

ERRATUM NOTICE
Important: There has been an erratum issued for this article. Read more …
Summary
Dieses Verfahren wurde für die Entwicklung fortgeschrittener 3D-Leberkulturen in vitro eingeführt, die eine physiologisch relevantere Bewertung der genotoxischen Gefahren im Zusammenhang mit Nanomaterial-Expositionen sowohl über ein akutes als auch ein langfristiges, wiederholtes Dosisregime ermöglichen können.
Abstract
Aufgrund der schnellen Entwicklung und Implementierung einer Vielzahl von technischen Nanomaterialien (ENM) ist eine Exposition gegenüber ENM unvermeidlich und die Entwicklung robuster, prädiktiver In-vitro-Testsysteme ist unerlässlich. Die Hepatische Toxikologie ist der Schlüssel bei der Betrachtung der ENM-Exposition, da die Leber eine wichtige Rolle bei der metabolischen Homöostase und Entgiftung spielt und ein wichtiger Ort der ENM-Akkumulation nach der Exposition ist. Basierend auf diesem und dem akzeptierten Verständnis, dass 2D-Hepatozytenmodelle die Komplexität komplizierter multizellulärer Wechselwirkungen und metabolischer Aktivität, die in vivo beobachtet werden, nicht genau imitieren, liegt ein größerer Fokus auf der Entwicklung physiologisch relevanter 3D-Lebermodelle, die auf ENM-Gefahrenbewertungszwecke in vitro zugeschnitten sind. In Übereinstimmung mit den Prinzipien der 3Rs, um Tierversuche zu ersetzen, zu reduzieren und zu verfeinern, wurde ein 3D HepG2 Zelllinien-basiertes Lebermodell entwickelt, das ein benutzerfreundliches, kostengünstiges System ist, das sowohl erweiterte als auch wiederholte ENM-Expositionsregime (≤14 Tage) unterstützen kann. Diese Sphäroidmodelle (≥500 m Durchmesser) behalten ihre proliferative Kapazität (d. h. Zellteilungsmodelle) und können so mit dem Mikrokernassay "Goldstandard" gekoppelt werden, um die Genotoxizität in vitro effektiv zu bewerten. Ihre Fähigkeit, über eine Reihe von toxikologischen Endpunkten zu berichten (z. B. Leberfunktion, (Pro-)Entzündungsreaktion, Zytotoxizität und Genotoxizität) wurde mit mehreren ENMs sowohl in akuten (24 h) als auch in langfristigen (120 h) Expositionsregimen charakterisiert. Dieses 3D-In-vitro-Hepatikmodell kann für die Bewertung realistischerer ENM-Expositionen genutzt werden und bietet damit einen zukünftigen In-vitro-Ansatz, um die ENM-Risikobewertung routinemäßig und leicht zugänglich zu unterstützen.
Introduction
Aufgrund der raschen Entwicklung und Umsetzung einer Vielzahl von technischen Nanomaterialien (ENM) in einer Vielzahl von anwendungen auf menschlicher Basis (z. B. Lebensmittel, Kosmetik, Bekleidung, Sportausrüstung, Elektronik, Transport und Medizin) ist es unvermeidlich, dass der Mensch regelmäßig ENM ausgesetzt wird. Damit wächst die Sorge, dass die neuartigen, größenspezifischen physiochemischen Eigenschaften, die diese Materialien in zahlreichen Anwendungen für vorteilhaft erachten, gleichzeitig negative Auswirkungen auf die menschliche Gesundheit und die Umwelt haben könnten. Derzeit gibt es viele internationale Aktivitäten, um physiologisch relevantere Expositionen gegenüber diesen ENM aktiv widerzuspiegeln und die potenzielle Toxizität dieser Materialien über akute, langfristige und wiederholte Szenarien mit niedriger Dosis exposition zu bewerten.
Hepatische Toxikologie ist der Schlüssel bei der Betrachtung der ENM-Exposition, da allgemein bekannt ist, dass die Leber ein wichtiger Ort der ENM-Akkumulation nach der Exposition1,2ist. Darüber hinaus ist die Leber das primäre Organsystem für den Stoffwechsel und die Entgiftung von Substanzen, die in den systemischen Kreislauf gelangen3. Basierend auf dem akzeptierten Verständnis, dass 2D-Hepatozytenmodelle die Komplexität komplizierter multizellulärer Wechselwirkungen nicht genau imitieren oder die in vivo beobachtete metabolische Aktivität angemessen darstellen, wurde ein größerer Fokus auf die Entwicklung robuster und physiologisch relevanter In-vitro-3D-Lebermodelle für In-vivo-Ersatztechnologien festgelegt4,5. Der Einsatz fortschrittlicher 3D-Kulturtechnologien verbessert die Langlebigkeit von In-vitro-Lebermodellen, so dass langfristige, wiederholte Expositionsregime untersucht werden können. Darüber hinaus fördert dieses fortschrittliche Kulturformat die Bildung verbesserter physiologischer, organotypischer Merkmale wie Gallenkanaliculi, aktive Transporterprozesse und verbesserte CYP450-Wirkstoffmetabolisierungsfähigkeiten, wodurch die Prädiktodibilität der Modelle6verbessert wird. Aktuelle 3D-In-vitro-Hepatikmodelle, die aus Monokulturen (nur Hepatozyten) oder Kokulturen (Hepatozyten mit nichtparenchymalen Zellen) bestehen, existieren in mehreren Formaten, von Mikrotissues oder Sphäroiden in ultraniedrigen Haftplatten, hängenden Tropfensphäroiden, In Matrizen und/oder Gerüsten eingebetteten Zellen und mikrofluidischen Zellkulturplattformen, die alle als effektive fortgeschrittene In-vitro-Modelle für die Beurteilung der Lebertoxizität6,7gelten. Die meisten dieser Modellsysteme sind jedoch wartungsarm, erfordern spezielle Ausrüstung und sind teuer. Darüber hinaus sind diese Modelle häufig statisch (d. h. nicht dividierende Zellmodelle), die ihre Verwendung bei der Bewertung von Gefahrenendpunkten verhindern, wie z. B. Genotoxizitätstests mit Methoden, die feste DNA-Schäden quantifizieren. Genotoxizität ist eine wesentliche Voraussetzung in der regulatorischen Toxikologie, und es ist ein wichtiger Bestandteil der Risikobewertung von giftem Mittel8. Es gibt keinen einzigen Test, der angewendet werden kann, um alle Formen von DNA-Schäden zu quantifizieren, die nach der Exposition gegenüber einem exogenen Mittel auftreten können. Ein Kernbestandteil der In-vitro-Genotoxizitätstestbatterie ist jedoch der Mikrokern-Assay, eine zuverlässige und facettenreiche Technik, die grobe Chromosomenschäden misst9. Es handelt sich um eine Goldstandardtechnik, die von der OECD-Testleitlinie 487 zur Beurteilung von In-vitro-DNA-Schäden und Genotoxizität beschrieben wird, und ist Teil der Prüfbatterieanforderung für die regulatorische Risikobewertung10,11.
Die humane hepatozelluläre Karzinom-Zelllinie, HepG2, wird häufig für das erste Risikobewertungsscreening verwendet, da die Zellen leicht verfügbar, relativ kostengünstig zu beschaffen, einfach zu kulturierbar und für ein Durchsatzscreening12,13zugänglich sind. Wenn sie in 3D-Sphärische Strukturen kultiviert werden, haben sie gezeigt, dass sie die Mikroumgebung der Leber gut rekapitulieren und ein Lebermodell mit ausreichenden proliferativen Fähigkeiten bieten, um den Mikrokern-Assay3zu unterstützen. Die Weiterentwicklung der HepG2-Sphäroidmodelle wurde etabliert, um die Langlebigkeit und Leber-ähnliche Funktionalität des Modells zu verbessern, um die Bewertung der Genotoxizitätsgefährdung über langfristige, wiederholte Expositionsregime (≤14 Tage) zu unterstützen. Daher wurde das vorliegende Protokoll gemäß den Grundsätzen der 3Rs, die Tierversuche ersetzen, reduzieren und verfeinern sollen, eingeführt, um ein fortschrittliches 3D-In-vitro-Hepatikmodell bereitzustellen, das in der Lage ist, mehrere toxikologische Endpunkte (z. B. Leberfunktionalität, (Pro-)Entzündungsmarker, Zytotoxizität und Genotoxizität) nach akuten, langfristigen und wiederholten chemischen und ENM-Expositionen routinemäßig und leicht zugänglich zuverlässig zu bewerten.
Hier stellen wir eine Methode zur Etablierung eines physiologisch relevanten 3D-Hepatozyten-Zelllinien-basierten In-vitro-Modellsystems zur Bewertung der Genotoxizitätsgefährdung nach akuten oder langfristigen, wiederholten ENM-Expositionen vor. Das Protokoll kann in 6 Schlüsselstufen unterteilt werden: Kultivierung kryokonservierter HepG2-Zellen; HepG2 Sphäroid Vorbereitung; HepG2 Sphäroid Transfer von hängenden Tropfen zu Agarose Suspension; HepG2 Sphäroid Ernte; Mikrokern-Assay und Scoring; datenanalyse.
Protocol
1.Culturing kryokonservierte HepG2-Zellen
HINWEIS: HepG2-Zellen, die aus der American Type Culture Collection (ATCC) gewonnen wurden, wurden in 1x Dulbecco es Modified Eagle Medium (DMEM) mit 4,5g/L D-Glucose und L-Glutamin kultiviert, ergänzt mit 10% fetalem Rinderserum (FBS) und 1% Penicillin/Streptomycin-Antibiotikum.
- Vorwarme DMEM Zellkultur Medium (einschließlich der Ergänzungen) in einem 37 °C Wasserbad für 30 min.
- Entfernen Sie eine Durchstechflasche mit HepG2-Zellen aus flüssigem Stickstoff und tauen Sie in einem 37 °C-Wasserbad für 2-3 min auf, während Sie die Durchstechflasche sanft wirbeln, um ein gleichmäßiges Auftauen der Zellsuspension zu ermöglichen. Achten Sie darauf, die Durchstechflasche über dem O-Ring nicht zu versenken, um das Kontaminationspotenzial zu verringern.
- Nach dem Auftauen die Durchstechflasche aus dem Wasserbad nehmen und großzügig mit 70% Ethanol besprühen, um die Außenfläche der Durchstechflasche zu dekontaminieren, bevor sie unter eine sterile, laminare Gewebekulturhaube der Klasse II gestellt wird.
- Den Inhalt des Kryovials von HepG2-Zellen sorgfältig in ein Zentrifugenrohr mit 9 ml vorgewärmtem DMEM-Zellkulturmedium (mit Nahrungsergänzungsmitteln) zu führen.
- Mit einem 10 ml Streifenpette 10 ml der Zellsuspension in einen 25 cm2 Einweg-Zellkulturkolben übertragen und die Kultur 3 Tage lang (von der Aussaat) bei 5%CO2 und 37 °C bebrüten, bis die Konfluenz von 80 % erreicht wird, bevor eine Subkultur in einen größeren 75 cm2 Einwegzellkulturkolben eingeflossen ist.
- Sobald 80% Konfluenz erreicht ist, subkulturzellen unter sterilen Bedingungen durch Trypsinisierung mit 0,05% Trypsin/EDTA-Lösung vorgewärmt in einem 37°C Wasserbad für 30 min. Zu keinem Zeitpunkt sollten die Zellen austrocknen dürfen.
- Wenn Zellen eine anhehrende Monoschicht bilden, entfernen Sie das Medium, indem Sie in einen Desinfektions-Abfalltopf kippen. Waschen Sie dann sofort die Monoschicht, um alle Spuren vorhandener Medien zu entfernen, indem Sie den Kolben zweimal mit 3 ml steriler 1x PBS-Lösung bei Raumtemperatur spülen. Entsorgen Sie PBS auch in den Desinfektionsmittel-Abfalltopf.
- Sobald PBS-Waschanlage entfernt ist, fügen Sie 5 ml vorgewärmte 0,05% Trypsin-EDTA-Lösung hinzu, um die gesamte Oberfläche der Zellen und Inkubationszellen für 6-8 min bei 37 °C und 5%CO2zu bedecken.
- Tippen Sie vorsichtig auf den Kolben, um die Zellen von der Unterseite des Kolbens zu entfernen und fügen Sie dann 5 ml DMEM Zellkulturmedium (mit Ergänzungen) hinzu, um das Trypsin-Enzym zu neutralisieren.
- Übertragen Sie die Zellsuspension in ein 50 ml Zentrifugenrohr und pipette die Zellsuspension gründlich nach oben und unten, um sicherzustellen, dass die Zellen vollständig getrennt sind.
- Zentrifugieren Sie die verdünnte Zellsuspension bei 230 x g für 5 min. Entsorgen Sie den Überstand in Desinfektionsmittel und suspendieren Sie das Zellpellet in 25 ml DMEM-Zellkulturmedium (mit Ergänzungen).
- Zellsuspension in einen 75 cm2 Einweg-Zellkulturkolben übertragen und bei 37 °C und 5%CO2 weitere 3 Tage vor der Sphäroidzubereitung inkubieren. Sobald die HepG2s Zeit hatten, sich zu akklimatisieren und wieder 80% Konfluenz zu erreichen, bestimmen Sie die Zellkonzentration in Vorbereitung auf die Sphäroid-Saat.
2. HepG2 Sphäroid-Vorbereitung
- Wiederholen Sie die oben genannten Subkulturschritte, außer nach der Zentrifugation, setzen Sie das Zellpellet in 1 ml DMEM-Kulturmedium wieder auf, das in einem 37 °C-Wasserbad vorgewärmt wird. Pipette-Zell-Suspension nach oben und unten gründlich.
- Score Zelllebensfähigkeit mit dem Trypan Blue Exclusion Assay (siehe OSHA SOP 3.21 Reproductive Toxins, Mutagens, Teratogens and Embryotoxins – Procedures for Safe Handling and Storage (2019) for health and safety guidance)14 with a 1:1 ratio of cell suspension to vor-filtered 0.4% Trypan blue solution.
- Vor der Zellzählung 1 ml Trypan-Blaulösung mit einer 1 ml Spritze und Filter mit einer 0,45-mm-Filtereinheit in ein steriles, 1 ml-Rohr nehmen.
- Übertragen Sie 10 l gefilterte, Trypan-blau-Lösung in ein 0,2 ml-Rohr und fügen Sie 10 l Zellsuspension hinzu. Die verbleibende gefilterte Trypan blue Lösung kann bis zu 3 Monate bei Raumtemperatur für die zukünftige Verwendung gelagert werden.
- Das Hämozytometer gründlich mit 70% Ethanol besprühen und mit einem sterilen Papiertuch trocken wischen, bevor der Deckelrutsch oben mit Atemdampf gesichert wird. Das Schieben des Deckelrutsches über die atmungsaktive Oberfläche induziert durch die Erzeugung von Newton-Ringen kohäsive Kräfte.
- Pipette der Trypan-Blauzellenaufhängung nach oben und unten mit einer 1000-L-Pipette (zur Reduzierung der Schiere-Beanspruchung), bevor dem Hämozytometer 10 l hinzugefügt werden. Stellen Sie sicher, dass die Lösung unter dem Deckelschlupf verteilt ist und das gesamte Netz ohne Luftblasen abdeckt.

Abbildung 1:Zählen von Zellen mit einem Hämozytometer. Diagrammmatische Darstellung eines Hämozytometers, der hervorhebt, aus welchem Quadranten Zellen gezählt werden sollen. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
- Zählen Sie unter dem Mikroskop die lebenden (unbefleckten) und toten (blau gefärbten) Zellen, die in den vier großen Eckquadraten gefunden wurden (Abbildung 1). Schließen Sie alle Zellen aus, die sich überlappen oder auf den inneren zwei Kanten der großen Eckquadrate (d. h. auf den Linien) in der Anzahl sitzen.
- Berechnen Sie anhand der folgenden Berechnung die durchschnittliche Anzahl lebender, lebensfähiger Zellen (unbefleckt) in der Stichprobe:
Gesamtzahl der Zellen/ml = Anzahl der Lebendzellen x x 10.000
x 10.000
wobei sich die Verdünnung darauf bezieht, wie oft die Stammlösung in Trypan blau verdünnt wurde (in diesem Fall 2x) und die Anzahl der gezählten Quadrate sich auf die vier großen Eckquadrate des haemozytometers bezieht, die gezählt wurden - Basierend auf der lebensfähigen HepG2-Zellzahl und mit der folgenden Formel:
C1V1=C2V2
wobeiC1 = die Konzentration lebensfähiger Zellen derzeit,
V1 = das Volumen der Zellsuspension derzeit,
C2 = Konzentration der Zellsuspension gewünscht,
V2 = volumender Zellsuspension gewünscht - Bereiten Sie eine 10 ml-Stammlösung der HepG2-Zellsuspension mit DMEM-Zellkulturmedium mit einer Konzentration von 2,0 x 105 Zellen/ml vor, um 4000 HepG2-Zellen pro 20 L hängendem Tropfen zu erreichen. Mischen Sie die Zellsuspension gründlich, indem Sie mit einer 1000-L-Pipette vorsichtig nach oben und unten pipetieren, um sicherzustellen, dass alle Zellen vollständig in den Medien aufgehängt sind.
- Zu den Brunnen einer 96-Well-Zellkulturplatte, fügen Sie 100 l sterile Raumtemperatur PBS, um zu verhindern, dass die hängenden Tropfen während der Inkubation austrocknen.
- Nehmen Sie den Deckel einer Standard-Flachboden 96-Well-Zellkulturplatte, invertieren Sie sie und pipette20 L Tropfen der Zellsuspension vorsichtig in die Mitte jeder Bohrung des Deckels, wie in Abbildung 2gezeigt. Verwenden Sie eine Mehrkanal-Pipette, aber fügen Sie nur 2 - 4 Tropfen auf einmal hinzu, da mehrere Aussaaten die Genauigkeit und Platzierung der Tropfen beeinflussen können.
- Zentrieren Sie die Tropfen innerhalb der Rillen der Brunnen auf dem Deckel ausgelegt; andernfalls hängen sie nicht in der Mitte der Brunnen, wenn der Deckel der Platte umgedreht wird und laufen Gefahr, in die Platte abzufallen. Drehen Sie den Deckel der 96-Well-Platte vorsichtig um, so dass die Tropfen nun hängen und vorsichtig auf die 96-Well-Platte legen.
- Legen Sie die gesamte 96-Wellplatte mit Deckel vorsichtig bei 37 °C und 5% CO2 3 Tage vor dem Sphäroidtransfer auf Agarose sanft in einen Inkubator.
HINWEIS: Beim Transport der Platten zu/von den Brutkästen ist besonders vorsichtig zu sein, sondern beim Öffnen und Schließen des Inkubators im Allgemeinen, da übermäßige Bewegung dazu führen kann, dass sich die Platten verschieben und die Sphäroide entweder fallen oder sich falsch bilden.
 x 10.000
x 10.000
Abbildung 2: 3D HepG2 in vitro Sphäroid Modellvorbereitung. (A) Die HepG2-Zellen in 20 l Samen fällt auf den Deckel einer 96-Well-Platte. (B) Die HepG2-Zellen nach dem Aussaat im hängenden Tropfenmodell, um eine Sphäroidbildung zu ermöglichen. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
3. HepG2 Sphäroidtransfer vom hängenden Tropfen zur Agarose-Suspension
HINWEIS: Am Tag 3 nach der Aussaat in hängende Tropfen werden die Sphäroide in die Brunnen der gleichen 96-Well-Platte übertragen, die alle zuvor mit einer feinen Schicht von 1,5% Agarose-Gel beschichtet wurden.
- Bereiten Sie Agarose-Gele und Autoklaven (d. h. Tag 2 nach der Aussaat) vor dem Tag der Plattenbeschichtung (d. h. Tag 3 nach der Aussaat).
- Um ein 1,5% Agarose-Gel zuzubereiten, 0,30 g Agarose in eine saubere Glasflasche wiegen und dann 20 ml phenolrotes DMEM-Medium hinzufügen. Autoklavieren Sie die Agarose für 1 h bei 230 °C zur Sterilisation. Die Agarose-Beschichtung verhindert, dass die HepG2-Sphäroide an der Basis von Brunnen haften und eine zelluläre Monoschicht bilden, anstatt ihre 3D-Sphäroidstruktur beizubehalten.
- Entfernen Sie am 3. Tag nach der Aussaat die 96-Well-Platte mit den HepG2 hängenden Tropfensphäroiden aus dem Inkubator und kippen Sie den Deckel vorsichtig um, damit die Sphäroide nicht mehr hängen.
- Entfernen und entsorgen Sie mit einer Mehrkanalpipette die 100 L PBS, die zuvor der Basis der 96-Well-Platte hinzugefügt wurden. Lassen Sie die Platten für 2-3 min lufttrocknen, während Sie die Agarose in Vorbereitung auf die Beschichtung erhitzen.
VORSICHT: Dieses Verfahren führt zu sehr heißem, flüssigem Agrose, das bei Vergossen auf die Haut brennen und Verletzungen verursachen kann. Darüber hinaus ist beim Umgang mit der Glasflasche, die die flüssige Agarose enthält, Vorsicht geboten, da auch diese sehr heiß sein kann. - Mit den zuvor hergestellten 1,5% Agarose-Gelen die Glasflasche mit dem 20 ml Agarose-Gel für 30 s in einer Mikrowelle mit maximal watt (d.h. 900 W) erhitzen. Um zwei 96-Well-Platten zu beschichten, sollte eine 20 ml Flasche vorgefertigtem 1,5% Agarose-Gel ausreichen.
- Nach dem Schmelzen die Agarose vorsichtig wirbeln, indem Sie die Glasflasche drehen, um Blasen zu entfernen, und dann 50 l Agarose in die Basis jedes Brunnens geben.
HINWEIS: Achten Sie beim Hinzufügen der Agarose darauf, die Platte nicht zu winkeln >45°, da sich die Agarose schnell festlegt und keine flache, ebene Schicht bildet, die das Sphäroidwachstum stören kann. Es ist wichtig, in diesem Stadium effizient zu arbeiten, um zu verhindern, dass sich die Agarose verfestigt, bevor die Platte vollständig beschichtet wird. - Lassen Sie die Platte für 2 min bei Raumtemperatur stehen, bevor Sie 100 l vorgewärmtes DMEM-Zellkulturmedium (mit Ergänzungen) auf die feste Agaroseschicht in jedem Brunnen hinzufügen.
- Drehen Sie den Deckel der 96-Well-Platte und legen Sie wieder auf die Oberseite der 96-Well-Platte, so dass die Sphäroide jetzt wieder hängen.
- Zentrifugieren Sie die Platte für 3 min bei 200 x g, um die Sphäroide aus dem hängenden Tröpfchen in die einzelnen Brunnen der 96-Well-Platte zu übertragen. Nach der Übertragung sollten die HepG2-Sphäroide nun im Zellkulturmedium ausgesetzt werden. Lassen Sie sie sich mit 24 h im Inkubator bei 37 °C und 5%CO2absetzen.
- HepG2-Sphäroide dieser Größe entweder chemischen oder ENM-Behandlungen am Tag 4 nach der Aussaat aussetzen (d. h. 24 h nach dem Transfer auf Agarose-beschichtete Platten).
- Um die Zelllebensfähigkeit über längere Kulturperioden zu erhalten, aktualisieren Sie das Zellkulturmedium alle 3 Tage. Um dies zu tun, saugen Sie sanft 50 l des Zellkulturmediums von der Oberfläche des Brunnens und ersetzen Sie durch ein frisches 50 l DMEM Zellkulturmedium. Achten Sie darauf, das Sphäroid nicht zu entfernen oder zu stören, wenn Sie einen Mittlerenwechsel durchführen.
4. Nanomaterial/Chemische Exposition
HINWEIS: Das HepG2-Lebersphäroidmodell kann sowohl ENM- als auch chemikalienbasierte Expositionsregime unterstützen, aber der Hauptfokus dieses Protokolls liegt auf ENM-Expositionen. Vor der Exposition muss die Prüfung ENM entsprechend dispergiert sein; dies kann nach anweisungsgemäß durch das NanoGenoTox Dispersion Protocol (Grant Agreement No. 20092101, 2018)15durchgeführt werden.
- Nach der Dispersion nach dem NanoGenoTox Dispersionsprotokoll die ENM-Suspension von der Anfangskonzentration von 2,56 mg/ml auf die endgültige gewünschte Konzentration im vorgewärmten DMEM-Zellkulturmedium (einschließlich der Ergänzungen) verdünnen. Zur Dosierung einer 96-Well-Platte ist ein Gesamtvolumen von 5 ml erforderlich.
- Um das HepG2-Sphäroid entweder einer Chemikalie oder einem ENM auszusetzen, indem man eine 200-L-Pipette verwendet, saugen Sie 50 l Zellkulturmedium von der Oberfläche jedes Brunnens ab (wobei 50 l im Brunnen bleiben, um die Sphäroide nicht zu stören) und durch 50 l Medium zu ersetzen, das das Testgiftmittel in der erforderlichen Dosis enthält.
- Nach dem Aufbringen des Prüfmaterials die Platten für die gewünschte Belichtungszeit bei 37 °C und 5%CO2inkubieren.
- Wenn ein langfristiges (≥24 h) Expositionsregime durchgeführt wird, dann ernten Sie unmittelbar nach Ablauf des gewünschten Expositionszeitraums die Sphäroide für die Mikrokernendpunktanalyse, wie unten in den Schritten 6.1 – 6.4 beschrieben.
- Bei akuten Expositionsregimen (z. B. ≤24 h) werden jedoch nach Ablauf der Expositionsperiode 50 L Überstand aus jedem Brunnen in der 96-Wellplatte bei -80 °C für weitere biochemische Analysen später geerntet und gelagert. Ersetzen Sie das Zellkulturmedium durch 50 l frisches Medium, das 6 g/ml Cytochalasin B enthält, und lassen Sie es für 1 – 1,5 Zellzyklen (d. h. 24 – 26 h für HepG2) in Vorbereitung auf die Zytokineseblock-Mikrokern-Assay-Ernte inkubieren.
HINWEIS: Bei akuten (≤24 h) Expositionsregimen kann der Zytokineseblock-Mikrokern-Assay mit Cytochalasin B angewendet werden, aber für langfristige (≥24 h) Expositionsregime muss die mononukleare Version (ohne Cytochalasin B) des Assays wie unten in Abbildung 4beschrieben verwendet werden.
5. HepG2 Sphäroid-Ernte
HINWEIS: Nach chemischen oder ENM-Expositionsbehandlungen können sowohl Zellkulturmedium als auch Sphäroidgewebe für die Analyse mehrerer Endpunkte geerntet werden. Je nach Endpunktanalyse können Sphäroide entweder einzeln geerntet (z.B. für die Bildanalyse) oder zusammengepoolt werden (z.B. für Zytokinese-Mikrokern-Assay).
- Entfernen Sie die 96-Well-Platte aus dem Inkubator.
- Mit einer 200-L-Pipette die 100 L Zellkulturmedium einschließlich des Sphäroidgewebes aus jedem Brunnen aspirieren und in einem sterilen, 15 ml Zentrifugenrohr sammeln. Achten Sie darauf, den Kontakt mit der Agarose zu vermeiden.
- Zentrifugieren Sie nach der Erhebung die Sphäroidsuspension bei 230 x g für 5 min. Entfernen Sie den Überstand und lagern Sie bei -80 °C für weitere Endpunktanalysen (z. B. Leberfunktionstests) später.
- Das Pellet der Sphäroide in 1 ml steriler Raumtemperatur PBS (1x) wieder aufhängen.
- Nach dem Waschen die Sphäroidsuspension wieder bei 230 x g für 3 min zentrieren. Den Überstand entsorgen, in 500 l mit 0,05% Trypsin-EDTA-Lösung erneut aussetzen und 6-8 min bei 37 °C und 5%CO2inkubieren.
- Nach der Inkubation pipettedien die Trypsinisierten Zellen sanft nach oben und unten, um die HepG2-Zellen vollständig zu trennen und wieder aufzuhängen, bevor sie mit 1 ml DMEM-Zellkulturmedium neutralisiert werden.
- Zentrifugieren Sie die verdünnte Zellsuspension bei 230 x g für 5 min. Entsorgen Sie den Überstand in desinfizierendes und setzen Sie das Zellpellet in 2 ml Raumtemperatur PBS (1x) wieder auf.
- Zentrifugieren Sie die Zellsuspension bei 230 x g für 5 min. Entsorgen Sie den Überstand in desinfizierend und setzen Sie das Zellpellet dann erneut in 2 ml kaltem PBS (1x) wieder auf. Stellen Sie sicher, dass die Zellen gut dispergiert sind, um zu verhindern, dass Zellklumpen das Sichtfeld verdecken, wenn sie auf Mikroskopschlitten montiert werden.
6. Mikrokern-Assay und Scoring
Für die manuelle Methode des Mikrokern-Assays ist eine Zytozentrifuge erforderlich, um ein Zytodot (eine definierte, konzentrierte Zellregion) in der Mitte des Mikroskopschlittens zu erzeugen. Dieser Prozess unterstützt eine effizientere Bewertung der Folie, da es dem Scorer ermöglicht, die Zellen von Interesse leicht zu lokalisieren, im Gegensatz zur Auswertung einer ganzen Folie, in der die Zellen weit verbreitet werden können.
- Tauchen Sie gefrostete Mikroskop-Dias (drei pro Dosis) in 70% Ethanol gefolgt von ddH2O und lassen Sie an der Luft für 5 min trocknen.
- Das präparierte Mikroskop gleitet in den Küvettentrichter, wie in Abbildung 3Adargestellt, wo der Glasschlitten (iii) mit einer Filterkarte (ii) und einem darüber befestigten Küvettentrichter (i) in den Metallträger (iv) gelegt wird.
- Ordnen Sie Küvettentrichter in der Zytozentrifuge mit nach oben gerichtetem Trichter an, so dass jeweils 100 l Zellsuspension direkt zugegeben werden können.
- Cytospin für 5 min bei 500 x g, um sicherzustellen, dass die Zellen gleichmäßig auf die Oberfläche des Dias verteilt sind.

Abbildung 3: Cytospin-Setup zur Vorbereitung behandelter Zellen auf Mikroskopschlitten. (A) Zeigt die einzelnen Komponenten, (i) Küvettentrichter, (ii) Filterkarte, (iii) Glasmikroskopschlitten und (iv) Metallunterstützung an, die erforderlich ist, um HepG2-Zellen auf Mikroskopschlitten zu verdrehen. (B) Der endgültige Küvettentrichter wurde aufgestellt. (C) Die korrekte Platzierung des Küvettentrichters innerhalb der Zytozentrifuge. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
- Lassen Sie die Dias vor der Fixierung in eiskalt, 90% Methanol für 10 min, an die Luft trocknen.
- Lassen Sie die Dias nach der Fixe über Nacht bei Raumtemperatur trocknen, bevor Sie sie bis zu 6 Monate bei -20 °C lagern.
- Entfernen Sie bei Bedarf die vorgefertigten Mikroskopschlitten aus dem Gefrierschrank von -20 °C und lassen Sie sich vor der Giemsa-Färbung auf Raumtemperatur erwärmen.
VORSICHT: Gemäß der Verordnung (EG) Nr. 1272/2008 [CLP] ist die Giemsa-Färbelösung eine leicht entzündliche Flüssigkeit, die bei Verschlucken giftig sein kann und bei Kontakt mit Denaugen, Haut oder beim Einatmen Schäden verursacht. Detaillierte Lagerungs-, Handhabungs-, Gesundheits- und Sicherheitshinweise zu dieser Chemikalie vor der Verwendung finden Sie auf dem zugehörigen SDS-Blatt. - Während die Dias auftauen, bereiten Sie eine 20% Giemsa-Färbungslösung (25 ml erforderlich, um 30 Dias zu färben) in Phosphatasepuffer (pH 6.8) verdünnt. Mischen Sie die Lösung gründlich, indem Sie die Lösung vorsichtig verwirbeln, bevor Sie mit gefaltetem Filterpapier filtern, das in einen Trichter gelegt wird.
- Mit einer Pasteur Pipette 3 – 5 Tropfen gefilterte Giemsa-Lösung auf jedem Schlitten zum Zytodot geben und 8 – 10 min lassen.
- Waschen Sie Dias in zwei aufeinanderfolgenden Phosphatase-Pufferwäschen, bevor Sie kurz unter kaltem Wasser spülen, um überschüssige Fleckenreste zu entfernen. Lassen Sie die Dias an der Luft trocken.
- Einmal trocken, in einer Dunstabzugshaube, tauchen Sie gebeizt Dias in Xylol für 10 s, bevor Sie einen Tropfen Desontes medium in die Mitte des Zytodots und einen Platz ein Glasabdeckungslip auf der Oberseite hinzufügen.
- Lassen Sie Mikroskop-Dias in der Dunstabzugshaube über Nacht trocknen, bevor sie manuell bewertet werden; sie können unbegrenzt bei Raumtemperatur gelagert werden.
7. Datenanalyse
- Wie in den OECD-Testrichtlinien 487 (2014)11beschrieben, verwenden Sie ein Lichtmikroskop (100x Objektiv mit Tauchöl) 2000 mononukleierte oder 1000 binukleierte Zellen pro biologischer Replikation, um das Vorhandensein von Mikrokernen zu bewerten, wie in Abbildung 4dargestellt.

Abbildung 4: Micronucleus Assay Scoring Entscheidungsbaum. Schematische Entscheidungsstruktur, um die Notwendigkeit unterschiedlicher Bewertungsverfahren für Scoring und Zytotoxizität bei der Verwendung des Mikrokern-Assays mit 3D-Modellen nach akuten oder langfristigen Expositionsregimen hervorzuheben. Akute (≤24 h) Expositionen ermöglichen die Verwendung des zytokinesisblockierten Mikrokerntests, während langfristige (≥24 h) Expositionen die mononukleare Version des Assays erfordern; beide sind in der OECD-Testleitlinie 487 beschrieben. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
- Berechnen Sie auf der Grundlage des Anteils der Mikronuklei pro Anzahl der bewerteten mononukleierten oder binucleatierten Zellen einen Prozentsatz des Genotoxizitätswertes.
- Um die beobachtete DNA-Schädigung nicht als Folge von Zellablagerungen durch einen hohen Anteil an apoptotischen Zellen verursacht, nehmen Sie ein Maß der Zytotoxizität neben. Verwenden Sie in diesem Fall je nach Vorhandensein von Cytochalasin B entweder CPBI- oder RVCC-Berechnung (wie in Abbildung 4beschrieben ). Die Genotoxizität darf nur in Proben bewertet werden, in denen die Zytotoxizität weniger als 55 % ± 5 % beträgt, wie in der OECD-Testleitlinie 48711definiert.
Representative Results
Die Eignung dieses zelllinienbasierten 3D-Lebersphäroidmodells für die Langzeitkultur und die genotoxische Gefährdungsbeurteilung wurde durch die Durchführung einer Basischarakterisierung bewertet, um die Lebensfähigkeit und Leber-ähnliche Funktionalität des Modells über die Dauer von 14 Tagen in der Kultur sowie seine Anwendbarkeit für den Mikrokern-Assay zu bestimmen.
Baseline Charakterisierung des 3D HepG2 Lebersphäroidmodells
Vor jeder in vitro toxikologischen Bewertung ist es wichtig zu überprüfen, ob sich die 3D HepG2 Sphäroide richtig gebildet haben, bevor der Agarosetransfer oder die chemische/ENM-Behandlung durchgeführt wurde. HepG2-Sphäroide, die mit der Hängetropfenmethode hergestellt werden, brauchen in der Regel 2 - 3 Tage nach der Aussaat (4000 Zellen/Sphäroid), um kompakte, kugelförmige Sphäroide mit einem durchschnittlichen Durchmesser von 495,52 w x 482,69 m H zu bilden, wie in Abbildung 5A-5Cdargestellt. HepG2-Sphäroide, die sich richtig gebildet haben und für die in vitro toxikologische Bewertung verwendet werden können, müssen eine kompakte, kugelförmige Struktur mit einer glatten Oberfläche und ohne visuelle Projektionen aufweisen. Abbildung 5 enthält Beispiele für gute Qualität (Abbildung 5D-F) und eine schlechte Qualität (Abbildung 5G-I) Sphäroide. Letzteres sollte verworfen werden. Typischerweise bilden sich 90-95% der pro Platte gebildeten Sphäroide richtig und sind für weitere Experimente lebensfähig.

Abbildung 5: Lichtmikroskopiebilder, die die natürliche Morphologie der HepG2-Sphäroide zeigen, die über die Hangtropfenmethode gebildetwerden. (A-C) zeigen Tag 2 und (D-I) Tag 4 HepG2 Lebersphäroide nach der Aussaat. (D-F) sind Beispiele für hochwertige HepG2-Sphäroide, während (G-I) schlecht geformte Sphäroide zeigt. Alle Bilder wurden auf einem X20-Objektiv mit einem Mikroskop aufgenommen. Die Maßstabsleiste steht für 20 m. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
Um die Lebensfähigkeit des HepG2-Sphäroids weiter zu bestätigen, kann ein farblich-ähnlicher Bromocresol Green Albumin (BCG) Assay oder Harnstoff-Assay durchgeführt werden, um deren Leber-ähnliche Funktionalität zu bewerten. Die leberähnliche Funktionalität wurde anhand des Trypan Blue Exclusion Assay über einen Zeitraum von 14 Tagen im Einklang mit der Lebensfähigkeit bewertet, um die Langlebigkeit des Lebersphäroidmodells zu bestimmen und festzustellen, ob es eine langfristige oder wiederholte ENM/chemische Risikobewertung unterstützen könnte (Abbildung 6). Die Albumin-Konzentration blieb während der gesamten Kulturperiode konstant. Die Harnstoffproduktion zeigt eine Erhöhung der Harnstoffkonzentration, die pro Sphäroid über eine Woche in der Kultur produziert wird, bevor sie am Tag 7 ein Plateau erreicht. Es ist wichtig zu beachten, dass die in den 3D HepG2 Sphäroiden produzierten Gehalte an Albumin und Harnstoff wesentlich höher sind als die in derselben Zelllinie in einem 2D-Format kultivierten. Tatsächlich betrugen die 2D-Kulturen von HepG2-Zellen, Peak-Albumin- und Harnstoffspiegeln 0,001 mg/ml bzw. 0,010 ng/l. Darüber hinaus heben die Autoren in früheren Arbeiten, die von Shah et al. unter Verwendung eines nahezu identischen HepG2-Sphäroidsystems veröffentlicht wurden, eine bemerkenswerte Verbesserung der metabolischen Aktivität (CYP1A1 und CYP1A2) in den 3D HepG2 In-vitro-Modellsystemen im Vergleich zu den 2D-kultivierten HepG2-Zellen5hervor.

Abbildung 6: 14-Tage-Basischarakterisierungsdaten für HepG2-Lebersphäroide. Nach der Übertragung von hängendem Tropfen hebt (A) die Lebensfähigkeit des HepG2 Sphäroidmodells über einen Zeitraum von 14 Tagen hervor, während (B) und (C) die leberähnliche Albumin- bzw. Harnstofffunktionalität hervorheben. Mittelwertdaten ± SEM dargestellt, n = 4. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
Mit der unvermeidlichen Entwicklung eines nekrotischen Kerns, einer bekannten Einschränkung von 3D-Lebersphäroidkulturen, musste die Lebensfähigkeit dieses HepG2-basierten Modells hergestellt werden, um zu zeigen, dass es in der Lage war, langfristige (5-10 Tage) Expositionsregime aufrechtzuerhalten und gleichzeitig die proliferative Fähigkeit aufrechtzuerhalten, die erforderlich ist, um den Mikrokern-Assay5zu unterstützen. Tatsächlich hat sich gezeigt, dass dieses 3D-Lebersphäroid-Modell über 10 Tage in der Kultur >70% Lebensfähigkeit behält. Basierend auf dieser und in Verbindung mit der anhaltenden Leber-ähnlichen Funktionalität, die während der ≥14-Tage-Kulturperiode beobachtet wurde, kann dieses 3D-Lebersphäroid-Modell so langfristige, wiederholte ENM-Expositionsregime von bis zu 10 Tagen unterstützen (d. h. bevor die Lebensfähigkeit der Sphäroide unter 70 fällt). Als Referenz wird empfohlen, dass die Albuminspiegel für HepG2-Sphäroide, die mit 4000 Zellen/Sphäroid ausgesät sind, ≥20,0 ng/L betragen sollten, während die Harnstoffproduktion ≥0,25 ng/L betragen sollte, bevor eine in vitro toxikologische Bewertung mit diesem Modell durchgeführt wird.
Genotoxizitätsbewertung von technischen Nanomaterialien
Für die Genotoxizitätsbewertung wurde der Mikrokerntest verwendet, um das Vorhandensein von Mikronuklei nach akuten (24 h) und langfristigen (120 h) ENM-Expositionen zu bestimmen. Aflatoxin B1 ist ein bekanntes Leberkarzinogen16,17 und ist eine empfohlene positive Kontrolle für den Mikrokern-Assay. Optimierungsexperimente haben gezeigt, dass 0,1 M Alfatoxin B1 eine signifikante positive (≥2,0-fache Zunahme) genotoxische Reaktion in 3D HepG2 Lebersphäroide induziert und somit in jedem Mikrokern-Assay verwendet wird, der mit diesem Modell durchgeführt wird. Um die Gültigkeit der Mikrokern-Assay-Ergebnisse mit dem HepG2-Sphäroidmodell zu gewährleisten, sollte die Hintergrundmikrokernfrequenz für HepG2-Zellen, die in diesem 3D-In-vitro-Modell verwendet werden, in einem Bereich von 0,6 % – 1,2 % liegen. Infolgedessen sollte Alfatoxin B1 eine genotoxische Reaktion auslösen, die mindestens doppelt so hoch ist als die mit der Negativkontrolle gesehene; Daher sollten 0,1 M Alfatoxin B1 eine Mikronuklei-Frequenz zwischen 1,5 % – 3,0 % induzieren. Mit diesen Kontrollparametern kann die ENM-assoziierte Genotoxizität in vitro zuverlässig beurteilt werden. Auf der Grundlage der OECD-Testleitlinie 487 ist zu beachten, dass bei der Prüfung eines ENM oder einer Chemikalie die ausgewählten Konzentrationen nicht mehr als 55 % ± 5 % Zytotoxizität (angezeigt durch eine Verringerung der CPBI- oder RVCC-Werte in Bezug auf die Negativkontrolle)11induzieren sollten. Abbildung 7 zeigt die Daten, die bei der Bewertung von Aflatoxin B1 und zwei ENMs (Titandioxid (TiO2) und Splitter (Ag)) nach akuten und langfristigen Expositionen in den HepG2-Sphäroiden und anschließender genotoxischer Potenziale mit hilfe des Mikrokern-Assays generiert wurden. Beide enMs wurden bei einer akuten (24 h) Exposition und einer langfristigen Exposition (120 h) mit einer nichtzytotoxischen, niedrigen Dosis von 5,00 g/ml getestet. Ein ähnlicher Trend zur Genotoxizität sowohl bei TiO2 als auch bei Ag ENMs ist zu beobachten, wobei die erhöhte Genotoxizitätsreaktion, die nach einer 24-h-Exposition resultierte, nach einer langfristigen Exposition von 5 Tagen nicht erkennbar war. Dies trotz anhaltender Genotoxizität, die durch die Aflatoxin B1-Positivekontrolle an beiden Zeitpunkten induziert wurde.

Abbildung 7: Genotoxizitätsbewertung nach TiO2 und Ag ENM-Exposition bei HepG2-Lebersphäroiden. Genozid (Mikrokernfrequenz) Bewertung mit dem Mikrokern-Assay-Post (A) akut (24 Stunden) und (B) Langzeit-(120-Stunden)-Exposition bei 5,00 g/ml TiO2 und Ag ENM. Negativkontrolle ist nur ein Medium, während die positive Kontrolle 0,1 m Aflatoxin B1 beträgt. Mittelwertdaten (n=2) ± SD dargestellt. Bedeutung, die in Bezug auf die Negative Kontrolle angegeben ist: * = p≤ 0,05. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
Discussion
Anwendungen für 3D-Lebermodelle variieren erheblich, je nachdem, auf den jeweiligen biochemischen Endpunkt oder den unerwünschten Ergebnisweg gezielt. Jedes Modell hat seine Vorteile und Einschränkungen, von Interdonor Variation in primären menschlichen Hepatozyten (PHH) Modelle zu reduzierten Cytochrom p450 Aktivität in Zelllinien-basierten Modellen, aber alle sind wertvoll in ihrem eigenen Recht6,12,18,19. Bei der Bewertung der Genotoxizität gibt es Einschränkungen in den Modellen Kompatibilität mit gesetzlich zugelassenen Endpunkten wie der In-vitro-Mikrokern-Assay, da aktive Proliferation erforderlich ist. Dies ist notwendig, da die Genotoxizitätsbewertung erfordert, dass die Quantifizierung von festen DNA-Schäden nach der Zellteilung bewertet wird, wenn die Möglichkeit besteht, dass die DNA-Reparatur vorübergehende Läsionen korrigiert. Leider bilden hochdifferenzierte Hepatozyten (d.h. HepaRG) sphäroide oder PHH-Mikrotissues, die als die physiologisch relevantesten leberähnlichen Eigenschaften gelten, statische (nicht-proliferative) Modelle12,19,20. Daher bietet das hier vorgestellte 3D HepG2 Sphäroid-Modell ein geeignetes, alternatives Modell, das Genotoxizitätstests unterstützen kann. HepG2-Zelllinien-basierte Sphäroide haben genügend aktiv teilende Zellen auf der außenoberfläche der Sphäroide unter Beibehaltung grundlegender leberähnlicher Eigenschaften, wie Albumin- und Harnstoffproduktion und einige CYP450-Aktivität5,12,19. Hauptsächlich wurde dieses In-vitro-Lebermodell entwickelt, um den Mikrokern-Assay zu ergänzen, da dies einer der beiden In-vitro-Assays ist, die in der Batterie für Genotoxizitätstests8,10,11,21empfohlen werden. Das Modell kann jedoch problemlos auf DNA-Sequenzierungsanalysen und Genexpressionstechnologien (RNA) angewendet werden, während es das Potenzial hat, weiter angepasst und für andere DNA-Schadensendpunkte wie den Kometentest genutzt zu werden. Nichtsdestotrotz ist es wichtig, die Rolle zu berücksichtigen, die ENM-Interferenzen bei einigen Endpunktanalysen spielen. Beispielsweise sind Strömungszytometrie-basierte Analysen möglicherweise nicht für die ENM-Genotoxizitätsbewertung geeignet, insbesondere aufgrund von Partikelstörungen22.
Ein begrenzender Faktor von Sphäroidmodellen, die sich aktiv einer Zellteilung unterziehen, ist ihre Größe. Die Optimierung der Saatdichte ist von entscheidender Bedeutung, da genügend Zellen vorhanden sein müssen, die es dem Modell ermöglichen, sich weiter auszumehren. aber nicht zu hoch eine Zellzahl, was dazu führt, dass das Sphäroid übermäßig kompakt wird, was zu einem erhöhten nekrotischen Kern führt. Die Ursache dieser Nekrose wird geglaubt, um eingeschränktsauerstoff- und Nährstoffdiffusion sein, da die Grenze dieser Diffusion wird angenommen, dass etwa 100 – 150 m Gewebe23,24. Dies hängt jedoch vom Zelltyp, der Zellnummer, den Gerüstinteraktionen und den Kulturbedingungen25ab. Seitdem hat sich gezeigt, dass etwa 700 m Durchmesser die Grenze zur Vermeidung eines vorzeitigen Beginns der Nekrose in der Mitte von C3A-Sphäroiden ist, die Aussaat von 4000 HepG2-Zellen pro Sphäroid stellt sicher, dass der Durchmesser des Modells zum Zeitpunkt der Exposition ≤500 m26beträgt. Darüber hinaus stellten Shah et al. fest, dass HepG2-Zellen mit einem Samen von mehr als 5000 Zellen pro Sphäroid nach 7 Tagen in der Kultur eine 25%ige Verringerung der Lebensfähigkeit aufwiesen, was sich auf den durchschnittlichen Durchmesser von 680 m und die begrenzte Verfügbarkeit von Nährstoffen in einem 20-L-Hängetropfen5beziehen konnte. Um dies zu überwinden, durchläuft das im vorliegenden Protokoll entwickelte Modell einen kritischen Schritt, bei dem der hängende Tropfen nach der Erstbildung des Sphäroids in Agarose-beschichtete Brunnen übertragen wird. Dies stellt sicher, dass ein größeres Volumen an Kulturmedium vorhanden ist, um die ständig wachsende Anzahl von Zellen innerhalb der Sphäroide zu erhalten. Infolgedessen bleibt das HepG2 Sphäroid-Modell nach 10 Tagen in der Kultur über 70% lebensfähig und kann für die langfristige Gefährdungsbeurteilung in vitro genutztwerden.
Während das HepG2 Sphäroid-Modell sowohl akute als auch langfristige Expositionsregime unterstützen kann, ist die Erfrischung des Zellkulturmediums während längerer Kulturperioden für dieses Modell eingeschränkt, da ein vollständiger Ersatz des Mediums aufgrund des potenziellen Verlusts der Sphäroide nicht empfohlen wird. Es wird vermutet, dass bei ENM-Expositionen die Tendenz zu homogenen ENM-Dispersionen zu Agglomerat und Sediment hoch ist. Es ist jedoch bemerkenswert, dass die Rate, mit der ein ENM-Sediment je nach Denplyse parametern (z. B. Größe, Form und Dichte) variieren kann und theoretisch anhand des In-vitro-Sedimentations-, Diffusions- und Dosimetriemodells (ISDD) oder seiner jüngsten Derivate bestimmt werden kann, auf die bei ENM-Expositionsansätzen oft Bezug genommen wird27,28. Dabei wird angenommen, dass, wenn nur 50% des Zellkulturmediums vorsichtig von der Oberfläche der Zellkultur entfernt werden, die Störung und anschließende Entfernung der ENM-Dosis theoretisch minimal sein sollte. Mit Brownian Bewegung im Spiel, kann dies jedoch nicht unbedingt der Fall sein und weitere Arbeiten in der Ablagerung und Sedimentation jedes einzelnen ENM getestet werden sollte durchgeführt werden, um sicherzustellen, dass die richtige Dosimetrie während der langfristigen Expositionsregime beibehalten wird27. Grundsätzlich ist dies eine potenzielle Einschränkung, die bei der Durchführung wiederholter Dosiersysteme zu berücksichtigen ist, da dies für die endgültige, akkumulierte Konzentration von entscheidender Bedeutung sein könnte. Chemische Expositionen hingegen bieten, auch wenn sie nicht ohne ihre eigenen Einschränkungen zu berücksichtigen sind, einen vereinfachteren Ansatz, da chemische Stoffe tendenziell in Lösung bleiben und somit ein direkter Ersatz der ursprünglichen chemischen Konzentration zusätzlich zur neu zugesetzten Konzentration dafür sorgt, dass alle Chemikalien, die während der Medienerfrischung verloren gehen, entsprechend ersetzt werden29. Zukünftige Anwendungen würden die Bewertung der Eignung des Modells für wiederholte Expositionsregime über langfristige Kulturperioden umfassen, da wiederholte Dosierstrategien von entscheidender Bedeutung für die Beurteilung der Fähigkeit eines bestimmten Organsystems sind, die durch die Bioakkumulation einer xenobiotischen Substanz verursachten nachteiligen Auswirkungen zu mildern oder zu überwinden.
Zusammenfassend lässt sich sagen, dass dieses 3D-In-vitro-Hepatikmodell in der Lage ist, eine Reihe realistischer Expositionsszenarien zu bewerten und damit einen zukünftigen In-vitro-Ansatz bereitzustellen, um sowohl enM als auch die Bewertung chemischer Gefahren routinemäßig und leicht zugänglich zu unterstützen.
Disclosures
Die Autoren haben nichts zu verraten.
Acknowledgments
Die Autoren möchten anerkennen, dass diese Forschung aus dem Forschungs- und Innovationsprogramm Horizont 2020 der Europäischen Union für das PATROLS-Projekt im Rahmen der Finanzhilfevereinbarung Nr. 760813 gefördert wurde.
Materials
| Name | Company | Catalog Number | Comments |
| Aflotoxin B1 | Sigma Aldrich, UK | A6636-5MG | |
| Agarose | Sigma Aldrich, UK | A9539-50G | |
| Autoclave Tape | |||
| BCG Albumin Assay | Sigma Aldrich, UK | MAK124 | |
| Bovine Serum Albumin Powder | Sigma Aldrich, UK | A9418 | |
| Cell Freezing Aid | Thermo Fisher Scientific, UK | 5100-0001 - Mr Frosty | |
| Centrifuge | Eppendorf | 5810 R | |
| Cytochalasin B | Merck, UK | 250233 | |
| Cytology Metal Clips | |||
| Cytospin 4 Centrifuge | ThermoFisher Scientific, UK | CM00730202 | |
| DMEM with 4.5g/L D-Glucose, L-Glutamine | GIBCO, Paisley, UK | 41965-039 | |
| DMEM, phenol-red free with 4.5g/L D-Glucose, L-Glutamine with Hepes | GIBCO, Paisley, UK | 21063-029 | |
| DPX Mounting Medium | FisherScientific, UK | D/5330/05 | |
| Ethanol | FisherScientific, UK | 10048291 | |
| FBS | GIBCO, Paisley, UK | 10270-106 | |
| Filter Cards for Shandon Cytospin | FisherScientific, UK | 15995742 | |
| Frosted Glass Slides | ThermoFisher Scientific, UK | ||
| Giemsa's Stain Improved R66 Solution, Gurr | VWR Chemicals, UK | MFCD00081642 | |
| Glass Coverslips (24 x 60) | Deckglaser, VWR | ECN631-1575 | |
| Haemocytometer and Coverslip | |||
| Immersion Oil for Microscope | Zeiss, UK | 518F, ISO8034 | |
| Laminar Class II Tissue Culture Hood | Scanlaf Mars | ||
| Light Microscope | Zeiss, UK | Axiovert 40C | |
| Liquid Nitrogen | |||
| Methanol | FisherScientific, UK | 10284580 | |
| Microwave | |||
| Non-Filtered, Sterile 200µl and 1000µl Pipette tips | Greiner-Bio-One, UK | ||
| NuncMicroWell 96-Well Microplates | ThermoFisher Scientific, Denmark | 167008 | |
| P1000 and P200 micropipettes | |||
| P300 and P50 multi-channel pipettes | |||
| PBS pH 7.4 1X, MgCl2 and CaCl2 Free | GIBCO, Paisley, UK | 14190-094 | |
| Pen/Strep | GIBCO, Paisley, UK | 15140-122, Penicillin/Strepmyocin 100X or 10,000U/ml | |
| Phosphatase Buffer Tablets | GIBCO, Paisley, UK | 10582-013 | |
| Pipette Boy | |||
| Simport Scientific CytoSep Funnels for Shandon Cytospin 4 Centrifuges | FisherScientific, UK | 11690581 | |
| Sonifier SFX 550 240V CE 1/2" - Probe | Branson, USA | 101-063-971R | |
| T-25 and T-75 Tissue Culture Flask | Greiner-Bio-One, UK | T-25 (690175) and T-75 (660175) | |
| Trypan Blue Solution | Sigma Aldrich, UK | T8154-100mL | |
| Urea Assay Kit | Sigma Aldrich, UK | MAK006 | |
| Virkon Disinfectant | DuPont, UK | Rely+On Virkon | |
| Water Bath (37?C) | Grant JBNova 18 | ||
| Weighing Balance | |||
| Xylene | FisherScientific, UK | 10588070 | |
| 0.05% Trypsin-EDTA | GIBCO, Paisley, UK | 5300-054 | |
| 0.2mL and 1.0mL Eppendorf Tubes | Greiner-Bio-One, UK | ||
| 0.45µm Filter Unit | Millex HA, MF-Millipore, UK | SLHA033SS | |
| 1.0mL Syringe | BD Plastipak, FisherScientific, UK | 300185 | |
| 20mL LS Scintillation Glass Vials, 22-400 Foil Lined PP Caps | DWK Life Sciences GmbH, Germany | WHEA986581 | |
| 37?C and 5% CO2 ISO Class 5 Hepa Filter Incubator | NUAIRE DHD Autoflow | ||
| 3mL Pasteur Pipette | Greiner-Bio-One, UK | ||
| 50mL Conical Falcon Tubes | Greiner-Bio-One, UK | ||
| 50mL or 100mL Glass Bottles | |||
| 50mL Skirted Falcon Tubes | Greiner-Bio-One, UK | ||
| 5mL, 10mL and 25mL Pipettes | Greiner-Bio-One, UK | ||
| 9.4cm Square, Petri Dish | Greiner-Bio-One, UK | 688161 |
References
- Geiser, M., Kreyling, W. G. Deposition and biokinetics of inhaled nanoparticles. Particle and Fibre Toxicology. 7, 2 (2010).
- Modrzynska, J. Toxicological effects of nanoparticle deposition in the liver. Kgs. Lyngby, Denmark: Technical University of Denmark. , (2018).
- Elje, E., et al. The comet assay applied to HepG2 liver spheroids. Mutation Research/Genetic Toxicology and Environmental Mutagenesis. 845, 403033 (2019).
- Breslin, S., O'Driscoll, L. Three-dimensional cell culture: the missing link in drug discovery. Drug Discovery Today. 18, 240-249 (2013).
- Shah, U. -K., et al. A three-dimensional in vitro HepG2 cells liver spheroid model for genotoxicity studies. Mutation Research/Genetic Toxicology and Environmental Mutagenesis. 825, 51-58 (2018).
- Lauschke, V. M., Hendriks, D. F. G., Bell, C. C., Andersson, T. B., Ingelman-Sundberg, M. Novel 3D Culture Systems for Studies of Human Liver Function and Assessments of the Hepatotoxicity of Drugs and Drug Candidates. Chemical Research in Toxicology. 29, 1936-1955 (2016).
- van Grunsven, L. A. 3D in vitro models of liver fibrosis. Advanced Drug Delivery Reviews. 121, 133-146 (2017).
- Corvi, R., Madia, F. In vitro genotoxicity testing - can the performance be enhanced. Food and Chemical Toxicology. 106, 600-608 (2017).
- Doak, S. H., Manshian, B., Jenkins, G. J. S., Singh, N. In vitro genotoxicity testing strategy for nanomaterials and the adaptation of current OECD guidelines. Mutation Research/Genetic Toxicology and Environmental Mutagenesis. 745, 104-111 (2012).
- Fenech, M. Cytokinesis-block micronucleus cytome assay. Nature Protocols. 2, 1084-1104 (2007).
- OECD. OECD Guidelines. Test 489: In vivo Mammalian Alkaline Comet Assay. , (2016).
- Gerets, H. H. J., et al. Characterization of primary human hepatocytes, HepG2 cells, and HepaRG cells at the mRNA level and CYP activity in response to inducers and their predictivity for the detection of human hepatotoxins. Cell Biology and Toxicology. 28, 69-87 (2012).
- Sison-Young, R. L., et al. A multicenter assessment of single-cell models aligned to standard measures of cell health for prediction of acute hepatotoxicity. Archives of Toxicology. 91, 1385-1400 (2017).
- European Guidelines 2019. European Agency for Safety and Health at Work. , Available from: https://osha.europa.eu/en/safety-and-health-legislation/european-guidelines (2019).
- Jensen, K. A. The NANOGENOTOX Dispersion Protocol for NANoREG. European Union Grant Agreement n° 2009. 21, 01 (2014).
- Marchese, S., et al. Aflatoxin B1 and M1: Biological Properties and Their Involvement in Cancer Development. Toxins. 10, 214 (2018).
- Rushing, B. R., Selim, M. I. Aflatoxin B1: A review on metabolism, toxicity, occurrence in food, occupational exposure, and detoxification methods. Food and Chemical Toxicology. 124, 81-100 (2019).
- Kermanizadeh, A., Brown, D. M., Moritz, W., Stone, V. The importance of inter-individual Kupffer cell variability in the governance of hepatic toxicity in a 3D primary human liver microtissue model. Scientific Reports. 9, 7295 (2019).
- Berger, B., et al. Comparison of Liver Cell Models Using the Basel Phenotyping Cocktail. Frontiers in Pharmacology. 7, 443 (2016).
- Ramaiahgari, S. C., et al. A 3D in vitro model of differentiated HepG2 cell spheroids with improved liver-like properties for repeated dose high-throughput toxicity studies. Archives of Toxicology. , (2014).
- Li, Y., et al. Factors affecting the in vitro micronucleus assay for evaluation of nanomaterials. Mutagenesis. 32 (1), 151-159 (2016).
- Kirkland, D., Reeve, L., Gatehouse, D., Vanparys, P. A core in vitro genotoxicity battery comprising the Ames test plus the in vitro micronucleus test is sufficient to detect rodent carcinogens and in vivo genotoxins. Mutation Research/Genetic Toxicology and Environmental Mutagenesis. 721 (1), 27-73 (2011).
- Curcio, E., et al. Mass transfer and metabolic reactions in hepatocyte spheroids cultured in rotating wall gas-permeable membrane system. Biomaterials. 28, 5487-5497 (2007).
- Glicklis, R., Merchuk, J. C., Cohen, S. Modeling mass transfer in hepatocyte spheroids via cell viability, spheroid size, and hepatocellular functions. Biotechnology and Bioengineering. 86, 672-680 (2004).
- Asthana, A., Kisaalita, W. S. Microtissue size and hypoxia in HTS with 3D cultures. Drug Discovery Today. 17, 810-817 (2012).
- Gaskell, H., et al. Characterization of a functional C3A liver spheroid model. Toxicology Research. 5, 1053-1065 (2016).
- Cho, E. C., Zhang, Q., Xia, Y. The effect of sedimentation and diffusion on cellular uptake of gold nanoparticles. Nature Nanotechnology. 6, 385-391 (2011).
- Hinderliter, P. M., et al. ISDD: A computational model of particle sedimentation, diffusion and target cell dosimetry for in vitro toxicity studies. Particle and Fiber Toxicology. 7, 36 (2010).
- Kramer, N. I., di Consiglio, E., Blaauboer, B. J., Testai, E. Biokinetics in repeated-dosing in vitro drug toxicity studies. Toxicology in vitro. 30, 217-224 (2015).
Tags
Bioengineering Ausgabe 160 In Vitro Lebermodelle Nanomaterialien Gefahrenbeurteilung Langzeitexposition Nano(Geno)Toxikologie DNA-SchädenErratum
Formal Correction: Erratum: Advanced 3D Liver Models for In vitro Genotoxicity Testing Following Long-Term Nanomaterial Exposure
Posted by JoVE Editors on 01/26/2021.
Citeable Link.
An erratum was issued for: Advanced 3D Liver Models for In vitro Genotoxicity Testing Following Long-Term Nanomaterial Exposure. The Representative Results section was updated.
Figure 6 in the Representative Results section was updated from:
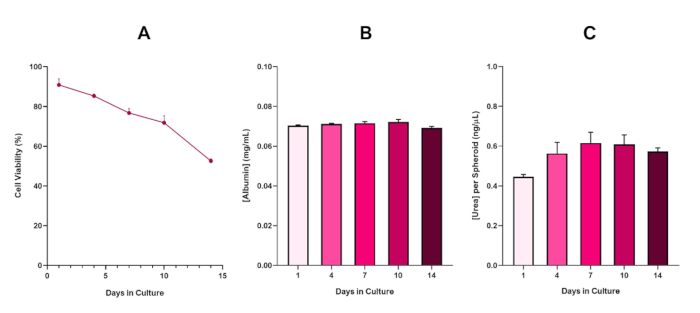
to:
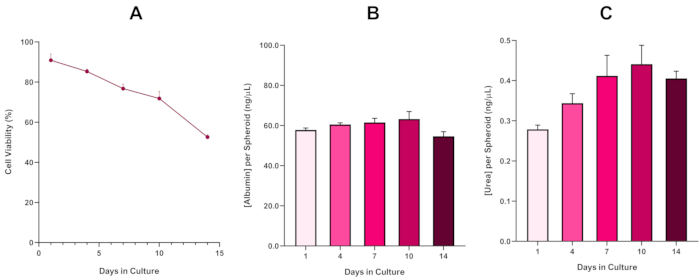
The fourth paragraph in the Representative Results section was updated from:
With the inevitable development of a necrotic core, a known limitation of 3D liver spheroid cultures, the viability of this HepG2 based model had to be established to demonstrate it was able to sustain long-term (5-10 day) exposure regimes whilst maintaining the proliferative capability required to support the micronucleus assay5. Indeed, this 3D liver spheroid model has been shown to retain >70% viability over 10 days in culture. Based on this and in conjunction with the sustained liver-like functionality observed over the ≥14 day culture period, this 3D liver spheroid model can thus support long-term, repeated ENM exposure regimes up to 10 days long (i.e., before viability of the spheroids drop below 70%). For reference, it is advised that albumin levels for HepG2 spheroids seeded at 4000 cells/spheroid should be ≥0.06 mg/mL whilst urea production should be ≥0.4 ng/µL before conducting an in vitro toxicological assessment with this model.
to:
With the inevitable development of a necrotic core, a known limitation of 3D liver spheroid cultures, the viability of this HepG2 based model had to be established to demonstrate it was able to sustain long-term (5-10 day) exposure regimes whilst maintaining the proliferative capability required to support the micronucleus assay5. Indeed, this 3D liver spheroid model has been shown to retain >70% viability over 10 days in culture. Based on this and in conjunction with the sustained liver-like functionality observed over the ≥14 day culture period, this 3D liver spheroid model can thus support long-term, repeated ENM exposure regimes up to 10 days long (i.e., before viability of the spheroids drop below 70%). For reference, it is advised that albumin levels for HepG2 spheroids seeded at 4000 cells/spheroid should be ≥50.0 ng/μL whilst urea production should be ≥0.25 ng/µL before conducting an in vitro toxicological assessment with this model.
