugleformet. Dette sikrer en større mængde af kultur medium er til stede for at opretholde det stadigt stigende antal celler i spheroids. Som følge heraf forbliver HepG2-kugleformet model over 70% levedygtig efter 10 dage i kultur og kan bruges til langsigtet farevurdering in vitro.
Mens HepG2 sfæroid model kan støtte både akut og langsigtet eksponering regimer, forfriskende cellekultur medium i længere kultur perioder er begrænset til denne model som fuldstændig udskiftning af mediet anbefales ikke på grund af det potentielle tab af spheroids. Det antages, at med ENM-eksponeringer er tendensen til homogene ENM-dispersioner til agglomerat og sediment høj. Det er imidlertid bemærkelsesværdigt, at den hastighed, hvormed en ENM-sedimenter kan variere afhængigt af partikelparametrene (f.eks. størrelse, form og tæthed) og kan bestemmes teoretisk ved hjælp af in vitro-sedimentations-, diffusions- og dosimetrimodellen (ISDD) eller dens nylige derivater, der ofte henvises til, når det drejer sig om ENM-eksponering (suspension) nærmer sig27,28. Med dette er sind, antages det, at hvis kun 50% af cellekulturmediet forsigtigt fjernes fra overfladen af cellekulturen, bør forstyrrelsen og den efterfølgende fjernelse af ENM-dosis i teorien være minimal. Med Brownian-bevægelse på spil er dette dog muligvis ikke strengt tilfældet, og der bør arbejdes videre med depositionen og sedimenteringen af hver enkelt ENM, der skal testes, for at sikre, at den korrekte dosimetri bevares under hele de langsigtede eksponeringsordninger27. Dette er hovedsagelig en potentiel begrænsning at overveje, når der udføres gentagne doseringsordninger, da dette kan være afgørende for den endelige akkumulerede koncentration. Kemisk baserede eksponeringer giver på den anden side, selv om de ikke er uden deres egne begrænsninger at overveje, en mere forenklet tilgang, idet kemiske stoffer har tendens til at forblive i opløsning, og en direkte erstatning af den oprindelige kemiske koncentration ud over den nyligt tilsatte koncentration sikrer, at ethvert kemikalie, der går tabt under medieforfriskning, erstattes i overensstemmelse hermed29. Fremtidige anvendelser vil omfatte en vurdering af modellens egnethed til gentagne eksponeringsordninger over længerevarende kulturperioder, da gentagne doseringsstrategier er af afgørende betydning for vurderingen af et bestemt organsystems evne til at forbedre eller overvinde eventuelle negative virkninger som følge af bioakkumulering af et xenobiotisk stof.
Afslutningsvis vil jeg sige, at denne 3D-in vitro-levermodel har kapacitet til at blive anvendt til at evaluere en række realistiske eksponeringsscenarier, hvilket giver en fremtidig in vitro-tilgang til bedre støtte til både ENM og kemikalierisikovurdering på en rutinemæssig og lettilgængelig måde.
Disclosures
Forfatterne har intet at afsløre.
Acknowledgments
Forfatterne vil gerne anerkende, at denne forskning har modtaget støtte fra EU's Horizon 2020-forsknings- og innovationsprogram for PATROLS-projektet i henhold til tilskudsaftale nr.
Materials
| Name | Company | Catalog Number | Comments |
| Aflotoxin B1 | Sigma Aldrich, UK | A6636-5MG | |
| Agarose | Sigma Aldrich, UK | A9539-50G | |
| Autoclave Tape | |||
| BCG Albumin Assay | Sigma Aldrich, UK | MAK124 | |
| Bovine Serum Albumin Powder | Sigma Aldrich, UK | A9418 | |
| Cell Freezing Aid | Thermo Fisher Scientific, UK | 5100-0001 - Mr Frosty | |
| Centrifuge | Eppendorf | 5810 R | |
| Cytochalasin B | Merck, UK | 250233 | |
| Cytology Metal Clips | |||
| Cytospin 4 Centrifuge | ThermoFisher Scientific, UK | CM00730202 | |
| DMEM with 4.5g/L D-Glucose, L-Glutamine | GIBCO, Paisley, UK | 41965-039 | |
| DMEM, phenol-red free with 4.5g/L D-Glucose, L-Glutamine with Hepes | GIBCO, Paisley, UK | 21063-029 | |
| DPX Mounting Medium | FisherScientific, UK | D/5330/05 | |
| Ethanol | FisherScientific, UK | 10048291 | |
| FBS | GIBCO, Paisley, UK | 10270-106 | |
| Filter Cards for Shandon Cytospin | FisherScientific, UK | 15995742 | |
| Frosted Glass Slides | ThermoFisher Scientific, UK | ||
| Giemsa's Stain Improved R66 Solution, Gurr | VWR Chemicals, UK | MFCD00081642 | |
| Glass Coverslips (24 x 60) | Deckglaser, VWR | ECN631-1575 | |
| Haemocytometer and Coverslip | |||
| Immersion Oil for Microscope | Zeiss, UK | 518F, ISO8034 | |
| Laminar Class II Tissue Culture Hood | Scanlaf Mars | ||
| Light Microscope | Zeiss, UK | Axiovert 40C | |
| Liquid Nitrogen | |||
| Methanol | FisherScientific, UK | 10284580 | |
| Microwave | |||
| Non-Filtered, Sterile 200µl and 1000µl Pipette tips | Greiner-Bio-One, UK | ||
| NuncMicroWell 96-Well Microplates | ThermoFisher Scientific, Denmark | 167008 | |
| P1000 and P200 micropipettes | |||
| P300 and P50 multi-channel pipettes | |||
| PBS pH 7.4 1X, MgCl2 and CaCl2 Free | GIBCO, Paisley, UK | 14190-094 | |
| Pen/Strep | GIBCO, Paisley, UK | 15140-122, Penicillin/Strepmyocin 100X or 10,000U/ml | |
| Phosphatase Buffer Tablets | GIBCO, Paisley, UK | 10582-013 | |
| Pipette Boy | |||
| Simport Scientific CytoSep Funnels for Shandon Cytospin 4 Centrifuges | FisherScientific, UK | 11690581 | |
| Sonifier SFX 550 240V CE 1/2" - Probe | Branson, USA | 101-063-971R | |
| T-25 and T-75 Tissue Culture Flask | Greiner-Bio-One, UK | T-25 (690175) and T-75 (660175) | |
| Trypan Blue Solution | Sigma Aldrich, UK | T8154-100mL | |
| Urea Assay Kit | Sigma Aldrich, UK | MAK006 | |
| Virkon Disinfectant | DuPont, UK | Rely+On Virkon | |
| Water Bath (37?C) | Grant JBNova 18 | ||
| Weighing Balance | |||
| Xylene | FisherScientific, UK | 10588070 | |
| 0.05% Trypsin-EDTA | GIBCO, Paisley, UK | 5300-054 | |
| 0.2mL and 1.0mL Eppendorf Tubes | Greiner-Bio-One, UK | ||
| 0.45µm Filter Unit | Millex HA, MF-Millipore, UK | SLHA033SS | |
| 1.0mL Syringe | BD Plastipak, FisherScientific, UK | 300185 | |
| 20mL LS Scintillation Glass Vials, 22-400 Foil Lined PP Caps | DWK Life Sciences GmbH, Germany | WHEA986581 | |
| 37?C and 5% CO2 ISO Class 5 Hepa Filter Incubator | NUAIRE DHD Autoflow | ||
| 3mL Pasteur Pipette | Greiner-Bio-One, UK | ||
| 50mL Conical Falcon Tubes | Greiner-Bio-One, UK | ||
| 50mL or 100mL Glass Bottles | |||
| 50mL Skirted Falcon Tubes | Greiner-Bio-One, UK | ||
| 5mL, 10mL and 25mL Pipettes | Greiner-Bio-One, UK | ||
| 9.4cm Square, Petri Dish | Greiner-Bio-One, UK | 688161 |
References
- Geiser, M., Kreyling, W. G. Deposition and biokinetics of inhaled nanoparticles. Particle and Fibre Toxicology. 7, 2 (2010).
- Modrzynska, J. Toxicological effects of nanoparticle deposition in the liver. Kgs. Lyngby, Denmark: Technical University of Denmark. , (2018).
- Elje, E., et al. The comet assay applied to HepG2 liver spheroids. Mutation Research/Genetic Toxicology and Environmental Mutagenesis. 845, 403033 (2019).
- Breslin, S., O'Driscoll, L. Three-dimensional cell culture: the missing link in drug discovery. Drug Discovery Today. 18, 240-249 (2013).
- Shah, U. -K., et al. A three-dimensional in vitro HepG2 cells liver spheroid model for genotoxicity studies. Mutation Research/Genetic Toxicology and Environmental Mutagenesis. 825, 51-58 (2018).
- Lauschke, V. M., Hendriks, D. F. G., Bell, C. C., Andersson, T. B., Ingelman-Sundberg, M. Novel 3D Culture Systems for Studies of Human Liver Function and Assessments of the Hepatotoxicity of Drugs and Drug Candidates. Chemical Research in Toxicology. 29, 1936-1955 (2016).
- van Grunsven, L. A. 3D in vitro models of liver fibrosis. Advanced Drug Delivery Reviews. 121, 133-146 (2017).
- Corvi, R., Madia, F. In vitro genotoxicity testing - can the performance be enhanced. Food and Chemical Toxicology. 106, 600-608 (2017).
- Doak, S. H., Manshian, B., Jenkins, G. J. S., Singh, N. In vitro genotoxicity testing strategy for nanomaterials and the adaptation of current OECD guidelines. Mutation Research/Genetic Toxicology and Environmental Mutagenesis. 745, 104-111 (2012).
- Fenech, M. Cytokinesis-block micronucleus cytome assay. Nature Protocols. 2, 1084-1104 (2007).
- OECD. OECD Guidelines. Test 489: In vivo Mammalian Alkaline Comet Assay. , (2016).
- Gerets, H. H. J., et al. Characterization of primary human hepatocytes, HepG2 cells, and HepaRG cells at the mRNA level and CYP activity in response to inducers and their predictivity for the detection of human hepatotoxins. Cell Biology and Toxicology. 28, 69-87 (2012).
- Sison-Young, R. L., et al. A multicenter assessment of single-cell models aligned to standard measures of cell health for prediction of acute hepatotoxicity. Archives of Toxicology. 91, 1385-1400 (2017).
- European Guidelines 2019. European Agency for Safety and Health at Work. , Available from: https://osha.europa.eu/en/safety-and-health-legislation/european-guidelines (2019).
- Jensen, K. A. The NANOGENOTOX Dispersion Protocol for NANoREG. European Union Grant Agreement n° 2009. 21, 01 (2014).
- Marchese, S., et al. Aflatoxin B1 and M1: Biological Properties and Their Involvement in Cancer Development. Toxins. 10, 214 (2018).
- Rushing, B. R., Selim, M. I. Aflatoxin B1: A review on metabolism, toxicity, occurrence in food, occupational exposure, and detoxification methods. Food and Chemical Toxicology. 124, 81-100 (2019).
- Kermanizadeh, A., Brown, D. M., Moritz, W., Stone, V. The importance of inter-individual Kupffer cell variability in the governance of hepatic toxicity in a 3D primary human liver microtissue model. Scientific Reports. 9, 7295 (2019).
- Berger, B., et al. Comparison of Liver Cell Models Using the Basel Phenotyping Cocktail. Frontiers in Pharmacology. 7, 443 (2016).
- Ramaiahgari, S. C., et al. A 3D in vitro model of differentiated HepG2 cell spheroids with improved liver-like properties for repeated dose high-throughput toxicity studies. Archives of Toxicology. , (2014).
- Li, Y., et al. Factors affecting the in vitro micronucleus assay for evaluation of nanomaterials. Mutagenesis. 32 (1), 151-159 (2016).
- Kirkland, D., Reeve, L., Gatehouse, D., Vanparys, P. A core in vitro genotoxicity battery comprising the Ames test plus the in vitro micronucleus test is sufficient to detect rodent carcinogens and in vivo genotoxins. Mutation Research/Genetic Toxicology and Environmental Mutagenesis. 721 (1), 27-73 (2011).
- Curcio, E., et al. Mass transfer and metabolic reactions in hepatocyte spheroids cultured in rotating wall gas-permeable membrane system. Biomaterials. 28, 5487-5497 (2007).
- Glicklis, R., Merchuk, J. C., Cohen, S. Modeling mass transfer in hepatocyte spheroids via cell viability, spheroid size, and hepatocellular functions. Biotechnology and Bioengineering. 86, 672-680 (2004).
- Asthana, A., Kisaalita, W. S. Microtissue size and hypoxia in HTS with 3D cultures. Drug Discovery Today. 17, 810-817 (2012).
- Gaskell, H., et al. Characterization of a functional C3A liver spheroid model. Toxicology Research. 5, 1053-1065 (2016).
- Cho, E. C., Zhang, Q., Xia, Y. The effect of sedimentation and diffusion on cellular uptake of gold nanoparticles. Nature Nanotechnology. 6, 385-391 (2011).
- Hinderliter, P. M., et al. ISDD: A computational model of particle sedimentation, diffusion and target cell dosimetry for in vitro toxicity studies. Particle and Fiber Toxicology. 7, 36 (2010).
- Kramer, N. I., di Consiglio, E., Blaauboer, B. J., Testai, E. Biokinetics in repeated-dosing in vitro drug toxicity studies. Toxicology in vitro. 30, 217-224 (2015).
Tags
Bioengineering Problem 160 In Vitro Levermodeller Nanomaterialer Farevurdering Langtidseksponering Nano(geno)toksikologi DNA-skaderErratum
Formal Correction: Erratum: Advanced 3D Liver Models for In vitro Genotoxicity Testing Following Long-Term Nanomaterial Exposure
Posted by JoVE Editors on 01/26/2021.
Citeable Link.
An erratum was issued for: Advanced 3D Liver Models for In vitro Genotoxicity Testing Following Long-Term Nanomaterial Exposure. The Representative Results section was updated.
Figure 6 in the Representative Results section was updated from:
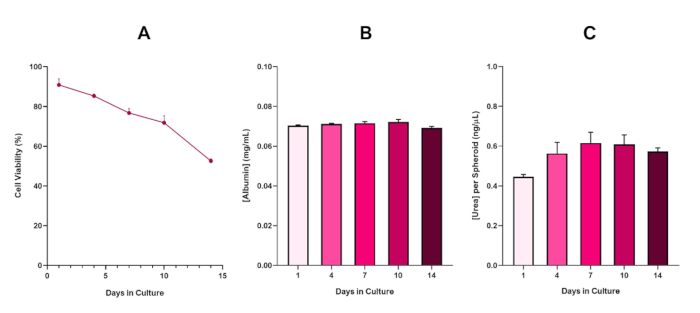
to:
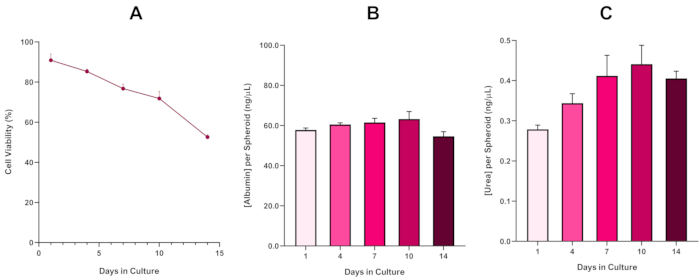
The fourth paragraph in the Representative Results section was updated from:
With the inevitable development of a necrotic core, a known limitation of 3D liver spheroid cultures, the viability of this HepG2 based model had to be established to demonstrate it was able to sustain long-term (5-10 day) exposure regimes whilst maintaining the proliferative capability required to support the micronucleus assay5. Indeed, this 3D liver spheroid model has been shown to retain >70% viability over 10 days in culture. Based on this and in conjunction with the sustained liver-like functionality observed over the ≥14 day culture period, this 3D liver spheroid model can thus support long-term, repeated ENM exposure regimes up to 10 days long (i.e., before viability of the spheroids drop below 70%). For reference, it is advised that albumin levels for HepG2 spheroids seeded at 4000 cells/spheroid should be ≥0.06 mg/mL whilst urea production should be ≥0.4 ng/µL before conducting an in vitro toxicological assessment with this model.
to:
With the inevitable development of a necrotic core, a known limitation of 3D liver spheroid cultures, the viability of this HepG2 based model had to be established to demonstrate it was able to sustain long-term (5-10 day) exposure regimes whilst maintaining the proliferative capability required to support the micronucleus assay5. Indeed, this 3D liver spheroid model has been shown to retain >70% viability over 10 days in culture. Based on this and in conjunction with the sustained liver-like functionality observed over the ≥14 day culture period, this 3D liver spheroid model can thus support long-term, repeated ENM exposure regimes up to 10 days long (i.e., before viability of the spheroids drop below 70%). For reference, it is advised that albumin levels for HepG2 spheroids seeded at 4000 cells/spheroid should be ≥50.0 ng/μL whilst urea production should be ≥0.25 ng/µL before conducting an in vitro toxicological assessment with this model.
