

ERRATUM NOTICE
Important: There has been an erratum issued for this article. Read more …
Summary
Cette procédure a été établie pour être utilisée pour développer des cultures hépatiques 3D avancées in vitro, ce qui peut fournir une évaluation plus physiologiquement pertinente des dangers génotoxiques associés aux expositions aux nanomatériaux sur un régime de dose répété aigu ou à long terme.
Abstract
En raison du développement et de la mise en œuvre rapides d’un large éventail de nanomatériaux d’ingénierie (ENM), l’exposition à l’ENM est inévitable et le développement de systèmes de test in vitro robustes et prédictifs est essentiel. La toxicologie hépatique est essentielle lors de l’examen de l’exposition à l’ENM, car le foie joue un rôle vital dans l’homéostasie métabolique et la désintoxication, en plus d’être un site majeur d’accumulation enM après exposition. Sur la base de ceci et de la compréhension acceptée que les modèles d’hépatocyte 2D n’imitent pas exactement les complexités des interactions multicellulaires complexes et de l’activité métabolique observée in vivo, il y a un plus grand accent sur le développement des modèles physiologiquement pertinents de foie 3D adaptés aux buts in vitro d’évaluation de danger d’ENM. Conformément aux principes des 3R pour remplacer, réduire et affiner l’expérimentation animale, un modèle hépatique à base de lignée cellulaire 3D HepG2 a été développé, qui est un système convivial et rentable qui peut prendre en charge les régimes d’exposition à la FOIS prolongés et répétés en MATIÈRE (≤14 jours). Ces modèles sphéroïdes (≥500 μm de diamètre) conservent leur capacité proliférative (c.-à-d. les modèles cellulaires de division) leur permettant d’être couplés à l’analyse micronucléus « étalon-or » pour évaluer efficacement la génotoxicité in vitro. Leur capacité de rendre compte d’une gamme de critères d’évaluation toxicologiques (p. ex., fonction hépatique, réponse (pro)inflammatoire, cytotoxicité et génotoxicité) a été caractérisée à l’aide de plusieurs ENM dans les régimes d’exposition aigus (24 h) et à long terme (120 h). Ce modèle hépatique in vitro 3D a la capacité d’être utilisé pour évaluer des expositions enM plus réalistes, offrant ainsi une approche in vitro future pour mieux soutenir l’évaluation des risques ENM d’une manière routinière et facilement accessible.
Introduction
En raison du développement et de la mise en œuvre rapides d’un large éventail de nanomatériaux d’ingénierie (ENM) dans une pléthore d’applications à base humaine (p. ex., alimentation, cosmétiques, vêtements, équipements sportifs, électronique, transport et médecine), il est inévitable que les humains soient exposés régulièrement à l’ENM. Avec cela, on s’inquiète de plus en plus du fait que les caractéristiques physiochimiques nouvelles et spécifiques à la taille qui jugent ces matériaux avantageux dans de nombreuses applications pourraient avoir des effets néfastes sur la santé humaine et l’environnement simultanément. À l’heure actuelle, de nombreuses activités internationales sont en place pour refléter activement des expositions plus pertinentes sur le plan physiologique à ces ENM et évaluer la toxicité potentielle de ces matériaux par rapport aux scénarios d’exposition aiguë, à long terme et répétées à faible dose.
La toxicologie hépatique est essentielle lors de l’examen de l’exposition à l’ENM, car il est largement connu que le foie est un site majeur de l’accumulation ENM aprèsexposition 1,2. En outre, le foie est le système principal d’organe pour le métabolisme et la désintoxication des substances qui entrent dans la circulation systémique3. Sur la base de la compréhension acceptée que les modèles d’hépatocytes 2D n’imitent pas exactement les complexités des interactions multicellulaires complexes ou représentent de manière appropriée l’activité métabolique observée in vivo, un plus grand accent a été mis sur le développement de modèles hépatiques 3D in vitro robustes et physiologiquement pertinents pour les technologies de substitution in vivo aété établi 4,5. L’utilisation de technologies avancées de culture 3D améliore la longévité des modèles hépatiques in vitro, ce qui permet d’étudier des régimes d’exposition répétés à long terme. En outre, ce format de culture avancée favorise la formation de caractéristiques physiologiques et organotypiques améliorées telles que le canaliculi biliaire, les processus de transport actif et l’amélioration des capacités de métabolisation des médicaments CYP450, améliorant ainsi la prévisibilité des modèles6. Les modèles hépatiques 3D actuels, composés de monocultures (hépatocytes seulement) ou de co-cultures (hépatocytes avec cellules non paroenchymales) existent sous plusieurs formats, allant des microtissues ou sphéroïdes dans des plaques d’adhérence ultralow, sphéroïdes suspendus goutte, cellules intégrées dans les matrices et / ou échafaudages et plates-formes de culture cellulaire microfluidique, qui sont tous considérés comme efficaces modèles avancés in vitro pour l’évaluation de la toxicité hépatique6,7. Cependant, la majorité de ces systèmes modèles sont à entretien élevé, nécessitent de l’équipement spécialisé et sont coûteux. En outre, ces modèles sont souvent statiques (c.-à-d. modèles cellulaires non divisés) qui empêchent leur utilisation dans l’évaluation des points de terminaison des dangers, tels que les tests de génotoxicité utilisant des méthodes qui quantifient les dommages fixes à l’ADN. La génotoxité est une condition essentielle à la toxicologie réglementaire, et elle est un élément essentiel de l’évaluation des risques de tout toxicant8. Il n’y a pas de seul essai qui peut être appliqué pour quantifier toutes les formes de dommages à l’ADN qui peuvent survenir après l’exposition à un agent exogène. Cependant, un composant central de la batterie d’essai de génotoxicité in vitro est l’essai de micronucléus, qui est une technique fiable et multifacetted qui mesure les dommages chromosomiques bruts9. Il s’agit d’une technique d’étalon-or décrite par la Ligne directrice 487 de l’OCDE sur les tests, pour évaluer les dommages et la génotoxicité de l’ADN in vitro, et elle fait partie de l’exigence relative à la batterie d’essaipour l’évaluation réglementaire des risques 10,11.
La lignée cellulaire du carcinome hépatocellulaire humain, HepG2, est largement utilisée pour le dépistage initial de l’évaluation des dangers, car les cellules sont facilement disponibles, relativement peu coûteuses à la source, simples à la culture et à un criblage à hautdébit 12,13. Lorsqu’ils sont cultivés dans des structures sphériques 3D, il a été démontré qu’ils récapitulent bien le microenvironnement hépatique et offrent un modèle hépatique avec des capacités prolifératives suffisantes pour soutenir l’analyse de micronucléus3. D’autres développements des modèles sphéroïdes HepG2 ont été mis en place pour améliorer la longévité et la fonctionnalité hépatique du modèle afin de soutenir l’évaluation des risques de génotoxicité sur les régimes d’exposition répétés à long terme (≤14 jours). Ainsi, conformément aux principes des 3R pour remplacer, réduire et affiner l’expérimentation animale, le protocole actuel a été établi pour fournir un modèle hépatique in vitro avancé en 3D capable d’évaluer de manière fiable de multiples critères toxicologiques (p. ex., fonctionnalité hépatique, marqueurs inflammatoires (pro)inflammatoires, cytotoxicité et génotoxicité) suivant des expositions chimiques aiguës, à long terme et répétées et enm d’une manière routinière et facilement accessible.
Ici, nous présentons une méthode pour établir une lignée physiologiquement pertinente de cellules d’hépatocyte 3D basée le système in vitro de modèle pour l’évaluation de danger de génotoxicité suivant les expositions aiguës ou à long terme, répétées d’ENM. Le protocole peut être décomposé en 6 étapes clés : la culture des cellules hepG2 cryopréservées; Préparation sphéroïde HepG2; Transfert de sphéroïdes HepG2 de la chute suspendue à la suspension agarose; Récolte de sphéroïdes HepG2; analyse et notation de micronucléus ; et l’analyse des données.
Protocol
cellules hepG2 cryopréservées 1.Culturing
REMARQUE : Les cellules HepG2, obtenues de l’American Type Culture Collection (ATCC) ont été cultivés dans 1x Dulbecco’s Modified Eagle Medium (DMEM) avec 4,5 g/L de D-glucose et de L-glutamine complétés par 10 % de sérum bovin fœtal (FBS) et 1 % d’antibiotique pénicilline/streptomicine.
- Milieu de culture cellulaire DMEM préchauffé (y compris les suppléments) dans un bain d’eau de 37 °C pendant 30 min.
- Retirer un flacon de cellules HepG2 de l’azote liquide et décongeler dans un bain d’eau de 37 °C pendant 2-3 minutes, tout en tourbillonnant doucement le flacon pour permettre un dégel uniforme de la suspension cellulaire. Prenez soin de ne pas submerger le flacon au-dessus de l’anneau O afin de réduire le risque de contamination.
- Une fois décongelé, retirer le flacon du bain d’eau et vaporiser généreusement avec 70 % d’éthanol pour décontaminer la surface extérieure du flacon avant de le placer sous une hotte stérile de culture de tissu laminaire de classe II.
- Pipette soigneusement le contenu du cryovial des cellules HepG2 dans un tube de centrifugeuse contenant 9 mL de milieu de culture cellulaire DMEM préchauffé (avec suppléments).
- À l’aide d’une bandepette de 10 mL, transférer 10 mL de la suspension cellulaire dans un flacon de culture cellulairejetable de 25 cm 2 et incuber la culture pendant 3 jours (à partir de l’ensemencement) à 5 % de CO2 et 37 °C jusqu’à ce qu’une confluence de ~80 % soit atteinte avant de subir une sous-culture dans un flacon de culture cellulairejetable de 75 cm 2 plus grand.
- Une fois que 80% de confluence est atteint, les cellules de sous-culture dans des conditions stériles par trypsinisation avec 0,05% trypsine / EDTA solution préchauffée dans un bain d’eau de 37 °C pendant 30 min. À aucun moment les cellules ne devraient être autorisées à se dessécher.
- Lorsque les cellules forment un monomouche adhérent, retirez le média en basculant dans une poubelle désinfectante. Lavez immédiatement la monocouche pour enlever toutes les traces de supports existants en rinçant le flacon deux fois avec 3 mL de solution stérile 1x PBS conservée à température ambiante. En outre, jetez PBS dans la casserole de déchets désinfectants.
- Une fois le lavage PBS enlevé, ajouter 5 mL de solution trypsine-EDTA préchauffée à 0,05 %, en veillant à couvrir toute la surface des cellules et à couver les cellules pendant 6-8 min à 37 °C et 5 % de CO2.
- Appuyez doucement sur le flacon pour déloger les cellules du fond du flacon, puis ajoutez 5 mL de milieu de culture cellulaire DMEM (avec suppléments) pour neutraliser l’enzyme trypsine.
- Transférer la suspension cellulaire dans un tube de centrifugeuse de 50 mL et pipette la suspension cellulaire de haut en bas à fond pour s’assurer que les cellules sont complètement dissociées.
- Centrifugeuse la suspension cellulaire diluée à 230 x g pendant 5 min. Jeter le supernatant dans le désinfectant et suspendre à nouveau les granulés cellulaires dans 25 mL de milieu de culture cellulaire DMEM (avec suppléments).
- Transférer la suspension cellulaire dans un flacon de culture cellulairejetable de 75 cm 2 et incuber à 37 °C et 5 % de CO2 pendant encore 3 jours avant de subir une préparation sphéroïde. Une fois que les HepG2 ont eu le temps de s’acclimater et d’atteindre une fois de plus ~80% de confluence, déterminez la concentration cellulaire en préparation de l’ensemencement sphéroïde.
2. Préparation sphéroïde HepG2
- Répétez les étapes de sous-culture indiquées ci-dessus, sauf après centrifugation, suspendez la pastille cellulaire dans 1 mL de milieu de culture DMEM préchauffé dans un bain d’eau de 37 °C. Suspension cellulaire pipette de haut en bas à fond.
- Score de viabilité cellulaire à l’aide de l’essai trypan blue exclusion assay (voir OSHA SOP 3.21 Reproductive Toxins, Mutagens, Teratogens and Embryotoxins – Procedures for Safe Handling and Storage (2019) for health and safety guidance)14 avec un rapport de 1:1 de suspension cellulaire à la solution bleue Trypan pré-filtrée de 0,4 %.
- Avant le comptage cellulaire, prenez 1 mL de solution bleue Trypan à l’aide d’une seringue de 1 mL et filtrez avec une unité de filtre de 0,45 μm dans un tube stérile de 1 mL.
- Transférer 10 μL de solution bleue trypan filtrée dans un tube de 0,2 mL et ajouter 10 μL de suspension cellulaire. La solution bleue Trypan filtrée restante peut être stockée jusqu’à 3 mois à température ambiante pour une utilisation future.
- Vaporiser l’héémocytomètre à fond avec 70% d’éthanol et essuyer à sec avec une serviette en papier stérile avant de fixer le coverslip sur le dessus à l’aide de vapeur d’haleine. Glisser le coverslip sur la surface humidifié de souffle induit des forces cohésives en générant des anneaux de Newton.
- Pipette doucement la suspension de cellules bleues Trypan de haut en bas à l’aide d’une pipette de 1000 μL (pour réduire le stress) avant d’ajouter 10 μL à l’héémocytomètre. Assurez-vous que la solution est dispersée sous le bordereau de couverture et couvre toute la grille sans bulles d’air.

Figure 1 :Compter les cellules à l’aide d’un héémocytomètre. Représentation schématique d’un héémocytomètre soulignant de quel quadrant compter les cellules. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.
- Au microscope, comptez les cellules vivantes (non tachées) et mortes (bleu teinté) trouvées dans les quatre grands carrés d’angle (figure 1). Exclure les cellules qui se chevauchent ou s’asseoir sur les deux bords intérieurs des grands carrés d’angle (c.-à-d. sur les lignes) dans le compte.
- À l’aide du calcul suivant, calculer le nombre moyen de cellules vivantes viables (non tachées) présentes dans l’échantillon :
Nombre total de cellules/mL = Nombre de cellules vivantes x x 10 000
vivantes x x 10 000
où la dilution se réfère au nombre de fois où la solution de stock a été diluée en bleu Trypan (2x dans ce cas) et # de carrés comptés se réfère aux quatre grands carrés d’angle de l’héémocytomètre compté - Basé sur le nombre viable de cellules HepG2 et en utilisant la formule suivante:
C1V1=C2V2
où C1 = la concentration de cellules viables actuellement,
V1 = le volume de suspension cellulaire actuellement,
C2 = la concentration de suspension cellulaire recherchée,
V2 = le volume de suspension cellulaire recherché - Préparer une solution de stock de 10 mL de suspension cellulaire HepG2 avec milieu de culture cellulaire DMEM à une concentration de 2,0 x 105 cellules/mL afin d’atteindre 4000 cellules HepG2 par 20 μL de chute suspendue. Mélangez soigneusement la suspension cellulaire en entcheminez doucement de haut en bas à l’aide d’une pipette de 1000 μL pour vous assurer que toutes les cellules sont complètement suspendues dans les médias.
- Aux puits d’une plaque de culture cellulaire de 96 puits, ajouter 100 μL de PBS stérile à température ambiante pour empêcher les gouttes suspendues de se dessécher pendant l’incubation.
- Prenez le couvercle d’une plaque de culture cellulaire à fond plat standard de 96 puits, inversionz-la et pipette soigneusement 20 gouttes de μL de la suspension cellulaire dans le centre de chaque rainure de puits du couvercle, comme le montre la figure 2. Utilisez une pipette multicanal, mais ajoutez seulement 2 à 4 gouttes à la fois car plusieurs semis peuvent affecter la précision et le placement des gouttes.
- Centrer les gouttes dans les rainures des puits disposés sur le couvercle; sinon, ils ne seront pas suspendus au centre des puits lorsque le couvercle de la plaque est retourné et risquent de tomber dans la plaque. Retournez doucement le couvercle de la plaque de 96 puits, de sorte que les gouttes sont maintenant suspendues et soigneusement placer sur le dessus de la plaque de 96 puits.
- Placer délicatement l’ensemble de la plaque de 96 puits avec couvercle dans un incubateur à 37 °C et 5 % de CO2 pendant 3 jours avant le transfert de sphéroïdes sur l’agarose.
REMARQUE : Il faut faire un soin supplémentaire non seulement lors du transport des plaques à l’entrée et à l’provenance des incubateurs, mais aussi lors de l’ouverture et de la fermeture de l’incubateur en général, car un mouvement excessif peut faire changer les plaques et les sphéroïdes tomber ou se former de façon incorrecte.
 vivantes x x 10 000
vivantes x x 10 000
Figure 2: Préparation in vitro du modèle sphéroïde 3D HepG2. (A) Les cellules HepG2 ensemencées en 20 μL tombent sur le couvercle d’une plaque de 96 puits. (B) Les cellules HepG2 post-ensemencement dans le modèle de chute suspendue pour permettre la formation de sphéroïdes. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.
3. Transfert sphéroïde HepG2 de la chute suspendue à la suspension agarose
REMARQUE : Le jour 3 après l’ensemencement en gouttes suspendues, les sphéroïdes sont transférés dans les puits de la même plaque de 96 puits qui ont tous déjà été recouverts d’une fine couche de gel d’agarose de 1,5 %.
- Préparer les gels d’agarose et l’autoclave (c.-à-d. le jour 2 après l’ensemencement) avant le jour du revêtement des plaques (c.-à-d. le jour 3 après l’ensemencement).
- Pour préparer un gel d’agarose de 1,5 %, peser 0,30 g d’agarose dans une bouteille en verre propre, puis ajouter 20 ml de dMEM sans phénol. Autoclave l’agarose pendant 1 h à 230 °C pour la stérilisation. Le revêtement agarose empêche les sphéroïdes HepG2 d’adhérer à la base des puits et de former un monocouche cellulaire au lieu de conserver leur structure sphéroïde 3D.
- Le jour 3 après l’ensemencement, retirez la plaque de 96 puits contenant les sphéroïdes de chute suspendus HepG2 hors de l’incubateur et retournez soigneusement le couvercle afin que les sphéroïdes ne soient plus suspendus.
- À l’aide d’une pipette multicanal, retirer et jeter les 100 μL de PBS précédemment ajoutés à la base de la plaque de 96 puits. Laisser les plaques aérer les airs pendant 2-3 min tout en chauffant l’agarose en préparation pour le revêtement.
MISE EN GARDE : Cette procédure entraîne une agarose liquide très chaude qui, si elle est renversée sur la peau, peut brûler et causer des blessures. En outre, le soin doit être pris lors de la manipulation de la bouteille en verre contenant l’agarose liquide que cela aussi peut être très chaud. - À l’aide des gels agarose de 1,5 % préalablement préparés, chauffer la bouteille en verre contenant le gel agarose de 20 ml pendant 30 s au micro-ondes au maximum (c.-à-d. 900 W). Pour enrober deux assiettes de 96 puits, une bouteille de 20 ml de gel agarose pré-préparé à 1,5 % devrait suffire.
- Une fois fondu, tourbillonnez doucement l’agarose en faisant pivoter la bouteille en verre pour enlever les bulles, puis ajoutez 50 μL d’agarose dans la base de chaque puits.
REMARQUE : Lors de l’ajout de l’agarose, assurez-vous de ne pas incliner la plaque >45° à mesure que l’agarose se couche rapidement et ne formera pas une couche plate et plane qui peut perturber la croissance des sphéroïdes. Il est important de travailler efficacement à ce stade pour empêcher l’agarose de se solidifier avant que la plaque ne soit complètement enduire. - Laisser reposer la plaque pendant 2 min à température ambiante avant d’ajouter 100 μL de milieu de culture cellulaire DMEM préchauffé (avec suppléments) sur la couche d’agarose solide dans chaque puits.
- Retournez le couvercle de la plaque de 96 puits et placez-le de nouveau sur la plaque de 96 puits afin que les sphéroïdes soient à nouveau suspendus.
- Centrifuger la plaque pendant 3 min à 200 x g afin de transférer les sphéroïdes de la gouttelette suspendue dans les puits individuels de la plaque de 96 puits. Après le transfert, les sphéroïdes HepG2 devraient maintenant être suspendus dans le milieu de culture cellulaire. Laissez-leur se contenter de 24 h dans l’incubateur à 37 °C et 5 % de CO2.
- Exposer les sphéroïdes HepG2 de cette taille à des traitements chimiques ou ENM le jour 4 après l’ensemencement (c.-à-d. 24 h après transfert dans des plaques enduites d’agarose).
- Afin de maintenir la viabilité cellulaire sur de longues périodes de culture, rafraîchir le milieu de culture cellulaire tous les 3 jours. Pour ce faire, aspirez doucement 50 μL du milieu de culture cellulaire de la surface du puits et remplacez-les par un nouveau milieu de culture cellulaire de 50 μL de DMEM. Prenez soin de ne pas enlever ou déranger le sphéroïde lors d’un changement moyen.
4. Exposition aux nanomatériaux et aux produits chimiques
REMARQUE : Le modèle sphéroïde hépatique HepG2 peut soutenir à la fois les régimes d’exposition à base d’ENM et de produits chimiques, mais l’objectif principal de ce protocole est l’exposition à l’ENM. Avant l’exposition, l’ENM d’essai doit être convenablement dispersé; cela peut être réalisé selon les directives du Protocole de dispersion NanoGenoTox (Accord de subvention n° 20092101, 2018)15.
- Après dispersion selon le protocole de dispersion NanoGenoTox, diluer la suspension ENM de la concentration de départ de 2,56 mg/mL à la concentration finale désirée dans le milieu de culture cellulaire DMEM préchauffé (y compris les suppléments). Un volume total de 5 mL est nécessaire pour doser une plaque de puits de 96.
- Pour exposer le sphéroïde HepG2 à un produit chimique ou à un ENM, à l’aide d’une pipette de 200 μL, aspirez 50 μL de milieu de culture cellulaire à partir de la surface de chaque puits (laissant 50 μL dans le puits afin de ne pas déranger les sphéroïdes) et remplacez-les par un milieu de 50 μL contenant le toxicant d’essai à la dose requise.
- Une fois que le matériau d’essai a été appliqué, incuber les plaques pour le temps d’exposition désiré à 37 °C et 5 % de CO2.
- Si un régime d’exposition à long terme (≥24 h) est effectué, alors immédiatement après l’écoulement du délai d’exposition souhaité, récolter les sphéroïdes pour l’analyse du point de terminaison des micronucléus décrite ci-dessous dans les étapes 6.1 - 6.4.
- Toutefois, avec les régimes d’exposition aiguë (p. ex., ≤24 h), une fois la période d’exposition terminée, récolter, mettre en commun et stocker 50 μL de supernatant de chaque puits dans la plaque de puits 96 à -80 °C pour une analyse biochimique plus poussée plus tard. Remplacer le milieu de culture cellulaire par 50 μL de milieu frais contenant 6 μg/mL de cytochalasine B et laisser incuber pendant 1 à 1,5 cycle cellulaire (c.-à-d. 24 à 26 h pour l’hepG2) en préparation de la récolte d’analyse de micronucléus de bloc de cytokinésie.
REMARQUE : Pour les régimes d’exposition aiguë (≤24 h), l’essai de micronucléus de bloc de cytokinésie avec la cytochalasine B peut être appliqué mais pour les régimes d’exposition à long terme (≥24 h), la version mononucléaire (sans cytochalasine B) du test doit être utilisée comme décrit ci-dessous dans la figure 4.
5. Récolte de sphéroïdes HepG2
REMARQUE : À la suite de traitements d’exposition chimiques ou ENM, le milieu de culture cellulaire ou le tissu sphéroïde peuvent être récoltés pour une analyse multiple des points de terminaison. Selon l’analyse du point de terminaison, les sphéroïdes peuvent être récoltés individuellement (p. ex., pour l’analyse d’images) ou mis en commun (p. ex., pour l’analyse micronucléaire du bloc de cytokinésie).
- Retirer la plaque de 96 puits de l’incubateur.
- À l’aide d’une pipette de 200 μL, aspirer les 100 μL de milieu de culture cellulaire, y compris le tissu sphéroïde de chaque puits, et les recueillir dans un tube stérile de centrifugeuse de 15 mL. Prenez soin d’éviter tout contact avec l’agarose.
- Une fois collectée, centrifuger la suspension sphéroïde à 230 x g pendant 5 min. Retirez le supernatant et rangez-le à -80 °C pour une analyse plus approfondie du point de terminaison (p. ex., tests de la fonction hépatique) plus tard.
- Suspendre à nouveau la pastille de sphéroïdes dans 1 mL de PBS stérile à température ambiante (1x).
- Une fois lavée, centrifugeuse la suspension sphéroïde à nouveau à 230 x g pendant 3 min. Jeter le supernatant, suspendre à nouveau en 500 μL de 0,05% trypsine-EDTA solution et incuber pendant 6-8 min à 37 °C et 5% CO2.
- Après l’incubation, pipette doucement les cellules trypsinisées de haut en bas pour dissocier complètement et re-suspendre les cellules HepG2 avant de neutraliser avec 1 mL de milieu de culture cellulaire DMEM.
- Centrifugeuse la suspension cellulaire diluée à 230 x g pendant 5 min. Jeter le supernatant dans le désinfectant et suspendre à nouveau les granulés cellulaires dans 2 mL de PBS à température ambiante (1x).
- Centrifugeuse de la suspension cellulaire à 230 x g pendant 5 min. Jeter le supernatant dans le désinfectant, puis suspendre à nouveau la pastille cellulaire dans 2 mL de PBS froid (1x). Assurez-vous que les cellules sont bien dispersées pour éviter que des amas de cellules obscurcissent le champ de vision lorsqu’elles sont montées sur des toboggans au microscope.
6. Analyse et notation de Micronucleus
Pour la méthode manuelle de l’analyse de micronucléus, une cytocentrifugeuse est nécessaire pour produire un cytodot (une région définie et concentrée des cellules) au centre de la lame du microscope. Ce processus prend en charge la notation plus efficace de la diapositive car il permet au marqueur de localiser facilement les cellules d’intérêt, plutôt que d’évaluer une diapositive entière où les cellules peuvent être largement répandues.
- Tremper les diapositives de microscope givrées (trois par dose) dans 70 %d’éthanolsuivies du ddH 2 O et laisser sécher à l’air pendant 5 min.
- Placez les glissières de microscope préparées dans l’entonnoir de cuvette comme indiqué dans la figure 3A,où la glissière en verre (iii) est placée dans le support métallique (iv) avec une carte filtr filtre (ii) et un entonnoir de cuvette (i) fixé sur le dessus.
- Disposer les entonnoirs de cuvette dans la cytocentrifugeuse avec l’entonnoir orienté vers le haut, de sorte que 100 μL de suspension cellulaire peuvent être directement ajoutés dans chacun d’eux.
- Cytospin pendant 5 min à 500 x g pour s’assurer que les cellules sont réparties uniformément sur la surface de la lame.

Figure 3: Configuration de cytospin pour préparer les cellules traitées sur des diapositives de microscope. (A) Affiche les composants individuels, (i) entonnoir de cuvette, (ii) carte de filtre, (iii) glissière de microscope en verre et (iv) support métallique requis pour cytospiner des cellules d’HepG2 sur des glissières de microscope. (B) L’entonnoir cuvette final mis en place. (C) Le placement correct de l’entonnoir de cuvette dans la cytocentrifugeuse. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.
- Laisser sécher les toboggans à l’air libre avant la fixation dans le froid glacial, 90% de méthanol pendant 10 min.
- Une fois fixées, laisser sécher les glissières à l’air sec toute la nuit à température ambiante avant de les stocker à -20 °C jusqu’à 6 mois.
- Au besoin, retirer les glissières de microscope pré-préparées du congélateur de -20 °C et laisser chauffer à température ambiante avant d’entreprendre la coloration giemsa.
MISE EN GARDE : Selon le règlement n° 1272/2008 [CLP], la solution de coloration Giemsa est un liquide hautement inflammable qui peut être toxique s’il est avalé et causer des dommages au contact des yeux, de la peau ou s’il est inhalé. Consultez la feuille SDS associée pour obtenir des conseils détaillés en matière d’entreposage, de manipulation et de santé et de sécurité sur ce produit chimique avant l’utilisation. - Pendant que les glissières sont décongelées, préparez une solution de coloration Giemsa de 20 % (25 mL nécessaire pour tacher ~30 diapositives) diluée dans un tampon de phosphatase (pH 6,8). Bien mélanger en faisant pivoter doucement la solution avant de filtrer à l’aide de papier filtre plié placé dans un entonnoir.
- À l’aide d’une pipette Pasteur, ajouter 3 à 5 gouttes de solution Giemsa filtrée au cytodot sur chaque toboggan et laisser de 8 à 10 minutes.
- Lavez les glissières dans deux lavages tampons successifs de phosphatase avant de rincer brièvement sous l’eau froide pour enlever les restes excédentaires de taches. Laisser sécher les glissières à l’air libre.
- Une fois sec, dans un capot de fumée, tremper les glissières tachées dans le xylène pendant 10 s avant d’ajouter une goutte de milieu de montage au centre du cytodot et un endroit un coverslip en verre sur le dessus.
- Laisser sécher les glissières au microscope dans le capot des vapeurs pendant la nuit avant de les marquer manuellement; ils peuvent être stockés indéfiniment à température ambiante.
7. Analyse des données
- Tel que décrit dans les Lignes directrices d’essai 487 (2014)11de l’OCDE, pour évaluer et quantifier les dommages causés par l’ADN causés par l’exposition à un ENM ou à un agent chimique, utilisez un microscope léger (objectif 100x avec huile d’immersion) 2000 cellules mononucléées ou 1000 cellules binucléées par réplique biologique pour marquer la présence de micronucléi, comme le montre la figure 4.

Figure 4 : Arbre de décision de notation de notation de micronucléus. Arbre de décision schématique pour souligner la nécessité de différentes procédures d’évaluation de notation et de cytotoxicité lors de l’utilisation de l’essai de micronucléus avec des modèles 3D suivant des régimes aigus ou à long terme d’exposition. Les expositions aiguës (≤24 h) permettent l’utilisation de l’essai de micronucléus bloqué par cytokinésie, tandis que les expositions à long terme (≥24 h) nécessitent la version mononucléaire de l’essai; tous deux sont décrits dans la Ligne directrice d’essai 487 de l’OCDE. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.
- Basé sur la proportion de micronucléi présents par nombre de cellules mononucléées ou binucléées notées, calculer un pourcentage de la valeur de génotoxique.
- Afin d’évaluer les dommages à l’ADN observés n’est pas à la suite de débris cellulaires causés par une forte proportion de cellules apoptotiques, prendre une mesure de cytotoxicité à côté. Dans ce cas, selon la présence de cytochalasine B, utilisez le calcul cpbi ou rvcc (tel que décrit dans la figure 4). La génototoxicité ne doit être évaluée que dans les échantillons dont la cytotoxicité est inférieure à 55 % ± 5 % telle que définie dans la Ligne directrice d’essai 48711 de l’OCDE.
Representative Results
L’adéquation de ce modèle sphéroïde hépatique 3D basé sur la lignée cellulaire pour la culture à long terme et l’évaluation des risques génotoxiques a été évaluée en effectuant une caractérisation de base pour déterminer la viabilité et la fonctionnalité hépatique du modèle sur une durée de 14 jours en culture ainsi que son applicabilité pour l’essai de micronucléus.
Caractérisation de base du modèle sphéroïde hépatique 3D HepG2
Avant toute évaluation toxicologique in vitro, il est important de vérifier que les sphéroïdes 3D HepG2 se sont formés correctement avant d’effectuer le transfert d’agarose ou le traitement chimique/ENM. Les sphéroïdes HepG2 produits selon la méthode de chute suspendue prennent habituellement de 2 à 3 jours après l’ensemencement (4 000 cellules/sphéroïdes) pour former des sphéroïdes compacts en forme de sphérique d’un diamètre moyen de 495,52 μm W x 482,69 μm H, comme le montre la figure 5A-5C. Les sphéroïdes HepG2 qui se sont formés correctement et qui sont acceptables pour l’évaluation toxicologique in vitro doivent avoir une structure compacte en forme sphérique avec une surface lisse et aucune projection visuelle. La figure 5 fournit des exemples de sphéroïdes de bonne qualité (figure 5D-F) et de mauvaise qualité (figure5G-I). Ces derniers devraient être jetés. En règle générale, 90-95% des sphéroïdes formés par plaque se formeront correctement et seront viables pour d’autres expérimentations.

Figure 5: Images de microscopie légère montrant la morphologie naturelle des sphéroïdes HepG2 formés par la méthode de la chute suspendue. (A-C) montrent jour 2 et (D-I) Jour 4 HépG2 sphéroïdes hépatiques après l’ensemencement. (D-F) sont des exemples de sphéroïdes HepG2 de bonne qualité tandis que (G-I) montre sphéroïdes mal formés. Toutes les images ont été prises sur un objectif X20 à l’aide d’un microscope. La barre d’échelle représente 20 μm. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
Pour confirmer davantage la viabilité sphéroïde hepG2, un test d’analyse ou d’urea de base de Bromocresol Green Albumin (BCG) peut être effectué pour évaluer leur fonctionnalité hépatique. La fonctionnalité de base du foie a été évaluée en fonction de la viabilité à l’aide de l’essai trypan blue exclusion sur une période de culture de 14 jours afin de déterminer la longévité du modèle sphéroïde hépatique et d’établir si elle pouvait soutenir l’évaluation à long terme ou répétée des risques à base d’ENM/produits chimiques (figure 6). La concentration d’albumine est restée constante pendant toute la durée de la période culturelle. La production d’urée affiche une augmentation de la concentration d’urée produite par sphéroïde sur une semaine en culture avant d’atteindre un plateau au jour 7. Il est important de noter que les niveaux d’albumine et d’urée produits dans les sphéroïdes 3D HepG2 sont sensiblement plus élevés que ceux observés dans la même lignée cellulaire cultivé dans un format 2D. En effet, les cultures 2D des cellules HepG2, des niveaux d’albumine et d’urée de pointe étaient respectivement de 0,001 mg/mL et de 0,010 ng/μL. En outre, dans des travaux antérieurs publiés par Shah et coll. utilisant un système sphéroïde HepG2 presque identique, les auteurs soulignent une amélioration notable de l’activité métabolique (CYP1A1 et CYP1A2) dans les systèmes de modèle in vitro 3D HepG2 par rapport aux cellules hepG22D cultivés 5.

Figure 6 : Données de caractérisation de base de 14 jours pour les sphéroïdes hépatiques HepG2. Après le transfert de la chute suspendue, (A) met en évidence la viabilité du modèle sphéroïde HepG2 sur une période de 14 jours tandis que (B) et (C) mettre en évidence le foie comme l’albumine et l’urée fonctionnalité respectivement. Données ± SEM présentées, n = 4. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.
Avec le développement inévitable d’un noyau nécrotique, une limitation connue des cultures sphéroïdes hépatiques 3D, la viabilité de ce modèle basé sur l’hépatite 2 a dû être établie pour démontrer qu’il était capable de maintenir des régimes d’exposition à long terme (5-10 jours) tout en maintenant la capacité proliférative nécessaire pour soutenir l’essai de micronucléus5. En effet, ce modèle sphéroïde hépatique 3D a été montré pour conserver >70% viabilité sur 10 jours en culture. Sur cette base et en conjonction avec la fonctionnalité soutenue de type foie observée au cours de la période de culture de 14 jours ≥, ce modèle sphéroïde hépatique 3D peut ainsi soutenir à long terme, les régimes répétés d’exposition en MEN jusqu’à 10 jours (c.-à-d., avant que la viabilité des sphéroïdes ne tombe en dessous de 70 %). À titre de référence, il est conseillé que les niveaux d’albumine pour les sphéroïdes HepG2 ensemencés à 4000 cellules/sphéroïdes devraient être ≥20,0 ng/μL tandis que la production d’urée devrait être de ≥0,25 ng/μL avant d’effectuer une évaluation toxicologique in vitro avec ce modèle.
Évaluation de la génotoxicité des nanomatériaux d’ingénierie
Pour l’évaluation de génotoxicité, l’essai de micronucléus a été employé pour déterminer la présence des micronucléi suivant des expositions aiguës (24 h) et à long terme (120 h) d’ENM. Aflatoxin B1 est un carcinogène connudu foie 16,17 et est un contrôle positif recommandé pour l’analyse micronucléus. Des expériences d’optimisation ont montré que 0,1 μM d’Alfatoxin B1 induit une réponse génotoxique positive significative (≥ 2,0 fois plus) dans les sphéroïdes hépatiques 3D HepG2 et est donc utilisé dans chaque analyse micronucléaire menée avec ce modèle. Pour assurer la validité des résultats de l’analyse micronucléus à l’aide du modèle sphéroïde HepG2, la fréquence micronucléus de fond pour les cellules HepG2 utilisées dans ce modèle in vitro 3D devrait se situer dans une fourchette de 0,6 % à 1,2 %. En conséquence, Alfatoxin B1 devrait induire une réponse génotoxique d’au moins deux fois plus élevé que celle observée avec le contrôle négatif; ainsi, 0,1 μM d’Alfatoxin B1 devrait induire une fréquence de micronucléi entre 1,5% - 3,0%. À l’aide de ces paramètres de contrôle, la génotoxicité associée à l’ENM in vitro peut ensuite être évaluée de manière fiable. D’après la Ligne directrice 487 de l’OCDE sur les essais, il est important de noter que lors de l’essai d’un ENM ou d’un produit chimique, les concentrations sélectionnées ne devraient pas induire plus de 55 % ± une cytotoxicité de 5 % (indiquée par une réduction des valeurs cpbi ou rvcc par rapport au contrôle négatif)11. La figure 7 illustre les données générées lorsque l’Aflatoxine B1 et deux ENM (dioxyde de titane (TiO2) et ruban (Ag)) ont été évalués à la suite d’expositions aiguës et à long terme dans les sphéroïdes HepG2, et le potentiel génotoxique subséquent a été analysé à l’aide de l’analyse du micronucléus. Les deux ENM évalués ont été testés à une faible dose noncytotoxique de 5,00 μg/mL au cours d’une exposition aiguë (24 h) et d’un régime d’exposition à long terme (120 h). On peut observer une tendance similaire à la génotoxicité chez les ENM TiO2 et Ag, selon laquelle la réponse élevée de génotoxicité qui a résulté après une exposition de 24 h n’était pas évidente après une exposition à long terme de 5 jours. Ceci en dépit de la génotoxicité soutenue induite par le contrôle positif d’Aflatoxin B1 aux deux points de temps.

Figure 7 :Évaluation de la génotoxité à la suite de l’exposition de TiO2 et ag ENM sur les sphéroïdes hépatiques HepG2. Évaluation de la génotoxité (fréquence micronucléus) à l’aide du poste d’analyse micronucléus (A) aiguë (24heures)et ( B ) exposition à long terme (120 heures) à 5,00 μg/mL de TiO2 et Ag ENM. Le contrôle négatif est un média seulement, tandis que le contrôle positif est de 0,1 μM d’Aflatoxine B1. Données moyennes (n=2) présentées ± SD. Importance indiquée par rapport au contrôle négatif : * = p≤ 0,05. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.
Discussion
Les applications pour les modèles hépatiques 3D varient considérablement selon le point de terminaison biochimique particulier ou la voie défavorable de résultats étant visée. Chaque modèle a ses avantages et ses limites, de la variation interdonor dans les modèles primaires d’hépatocyte humain (PHH) à l’activité réduite de cytochrome p450 dans les modèles basés sur la ligne cellulaire, mais tous sont valables dans leur propredroit 6,12,18,19. Lors de l’évaluation de la génotoxicité, il y a des limites dans la compatibilité des modèles avec les critères d’évaluation approuvés par la réglementation tels que l’analyse in vitro du micronucléus, car la prolifération active est nécessaire. Ceci est nécessaire, car l’évaluation de génotoxicité exige la quantification des dommages fixes d’ADN pour être évaluée division de poteau de cellule quand il y a l’occasion pour la réparation d’ADN pour corriger des lésions passagères. Malheureusement, les hépatocytes à base d’hépatocytes hautement différenciés (c.-à-d. les sphéroïdes à base d’HépaRG) ou microtissues PHH, qui sont réputés présenter les caractéristiques hépatiques les plus pertinentes sur le plan physiologique, forment des modèles statiques (non prolifératifs)12,19,20. En conséquence, le modèle sphéroïde 3D HepG2 présenté ici fournit un modèle alternatif approprié capable de soutenir les tests de génotoxicité. Les sphéroïdes à base de lignée cellulaire HepG2 ont suffisamment de cellules de division active sur la surface externe des sphéroïdes tout en conservant des caractéristiques hépatiques de base, telles que la production d’albumine et d’urée et certaines activités CYP4505,12,19. Principalement ce modèle de foie in vitro a été développé pour compléter l’analyse de micronucléus, car c’est l’un des deux essais in vitro recommandés dans la batterie pour l’essai de génotoxicité8,10,11,21. Cependant, le modèle peut être facilement appliqué aux technologies d’analyse du séquençage de l’ADN et d’expression des gènes (ARN), alors qu’il a le potentiel d’être davantage adapté et utilisé pour d’autres critères d’évaluation des dommages à l’ADN, tels que l’analyse de la comète. Néanmoins, il est important de tenir compte du rôle que joue l’interférence ENM dans certaines analyses de points de terminaison. Par exemple, les analyses basées sur la cytométrie de flux peuvent ne pas convenir à l’évaluation de la génotoxicité enM spécifiquement en raison de l’interférence desparticules 22.
Un facteur limitant des modèles sphéroïdes qui subissent activement la division cellulaire est leur taille. L’optimisation de la densité d’ensemencement est essentielle car il doit y avoir suffisamment de cellules qui permettent au modèle de continuer à proliférer; mais pas trop élevé un nombre de cellules, ce qui entraîne le sphéroïde devient trop compact, conduisant à un noyau nécrotique accru. La cause de cette nécrose est considérée comme limitant la diffusion de l’oxygène et des nutriments, car la limite de cette diffusion est considérée comme environ 100 - 150 μm de tissu23,24. Toutefois, cela dépend du type de cellule, du numéro de cellule, des interactions d’échafaudage et des conditions de culture25. Depuis, il a été démontré qu’environ 700 μm de diamètre est la limite pour éviter l’apparition prématurée de la nécrose au centre des sphéroïdes C3A, l’ensemencement de 4000 cellules HepG2 par sphéroïde assure le diamètre du modèle au moment de l’exposition est de ≤500 μm26. En outre, Shah et coll. ont établi que les cellules HepG2 ensemencées au-dessus de 5000 cellules par sphéroïde présentaient une réduction de 25 % de la viabilité après 7 jours de culture, ce qui pourrait se rapporter au diamètre moyen de 680 μm et à la disponibilité limitée des nutriments dans une goutte suspendue de 20 μL5. Pour surmonter cela, le modèle conçu dans le présent protocole subit une étape critique où la goutte suspendue est transférée dans des puits recouverts d’agarose après la formation initiale du sphéroïde. Cela garantit un plus grand volume de milieu de culture est présent pour soutenir le nombre sans cesse croissant de cellules dans les sphéroïdes. En conséquence, le modèle sphéroïde HepG2 reste plus de 70% viable après 10 jours en culture et peut être utilisé pour l’évaluation des risques à long terme in vitro.
Tandis que le modèle sphéroïde d’HepG2 peut soutenir des régimes aigus et à long terme d’exposition, le milieu rafraîchissant de culture cellulaire pendant des périodes prolongées de culture est limité pour ce modèle car le remplacement complet du milieu n’est pas conseillé en raison de la perte potentielle des sphéroïdes. On présume qu’avec les expositions à l’ENM, la tendance à des dispersions homogènes de l’ENM à l’aggloméate et aux sédiments est élevée. Toutefois, il est à noter que la vitesse à laquelle un ENM sédimente peut varier en fonction des paramètres des particules (p. ex., taille, forme et densité) et peut être déterminée théoriquement à l’aide du modèle de sédimentation, de diffusion et de dosimétrie in vitro (DISD), ou de ses dérivés récents, souvent mentionnés lorsqu’il s’agit de l’exposition à l’ENM (suspension),approche 27,28. Avec ceci est esprit, on suppose que si seulement 50% du milieu de culture cellulaire est soigneusement enlevé de la surface de la culture cellulaire, la perturbation et l’enlèvement ultérieur de la dose d’ENM devraient en théorie être minimes. Toutefois, avec le mouvement brownien en jeu, ce n’est peut-être pas strictement le cas et d’autres travaux de dépôt et de sédimentation de chaque ENM à tester devraient être entrepris pour s’assurer que la dosimétrie correcte est conservée tout au long des régimes d’exposition à long terme27. Il s’agit principalement d’une limitation potentielle à considérer lors de l’exécution de régimes de dosage répétés, car cela pourrait être essentiel à la concentration finale accumulée. Les expositions à base de produits chimiques, d’autre part, bien que non sans leurs propres limites à considérer, offrent une approche plus simpliste en ce que les substances chimiques ont tendance à rester en solution et donc un remplacement direct de la concentration chimique d’origine en plus de la concentration nouvellement ajoutée garantit que tout produit chimique perdu lors du rafraîchissement des médias est remplacéen conséquence 29. Les applications futures comprendraient l’évaluation de l’adéquation du modèle pour les régimes d’exposition répétés sur des périodes de culture à long terme, car les stratégies de dosage répétées sont d’une importance cruciale pour évaluer la capacité d’un système d’organes particulier à améliorer ou à surmonter les effets indésirables, le cas échéant, induits par la bioaccumulation d’une substance xénobiobiotique.
En conclusion, ce modèle hépatique in vitro 3D a la capacité d’être utilisé pour évaluer une gamme de scénarios d’exposition réalistes, offrant ainsi une approche in vitro future pour mieux soutenir l’ENM et l’évaluation des risques chimiques d’une manière routinière et facilement accessible.
Disclosures
Les auteurs n’ont rien à divulguer.
Acknowledgments
Les auteurs aimeraient reconnaître que cette recherche a reçu des fonds du programme de recherche et d’innovation Horizon 2020 de l’Union européenne pour le projet PATROLS, en vertu de l’accord de subvention n° 760813
Materials
| Name | Company | Catalog Number | Comments |
| Aflotoxin B1 | Sigma Aldrich, UK | A6636-5MG | |
| Agarose | Sigma Aldrich, UK | A9539-50G | |
| Autoclave Tape | |||
| BCG Albumin Assay | Sigma Aldrich, UK | MAK124 | |
| Bovine Serum Albumin Powder | Sigma Aldrich, UK | A9418 | |
| Cell Freezing Aid | Thermo Fisher Scientific, UK | 5100-0001 - Mr Frosty | |
| Centrifuge | Eppendorf | 5810 R | |
| Cytochalasin B | Merck, UK | 250233 | |
| Cytology Metal Clips | |||
| Cytospin 4 Centrifuge | ThermoFisher Scientific, UK | CM00730202 | |
| DMEM with 4.5g/L D-Glucose, L-Glutamine | GIBCO, Paisley, UK | 41965-039 | |
| DMEM, phenol-red free with 4.5g/L D-Glucose, L-Glutamine with Hepes | GIBCO, Paisley, UK | 21063-029 | |
| DPX Mounting Medium | FisherScientific, UK | D/5330/05 | |
| Ethanol | FisherScientific, UK | 10048291 | |
| FBS | GIBCO, Paisley, UK | 10270-106 | |
| Filter Cards for Shandon Cytospin | FisherScientific, UK | 15995742 | |
| Frosted Glass Slides | ThermoFisher Scientific, UK | ||
| Giemsa's Stain Improved R66 Solution, Gurr | VWR Chemicals, UK | MFCD00081642 | |
| Glass Coverslips (24 x 60) | Deckglaser, VWR | ECN631-1575 | |
| Haemocytometer and Coverslip | |||
| Immersion Oil for Microscope | Zeiss, UK | 518F, ISO8034 | |
| Laminar Class II Tissue Culture Hood | Scanlaf Mars | ||
| Light Microscope | Zeiss, UK | Axiovert 40C | |
| Liquid Nitrogen | |||
| Methanol | FisherScientific, UK | 10284580 | |
| Microwave | |||
| Non-Filtered, Sterile 200µl and 1000µl Pipette tips | Greiner-Bio-One, UK | ||
| NuncMicroWell 96-Well Microplates | ThermoFisher Scientific, Denmark | 167008 | |
| P1000 and P200 micropipettes | |||
| P300 and P50 multi-channel pipettes | |||
| PBS pH 7.4 1X, MgCl2 and CaCl2 Free | GIBCO, Paisley, UK | 14190-094 | |
| Pen/Strep | GIBCO, Paisley, UK | 15140-122, Penicillin/Strepmyocin 100X or 10,000U/ml | |
| Phosphatase Buffer Tablets | GIBCO, Paisley, UK | 10582-013 | |
| Pipette Boy | |||
| Simport Scientific CytoSep Funnels for Shandon Cytospin 4 Centrifuges | FisherScientific, UK | 11690581 | |
| Sonifier SFX 550 240V CE 1/2" - Probe | Branson, USA | 101-063-971R | |
| T-25 and T-75 Tissue Culture Flask | Greiner-Bio-One, UK | T-25 (690175) and T-75 (660175) | |
| Trypan Blue Solution | Sigma Aldrich, UK | T8154-100mL | |
| Urea Assay Kit | Sigma Aldrich, UK | MAK006 | |
| Virkon Disinfectant | DuPont, UK | Rely+On Virkon | |
| Water Bath (37?C) | Grant JBNova 18 | ||
| Weighing Balance | |||
| Xylene | FisherScientific, UK | 10588070 | |
| 0.05% Trypsin-EDTA | GIBCO, Paisley, UK | 5300-054 | |
| 0.2mL and 1.0mL Eppendorf Tubes | Greiner-Bio-One, UK | ||
| 0.45µm Filter Unit | Millex HA, MF-Millipore, UK | SLHA033SS | |
| 1.0mL Syringe | BD Plastipak, FisherScientific, UK | 300185 | |
| 20mL LS Scintillation Glass Vials, 22-400 Foil Lined PP Caps | DWK Life Sciences GmbH, Germany | WHEA986581 | |
| 37?C and 5% CO2 ISO Class 5 Hepa Filter Incubator | NUAIRE DHD Autoflow | ||
| 3mL Pasteur Pipette | Greiner-Bio-One, UK | ||
| 50mL Conical Falcon Tubes | Greiner-Bio-One, UK | ||
| 50mL or 100mL Glass Bottles | |||
| 50mL Skirted Falcon Tubes | Greiner-Bio-One, UK | ||
| 5mL, 10mL and 25mL Pipettes | Greiner-Bio-One, UK | ||
| 9.4cm Square, Petri Dish | Greiner-Bio-One, UK | 688161 |
References
- Geiser, M., Kreyling, W. G. Deposition and biokinetics of inhaled nanoparticles. Particle and Fibre Toxicology. 7, 2 (2010).
- Modrzynska, J. Toxicological effects of nanoparticle deposition in the liver. Kgs. Lyngby, Denmark: Technical University of Denmark. , (2018).
- Elje, E., et al. The comet assay applied to HepG2 liver spheroids. Mutation Research/Genetic Toxicology and Environmental Mutagenesis. 845, 403033 (2019).
- Breslin, S., O'Driscoll, L. Three-dimensional cell culture: the missing link in drug discovery. Drug Discovery Today. 18, 240-249 (2013).
- Shah, U. -K., et al. A three-dimensional in vitro HepG2 cells liver spheroid model for genotoxicity studies. Mutation Research/Genetic Toxicology and Environmental Mutagenesis. 825, 51-58 (2018).
- Lauschke, V. M., Hendriks, D. F. G., Bell, C. C., Andersson, T. B., Ingelman-Sundberg, M. Novel 3D Culture Systems for Studies of Human Liver Function and Assessments of the Hepatotoxicity of Drugs and Drug Candidates. Chemical Research in Toxicology. 29, 1936-1955 (2016).
- van Grunsven, L. A. 3D in vitro models of liver fibrosis. Advanced Drug Delivery Reviews. 121, 133-146 (2017).
- Corvi, R., Madia, F. In vitro genotoxicity testing - can the performance be enhanced. Food and Chemical Toxicology. 106, 600-608 (2017).
- Doak, S. H., Manshian, B., Jenkins, G. J. S., Singh, N. In vitro genotoxicity testing strategy for nanomaterials and the adaptation of current OECD guidelines. Mutation Research/Genetic Toxicology and Environmental Mutagenesis. 745, 104-111 (2012).
- Fenech, M. Cytokinesis-block micronucleus cytome assay. Nature Protocols. 2, 1084-1104 (2007).
- OECD. OECD Guidelines. Test 489: In vivo Mammalian Alkaline Comet Assay. , (2016).
- Gerets, H. H. J., et al. Characterization of primary human hepatocytes, HepG2 cells, and HepaRG cells at the mRNA level and CYP activity in response to inducers and their predictivity for the detection of human hepatotoxins. Cell Biology and Toxicology. 28, 69-87 (2012).
- Sison-Young, R. L., et al. A multicenter assessment of single-cell models aligned to standard measures of cell health for prediction of acute hepatotoxicity. Archives of Toxicology. 91, 1385-1400 (2017).
- European Guidelines 2019. European Agency for Safety and Health at Work. , Available from: https://osha.europa.eu/en/safety-and-health-legislation/european-guidelines (2019).
- Jensen, K. A. The NANOGENOTOX Dispersion Protocol for NANoREG. European Union Grant Agreement n° 2009. 21, 01 (2014).
- Marchese, S., et al. Aflatoxin B1 and M1: Biological Properties and Their Involvement in Cancer Development. Toxins. 10, 214 (2018).
- Rushing, B. R., Selim, M. I. Aflatoxin B1: A review on metabolism, toxicity, occurrence in food, occupational exposure, and detoxification methods. Food and Chemical Toxicology. 124, 81-100 (2019).
- Kermanizadeh, A., Brown, D. M., Moritz, W., Stone, V. The importance of inter-individual Kupffer cell variability in the governance of hepatic toxicity in a 3D primary human liver microtissue model. Scientific Reports. 9, 7295 (2019).
- Berger, B., et al. Comparison of Liver Cell Models Using the Basel Phenotyping Cocktail. Frontiers in Pharmacology. 7, 443 (2016).
- Ramaiahgari, S. C., et al. A 3D in vitro model of differentiated HepG2 cell spheroids with improved liver-like properties for repeated dose high-throughput toxicity studies. Archives of Toxicology. , (2014).
- Li, Y., et al. Factors affecting the in vitro micronucleus assay for evaluation of nanomaterials. Mutagenesis. 32 (1), 151-159 (2016).
- Kirkland, D., Reeve, L., Gatehouse, D., Vanparys, P. A core in vitro genotoxicity battery comprising the Ames test plus the in vitro micronucleus test is sufficient to detect rodent carcinogens and in vivo genotoxins. Mutation Research/Genetic Toxicology and Environmental Mutagenesis. 721 (1), 27-73 (2011).
- Curcio, E., et al. Mass transfer and metabolic reactions in hepatocyte spheroids cultured in rotating wall gas-permeable membrane system. Biomaterials. 28, 5487-5497 (2007).
- Glicklis, R., Merchuk, J. C., Cohen, S. Modeling mass transfer in hepatocyte spheroids via cell viability, spheroid size, and hepatocellular functions. Biotechnology and Bioengineering. 86, 672-680 (2004).
- Asthana, A., Kisaalita, W. S. Microtissue size and hypoxia in HTS with 3D cultures. Drug Discovery Today. 17, 810-817 (2012).
- Gaskell, H., et al. Characterization of a functional C3A liver spheroid model. Toxicology Research. 5, 1053-1065 (2016).
- Cho, E. C., Zhang, Q., Xia, Y. The effect of sedimentation and diffusion on cellular uptake of gold nanoparticles. Nature Nanotechnology. 6, 385-391 (2011).
- Hinderliter, P. M., et al. ISDD: A computational model of particle sedimentation, diffusion and target cell dosimetry for in vitro toxicity studies. Particle and Fiber Toxicology. 7, 36 (2010).
- Kramer, N. I., di Consiglio, E., Blaauboer, B. J., Testai, E. Biokinetics in repeated-dosing in vitro drug toxicity studies. Toxicology in vitro. 30, 217-224 (2015).
Tags
Bioingénierie numéro 160 Modèles hépatiques in vitro Nanomatériaux Évaluation des dangers Exposition à long terme Nano(geno)toxicologie Dommages à l’ADNErratum
Formal Correction: Erratum: Advanced 3D Liver Models for In vitro Genotoxicity Testing Following Long-Term Nanomaterial Exposure
Posted by JoVE Editors on 01/26/2021.
Citeable Link.
An erratum was issued for: Advanced 3D Liver Models for In vitro Genotoxicity Testing Following Long-Term Nanomaterial Exposure. The Representative Results section was updated.
Figure 6 in the Representative Results section was updated from:
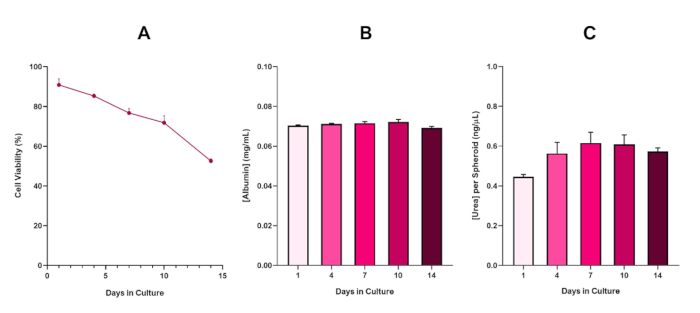
to:
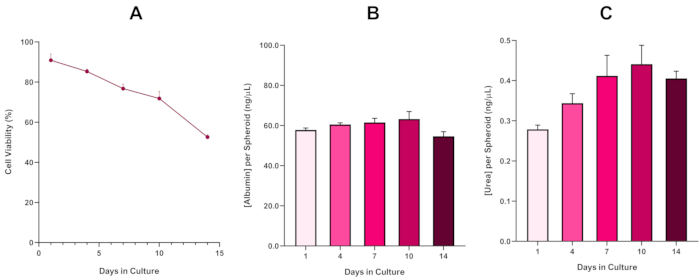
The fourth paragraph in the Representative Results section was updated from:
With the inevitable development of a necrotic core, a known limitation of 3D liver spheroid cultures, the viability of this HepG2 based model had to be established to demonstrate it was able to sustain long-term (5-10 day) exposure regimes whilst maintaining the proliferative capability required to support the micronucleus assay5. Indeed, this 3D liver spheroid model has been shown to retain >70% viability over 10 days in culture. Based on this and in conjunction with the sustained liver-like functionality observed over the ≥14 day culture period, this 3D liver spheroid model can thus support long-term, repeated ENM exposure regimes up to 10 days long (i.e., before viability of the spheroids drop below 70%). For reference, it is advised that albumin levels for HepG2 spheroids seeded at 4000 cells/spheroid should be ≥0.06 mg/mL whilst urea production should be ≥0.4 ng/µL before conducting an in vitro toxicological assessment with this model.
to:
With the inevitable development of a necrotic core, a known limitation of 3D liver spheroid cultures, the viability of this HepG2 based model had to be established to demonstrate it was able to sustain long-term (5-10 day) exposure regimes whilst maintaining the proliferative capability required to support the micronucleus assay5. Indeed, this 3D liver spheroid model has been shown to retain >70% viability over 10 days in culture. Based on this and in conjunction with the sustained liver-like functionality observed over the ≥14 day culture period, this 3D liver spheroid model can thus support long-term, repeated ENM exposure regimes up to 10 days long (i.e., before viability of the spheroids drop below 70%). For reference, it is advised that albumin levels for HepG2 spheroids seeded at 4000 cells/spheroid should be ≥50.0 ng/μL whilst urea production should be ≥0.25 ng/µL before conducting an in vitro toxicological assessment with this model.
