

ERRATUM NOTICE
Important: There has been an erratum issued for this article. Read more …
Summary
Este procedimento foi estabelecido para ser utilizado para o desenvolvimento de culturas hepáticas 3D avançadas in vitro, que podem fornecer uma avaliação mais fisiologicamente relevante dos perigos genotoxicos associados a exposições nanomateriais em regimes de dose aguda ou a longo prazo.
Abstract
Devido ao rápido desenvolvimento e implementação de uma variedade diversificada de nanomateriais projetados (ENM), a exposição ao ENM é inevitável e o desenvolvimento de sistemas de teste in vitro robustos e preditivos é essencial. A toxicologia hepática é fundamental quando se considera a exposição ao ENM, pois o fígado serve a um papel vital na homeostase metabólica e na desintoxicação, além de ser um dos principais locais de acumulação de ENM após a exposição. Com base nisso e no entendimento aceito de que os modelos de hepatócitos 2D não imitam com precisão as complexidades das interações multicelulares intrincadas e da atividade metabólica observada in vivo, há um maior foco no desenvolvimento de modelos hepáticos 3D fisiologicamente relevantes sob medida para fins de avaliação de riscos ENM in vitro. De acordo com os princípios dos 3Rs para substituir, reduzir e refinar a experimentação animal, foi desenvolvido um modelo hepático baseado em células 3D HepG2, que é um sistema fácil de usar e econômico que pode suportar regimes de exposição enm estendidos e repetidos (≤14 dias). Esses modelos esferoides (≥500 μm de diâmetro) mantêm sua capacidade proliferativa (ou seja, modelos celulares divisórias) permitindo que eles sejam acoplado ao ensaio de micronucleo 'padrão ouro' para avaliar efetivamente a genotoxicidade in vitro. Sua capacidade de relatar uma gama de pontos finais toxicológicos (por exemplo, função hepática, (pro-)resposta inflamatória, citotoxicidade e genotoxicidade) tem sido caracterizada usando vários ENMs em regimes de exposição agudos (24 h) e de longo prazo (120 h). Este modelo hepático in vitro 3D tem a capacidade de ser utilizado para avaliar exposições enm mais realistas, fornecendo assim uma futura abordagem in vitro para melhor suportar a avaliação de riscos ENM de forma rotineira e de fácil acesso.
Introduction
Devido ao rápido desenvolvimento e implementação de uma variedade diversificada de nanomateriais projetados (ENM) em uma infinidade de aplicações de base humana (por exemplo, alimentos, cosméticos, vestuário, equipamentos esportivos, eletrônicos, transporte e medicina), é inevitável que os seres humanos sejam expostos ao ENM regularmente. Com isso, há preocupações crescentes de que o novo, tamanho de características fisio-químicas específicas que considerem esses materiais vantajosos em inúmeras aplicações, possa causar efeitos adversos sobre a saúde humana e o meio ambiente concomitantemente. Atualmente, muitas atividades internacionais estão em vigor para refletir ativamente exposições mais fisiologicamente relevantes a esses ENM e avaliar a toxicidade potencial desses materiais em cenários de exposição aguda, de longo prazo e repetidos de baixa dose.
Toxicologia hepática é fundamental quando se considera a exposição ao ENM, pois é amplamente conhecido que o fígado é um dos principais locais de acumulação de ENM pós exposição1,2. Além disso, o fígado é o sistema de órgãos primários para metabolismo e desintoxicação de substâncias que entram em circulação sistêmica3. Com base no entendimento aceito de que modelos hepatócitos 2D não imitam com precisão as complexidades de interações multicelulares intrincadas ou representam adequadamente a atividade metabólica observada in vivo, um maior foco no desenvolvimento de modelos hepáticos in vitro 3D robustos e fisiologicamente relevantes para tecnologias substitutas in vivo foi estabelecido4,5. A utilização de tecnologias avançadas de cultura 3D melhora a longevidade de modelos hepáticos in vitro, permitindo que regimes de exposição repetidas e de longo prazo sejam investigados. Além disso, esse formato de cultura avançada promove a formação de características fisiológicas e organotípicas aprimoradas, como bile canaliculi, processos de transporte ativo e capacidades de metabolização de medicamentos CYP450 aprimorados, melhorando assim a previsibilidade dos modelos6. Modelos hepáticos in vitro atuais 3D constituídos por monoculturas (somente hepatócitos) ou co-culturas (hepatócitos com células não equilíquicas) existem em vários formatos, desde microtissues ou esferoides em placas de aderência ultralow, esferoides de gota suspensa, células embutidas em matrizes e/ou andaimes e plataformas de cultura celular microfluida, todos os quais são considerados modelos avançados in vitro eficazes para avaliação de toxicidade hepática6,7. No entanto, a maioria desses sistemas de modelos são de alta manutenção, exigem equipamentos especializados e são caros. Além disso, esses modelos são frequentemente estáticos (ou seja, modelos de células nãondividing) que impedem seu uso na avaliação de pontos finais de perigo, como testes de genotoxicidade utilizando métodos que quantificam danos consertos de DNA. A genotoxicidade é um pré-requisito central na toxicologia regulatória, e é um componente vital da avaliação de risco de qualquer toxicante8. Não há um único ensaio que possa ser aplicado para quantificar todas as formas de dano de DNA que possam surgir após a exposição a um agente exógeno. No entanto, um componente central da bateria de teste de genotoxicidade in vitro é o ensaio de micronucleus, que é uma técnica confiável e multifacetada que mede danos cromossômicos brutos9. É uma técnica padrão-ouro descrita pela Diretriz de Teste da OCDE 487, para avaliação de danos in vitro de DNA e genotoxicidade e faz parte do requisito da bateria de teste para avaliação de risco regulatório10,11.
A linha celular hepatocelular humana, HepG2, é amplamente utilizada para a triagem inicial de avaliação de riscos, pois as células estão prontamente disponíveis, relativamente baratas à fonte, simples à cultura e amenáveis à triagem de alto rendimento12,13. Quando cultivadas em estruturas esféricas 3D, elas têm sido demonstradas para recapitular bem o microambiente hepático e oferecer um modelo hepático com capacidades proliferativas suficientes para suportar o ensaio de micronucleus3. Foi estabelecido mais desenvolvimento dos modelos esferoides HepG2 para melhorar a longevidade e a funcionalidade hepática do modelo, a fim de apoiar a avaliação de risco genotoxicidade em regimes de exposição repetidas a longo prazo (≤14 dias). Assim, em consonância com os princípios dos 3Rs para substituir, reduzir e refinar a experimentação animal, o presente protocolo foi estabelecido para fornecer um modelo hepático in vitro avançado 3D capaz de avaliar de forma confiável múltiplos pontos finais toxicológicos (por exemplo, funcionalidade hepática, (pro-)marcadores inflamatórios, citotoxicidade e genotoxicidade) seguindo exposições químicas agudas, de longo prazo e repetidas e ENM em uma rotina e de forma facilmente acessível.
Aqui, apresentamos um método para estabelecer uma linha de células hepatócitos 3D fisiologicamente relevante baseada em sistema de modelo in vitro para avaliação de risco de genotoxicidade após exposições agudas ou de longo prazo do ENM. O protocolo pode ser dividido em 6 estágios-chave: acultura de células HepG2 criopreservadas; Preparação de sferóide hepG2; Transferência de spheróide hepG2 de queda de suspensão para suspensão de agarose; Colheita de sferóides hepG2; ensaio micronucleus e pontuação; e análise de dados.
Protocol
1.Culturing criopreserved células HepG2
NOTA: As células HepG2, obtidas da American Type Culture Collection (ATCC) foram cultivadas no 1x Dulbecco's Modified Eagle Medium (DMEM) com 4,5g/L D-glicose e L-glutamina suplementadas com 10% de soro bovino fetal (FBS) e 1% de penicilina/antibiótico de estreptomicina.
- Meio de cultura celular DMEM pré-quente (incluindo os suplementos) em um banho de água de 37 °C por 30 min.
- Remova um frasco de células HepG2 de nitrogênio líquido e descongele em um banho de água de 37 °C por 2-3 min, enquanto gira suavemente o frasco para permitir o descongelamento uniforme da suspensão da célula. Tome cuidado para não submergir o frasco acima do anel O, a fim de reduzir o potencial de contaminação.
- Uma vez descongelado, remova o frasco do banho de água e pulverize generosamente com 70% de etanol para descontaminar a superfície externa do frasco antes de colocar sob uma estéril capa de cultura de tecido laminar classe II.
- Pipeta cuidadosamente o conteúdo do criovial das células HepG2 em um tubo de centrífuga contendo 9 mL de meio de cultura celular DMEM pré-aquecido (com suplementos).
- Usando um stripette de 10 mL, transfira 10 mL de suspensão celular para um frasco de cultura celular descartável de25 cm 2 e incubar a cultura por 3 dias (a partir da semeadura) a 5% de CO2 e 37 °C até que ~80% de confluência seja alcançada antes de se submeter à subcultura em um frasco de cultura celular descartável maior de 75 cm2.
- Uma vez alcançada a confluência de 80%, as células subcultura em condições estéreis por experimentação com solução de trippsina/EDTA de 0,05% pré-aquecidas em um banho de água de 37°C por 30 minutos. Em nenhum momento as células devem ser permitidas a secar.
- À medida que as células formam uma monocamada aderente, remova a mídia derrubando-a em um pote de resíduo desinfetante. Em seguida, lave imediatamente a monocamheira para remover todos os traços da mídia existente enxaguando o frasco duas vezes com 3 mL de solução estéril de 1x PBS mantida à temperatura ambiente. Além disso, descarte pbs em pote de resíduos desinfetante.
- Uma vez que a lavagem pbs é removida, adicione 5 mL de solução pré-aquecida de 0,05% trypsin-EDTA, garantindo cobrir toda a superfície das células e incubar células por 6-8 min a 37 °C e 5% CO2.
- Bata suavemente no frasco para desalojar as células da parte inferior do frasco e, em seguida, adicione 5 mL de meio de cultura celular DMEM (com suplementos) para neutralizar a enzima trypsin.
- Transfira a suspensão da célula para um tubo de centrífuga de 50 mL e pipete a suspensão da célula para cima e para baixo completamente para garantir que as células sejam completamente dissociadas.
- Centrifugar a suspensão da célula diluída a 230 x g por 5 min. Descarte o supernasal em desinfetante e suspenda a pelota celular em 25mL de meio de cultura celular DMEM (com suplementos).
- Transfira a suspensão celular para um frasco de cultura celular descartável de 75 cm2 e incubar a 37 °C e 5% de CO2 por mais 3 dias antes de se submeter à preparação de esferoides. Uma vez que os HepG2s tenham tido tempo para aclimatar e mais uma vez atingir ~80% de confluência, determine a concentração celular em preparação para a semeadura esferoide.
2. Preparação esferoide hepG2
- Repita as etapas de subcultura indicadas acima, exceto após a centrifugação, suspenda a pelota de célula em 1 mL de cultura DMEM pré-aquecida em um banho de água de 37 °C. Suspensão de célula pipeta para cima e para baixo completamente.
- Escore a viabilidade celular usando o Ensaio de Exclusão Azul trypan (ver OSHA SOP 3.21 Toxinas Reprodutivas, Mutagens, Teratogens e Embri toxinas – Procedimentos para Manuseio e Armazenamento Seguro (2019) para orientação de saúde e segurança)14 com uma razão de 1:1 de suspensão celular para solução azul trypan pré-filtrada de 0,4%.
- Antes da contagem celular, pegue 1 mL de solução azul Trypan usando uma seringa de 1 mL e filtro com uma unidade de filtro de 0,45 μm em um tubo estéril de 1 mL.
- Transfira 10 μL de solução filtrada, azul Trypan em um tubo de 0,2 mL e adicione 10 μL de suspensão celular. A solução azul Trypan filtrada restante pode ser armazenada até 3 meses em temperatura ambiente para uso futuro.
- Pulverize o hemoocítômetro completamente com 70% de etanol e limpe com uma toalha de papel estéril antes de fixar a mancha de cobertura em cima usando vapor de respiração. Deslizar a mancha de cobertura através da superfície umedeçada da respiração induz forças coesas gerando anéis de Newton.
- Pipeta suavemente a suspensão da célula azul Trypan para cima e para baixo usando uma pipeta de 1000 μL (para reduzir o estresse total) antes de adicionar 10 μL ao hemoocímetro. Certifique-se de que a solução está dispersa sob o deslizamento da tampa e cubra toda a grade sem bolhas de ar.

Figura 1: Contar células usando um hemoocítômetro. Representação diagramática de um hemoocítômetro destacando de qual quadrante contar células. Clique aqui para ver uma versão maior desta figura.
- Sob o microscópio, conte as células vivas (não manchadas) e mortas (azul manchado) encontradas nos quatro grandes quadrados de canto(Figura 1). Exclua todas as células encontradas para se sobrepor ou sentar-se no interior duas bordas dos grandes quadrados de canto (ou seja, nas linhas) na contagem.
- Utilizando o cálculo a seguir, calcule o número médio de células vivas e viáveis (não manchadas) presentes na amostra:
Número total de células/mL = Contagem de Células Vivas x x 10.000
x 10.000
onde a diluição refere-se a quantas vezes a solução de estoque foi diluída em azul Trypan (2x neste caso) e # de quadrados contados refere-se aos quatro grandes quadrados de canto do hemoocímetro contado - Com base na contagem de células HepG2 viável e usando a seguinte fórmula:
C1V1=C2V2
onde C1 = a concentração de células viáveis atualmente,
V1 = o volume de suspensão celular atualmente,
C2 = a concentração de suspensão celular desejada,
V2 = o volume de suspensão celular desejado - Prepare uma solução de estoque de 10 mL de suspensão celular HepG2 com meio de cultura celular DMEM a uma concentração de 2,0 x 105 células/mL, a fim de alcançar 4000 células HepG2 por 20 μL de queda suspensa. Misture bem a suspensão da célula, pipetando suavemente para cima e para baixo usando uma pipeta de 1000 μL para garantir que todas as células estejam totalmente suspensas dentro da mídia.
- Aos poços de uma placa de cultura celular de 96 poços, adicione 100 μL de PBS estéril e temperatura ambiente para evitar que as gotas de enforcamento sequem durante a incubação.
- Pegue a tampa de uma placa de cultura celular de fundo plano padrão de 96 poços, inverta-a e cuidadosamente pipeta 20 gotas de μL da suspensão da célula no centro de cada sulco de poço da tampa, como mostrado na Figura 2. Use uma pipeta multicanal, mas adicione apenas 2 - 4 gotas de uma só vez, pois a semeadura múltipla pode afetar a precisão e a colocação das gotas.
- Centralizar as gotas dentro das ranhuras dos poços dispostos na tampa; caso contrário, eles não vão pendurar no centro dos poços quando a tampa da placa é virada e correm o risco de cair na placa. Vire suavemente a tampa da placa de 96 poços, de modo que as gotas estão agora penduradas e coloque cuidadosamente em cima da placa de 96 poços.
- Coloque toda a placa 96 com tampa suavemente em uma incubadora a 37 °C e 5% de CO2 por 3 dias antes da transferência de spheroid para agarose.
NOTA: Deve-se tomar cuidado extra não apenas ao transportar as placas de/para as incubadoras, mas ao abrir e fechar a incubadora em geral, pois o movimento excessivo pode fazer com que as placas mudem e os esferoides caiam ou se formem incorretamente.
 x 10.000
x 10.000
Figura 2: 3D HepG2 in vitro spheroid model preparation. (A) As células HepG2 semeadas em 20 μL cai na tampa de uma placa de 96 poços. (B) As células HepG2 pós-semeadura no modelo de queda suspensa permitem a formação de esferoides. Clique aqui para ver uma versão maior desta figura.
3. Transferência de spheróide hepG2 de queda de suspensão para suspensão agarose
NOTA: No dia 3, postando semeadura em gotas penduradas, os esferoides são transferidos para os poços da mesma placa de 96 poços, todos os quais foram previamente revestidos com uma fina camada de gel de 1,5% de agarose.
- Prepare géis de agarose e autoclave (ou seja, dia 2 pós semeadura) antes do dia do revestimento de placas (ou seja, dia 3 pós semeadura).
- Para preparar um gel de 1,5% de agarose, pese 0,30 g de agarose em uma garrafa de vidro limpa e, em seguida, adicione 20 mL de meio DMEM livre de fenol-vermelho. Autoclave a agarose por 1 h a 230 °C para esterilização. O revestimento de agarose impede que os esferoides HepG2 adricarem a base dos poços e formam uma monocamide celular em vez de manter sua estrutura esferoide 3D.
- No 3º dia, retire a placa de 96 poços contendo os esferoides hepG2 pendurados para fora da incubadora e vire cuidadosamente a tampa para que os esferoides não estejam mais pendurados.
- Usando uma pipeta multicanal, remova e descarte os 100 μL de PBS previamente adicionados à base da placa de 96 poços. Deixe as placas arejadas por 2-3 minutos enquanto aquecem a agarose em preparação para o revestimento.
ATENÇÃO: Este procedimento resulta em uma agarose muito quente e líquida que, se derramada na pele, pode queimar e causar ferimentos. Além disso, deve-se tomar cuidado ao manusear a garrafa de vidro contendo a agarose líquida, pois isso também pode ser muito quente. - Usando os géis de agarose de 1,5% previamente preparados, aqueça a garrafa de vidro contendo o gel de agarose de 20 mL por 30 s em um micro-ondas no máximo watt (ou seja, 900 W). Para revestir duas placas de 96 poços, uma garrafa de 20 mL de gel de 1,5% de agarose pré-preparado deve ser suficiente.
- Uma vez derretido, gire suavemente a agarose girando a garrafa de vidro para remover quaisquer bolhas e, em seguida, adicione 50 μL de agarose na base de cada poço.
NOTA: Ao adicionar a agarose, certifique-se de não inclinar a placa >45° à medida que a agarose se fixa rapidamente e não formará uma camada plana e nivelada que pode interromper o crescimento esferoide. É importante trabalhar de forma eficiente nesta fase para evitar que a agarose se solidifique antes que a placa esteja completamente revestida. - Deixe a placa ficar por 2 minutos à temperatura ambiente antes de adicionar 100 μL de meio de cultura celular DMEM pré-aquecida (com suplementos) em cima da camada de agarose sólida em cada poço.
- Vire a tampa da placa de 96 poços e coloque de volta em cima da placa de 96 poços para que os esferoides estejam pendurados mais uma vez.
- Centrifugar a placa por 3 min a 200 x g, a fim de transferir os esferoides da gotícula pendurada para os poços individuais da placa de 96 poços. Após a transferência, os esferoides HepG2 devem agora ser suspensos no meio de cultura celular. Permita que eles se contentem com 24h na incubadora a 37 °C e 5% de CO2.
- Exponha os esferoides HepG2 deste tamanho a tratamentos químicos ou ENM no dia 4 pós semeadura (ou seja, 24h após a transferência para placas revestidas de agarose).
- Para manter a viabilidade celular durante longos períodos de cultura, refrescar o meio de cultura celular a cada 3 dias. Para isso, aspire suavemente 50 μL do meio de cultura celular da superfície do poço e substitua por um novo meio de cultura celular DMEM de 50 μL. Tome cuidado para não remover ou perturbar o esferoide ao realizar uma mudança média.
4. Exposição nanomaterial/química
NOTA: O modelo de spheroid hepG2 pode suportar regimes de exposição enm e químicos, mas o foco principal deste protocolo são as exposições ENM. Antes da exposição, o ENM de teste deve ser adequadamente dispersado; isso pode ser executado conforme o Protocolo de Dispersão NanoGenoTox (Grant Agreement nº 20092101, 2018)15.
- Após a dispersão de acordo com o Protocolo de Dispersão NanoGenoTox, diluir a suspensão ENM da concentração inicial de 2,56 mg/mL para a concentração final desejada no meio de cultura celular DMEM pré-aquecido (incluindo os suplementos). Um volume total de 5 mL é necessário para dosar uma placa de 96 poços.
- Para expor o esferoide HepG2 a um produto químico ou ENM, utilizando uma pipeta de 200 μL, aspire 50 μL de meio de cultura celular a partir da superfície de cada poço (deixando 50 μL no poço para não perturbar os esferoides) e substituir por 50 μL médio contendo o toxicante de teste na dose necessária.
- Uma vez aplicado o material de teste, incubar as placas para o tempo de exposição desejado a 37 °C e 5% de CO2.
- Se for realizado um regime de exposição de longo prazo (≥24 h), imediatamente após o prazo de exposição desejado tiver transcorrido, colher os esferoides para análise de ponto final de micronucleus conforme descrito abaixo nas etapas 6.1 – 6.4.
- No entanto, com regimes de exposição aguda (por exemplo, ≤24 h), uma vez que o período de exposição tenha terminado, a colheita, a piscina e a loja de 50 μL de supernascimento de cada poço na placa de poço 96 a -80 °C para análise bioquímica posterior. Substitua o meio de cultura celular por 50 μL de meio fresco contendo 6 μg/mL de Citochalasina B e deixe incubar por 1 – 1,5 ciclos celulares (ou seja, 24 – 26 h para HepG2) em preparação para a colheita de micronucleos do bloco de citocinas.
NOTA: Para regimes de exposição agudos (≤24 h), o ensaio de micronucleo de bloco citocina com Citochalasina B pode ser aplicado, mas para regimes de exposição a longo prazo (≥24 h), a versão mononuclear (sem Citochalasin B) do ensaio deve ser utilizada conforme descrito abaixo na Figura 4.
5. Colheita de spheróides hepG2
NOTA: Seguindo os tratamentos químicos ou de exposição ao ENM, tanto o meio de cultura celular quanto o tecido esferoide podem ser colhidos para análise múltipla do ponto final. Dependendo da análise do ponto final, os esferoides podem ser colhidos individualmente (por exemplo, para análise de imagem) ou agrupados (por exemplo, para ensaio de micronucleus de bloco de citocinas).
- Remova a placa de 96 poços da incubadora.
- Usando uma pipeta de 200 μL, aspire os 100 μL de meio de cultura celular, incluindo o tecido esferoide de cada poço e colete em um tubo de centrífuga de 15 mL estéril. Tome cuidado para evitar contato com a agarose.
- Uma vez coletado, centrifugar a suspensão esferoide a 230 x g por 5 min. Remova o supernasciente e armazene a -80 °C para uma análise mais aprofundada do ponto final (por exemplo, testes de função hepática) posteriormente.
- Suspenda a pelota de esferoides em 1 mL de PBS estéril e temperatura ambiente (1x).
- Uma vez lavado, centrifugar a suspensão esferoide novamente a 230 x g por 3 min. Descarte a supernasce, suspenda-se em 500 μL de solução trypsin-EDTA de 0,05% e incubar por 6-8 min a 37 °C e 5% de CO2.
- Após a incubação, pipeta suavemente as células experimentpsinizadas para cima e para baixo para desassociar e suspender totalmente as células HepG2 antes de neutralizar com 1 mL de meio de cultura celular DMEM.
- Centrifugar a suspensão da célula diluída a 230 x g por 5 min. Descarte o supernasciente em desinfetante e suspenda a pelota celular em 2mL de PBS de temperatura ambiente (1x).
- Centrifugar a suspensão da célula a 230 x g por 5 min. Descarte o supernatante em desinfetante e, em seguida, suspenda novamente a pelota celular em 2 mL de PBS frio (1x). Certifique-se de que as células estão bem dispersas para evitar que aglomerados de células obscurecem o campo de visão quando montados em slides de microscópio.
6. Ensaio e pontuação de microcússo
Para o método manual do ensaio micronucleus, é necessário um citocentrifuuge para produzir um citodoto (uma região definida e concentrada de células) no centro do slide do microscópio. Este processo suporta uma pontuação mais eficiente do slide, pois permite que o marcador localize facilmente as células de interesse, em vez de avaliar um slide inteiro onde as células podem ser amplamente difundidas.
- Mergulhe slides de microscópio fosco (três por dose) em 70% de etanol seguido de ddH2O e deixe secar ao ar por 5 min.
- Coloque slides de microscópio preparados no funil cuvette, como mostrado na Figura 3A,onde o slide de vidro (iii) é colocado no suporte metálico (iv) com uma placa de filtro (ii) e funil cuvette (i) fixado em cima.
- Disponha os funis cuvette no cytocentrifuge com o funil voltado para cima, para que 100 μL de suspensão celular possam ser adicionados diretamente em cada um deles.
- Citospin por 5 min a 500 x g para garantir que as células sejam distribuídas uniformemente na superfície do slide.

Figura 3: Configuração de citospin para preparar células tratadas em slides de microscópio. (A) Exibe os componentes individuais, (i) funil cuvette, (ii) cartão de filtro, (iii) deslizamento de microscópio de vidro e (iv) suporte metálico necessário para citospin hepG2 células em slides de microscópio. (B) O funil cuvette final configurado. (C) A colocação correta do funil cuvette dentro do citocentrifuge. Clique aqui para ver uma versão maior desta figura.
- Deixe slides para secar o ar antes da fixação no gelo frio, 90% de metanol por 10 minutos.
- Uma vez fixado, deixe os slides para secar o ar durante a noite à temperatura ambiente antes de armazenar a -20 °C por até 6 meses.
- Quando necessário, remova os slides do microscópio pré-preparado do congelador de -20 °C e deixe aquecer até a temperatura ambiente antes de realizar a coloração de Giemsa.
ATENÇÃO: De acordo com o Regulamento (CE) nº 1272/2008 [CLP], a solução de coloração Giemsa é um líquido altamente inflamável que pode ser tóxico se engolido e causar danos no contato com os olhos, pele ou se inalado. Consulte a folha SDS associada para obter conselhos detalhados de armazenamento, manuseio e saúde e segurança sobre este produto químico antes de usar. - Enquanto os slides estão descongelando, prepare uma solução de coloração Giemsa de 20% (25 mL necessária para manchar ~30 slides) diluída em tampão fosfatado (pH 6.8). Misture bem girando suavemente a solução antes de filtrar usando papel filtro dobrado colocado em um funil.
- Usando uma pipeta Pasteur, adicione 3 – 5 gotas de solução Giemsa filtrada ao citodoto em cada slide e deixe por 8 – 10 min.
- Lave slides em duas lavagens sucessivas de tampão fosfates antes de enxaguar brevemente sob água fria para remover qualquer excesso de sobra de mancha. Deixe os slides para secar o ar.
- Uma vez seco, em um capô de fumaça, mergulhe lâminas de vitrais em xileno por 10 s antes de adicionar uma gota de meio de montagem ao centro do citodoto e um lugar um deslizamento de vidro em cima.
- Deixe os slides do microscópio no capô da fumaça durante a noite para secar antes da pontuação manual; eles podem ser armazenados indefinidamente à temperatura ambiente.
7. Análise de dados
- Conforme descrito nas Diretrizes de Teste da OCDE 487 (2014)11, para avaliar e quantificar danos de DNA induzidos como resultado da exposição a um AGENTE QUÍMICO ou ENM, utilize um microscópio leve (objetivo 100x com óleo de imersão) 2000 mononucleados ou 1000 células binucleadas por réplica biológica para pontuação para a presença de micronucleís, como mostra a Figura 4.

Figura 4: Árvore de decisão de pontuação de ensaio micronucleus. Árvore de decisão esquemática para destacar a necessidade de diferentes procedimentos de pontuação e avaliação de citotoxicidade ao utilizar o ensaio micronucleus com modelos 3D seguindo regimes de exposição aguda ou de longo prazo. Exposições agudas (≤24 h) permitem o uso do ensaio micronucleo bloqueado por citocinas, enquanto exposições de longo prazo (≥24 h) requerem a versão mononuclear do ensaio; ambos estão descritos na Diretriz de Testes da OCDE 487. Clique aqui para ver uma versão maior desta figura.
- Com base na proporção de micronuclei presentes por número de células mononucleadas ou binucleadas pontuadas, calcule uma porcentagem do valor da genotoxicidade.
- Para avaliar os danos de DNA observados não é como resultado de detritos celulares causados por uma alta proporção de células apoptóticas, tome uma medida de citotoxicidade ao lado. Neste caso, dependendo da presença de Cytochalasin B, utilize o cálculo cpbi ou rvcc (conforme descrito na Figura 4). A genotoxicidade só deve ser avaliada em amostras em que a citotoxicidade seja inferior a 55% ± 5% conforme definido na Diretriz de Testes da OCDE 48711.
Representative Results
A adequação deste modelo spheróide hepático 3D baseado em linha celular para cultura de longo prazo e avaliação de risco genotipico foi avaliada pela realização da caracterização da linha de base para determinar a viabilidade e funcionalidade hepática do modelo ao longo da duração de 14 dias na cultura, bem como sua aplicabilidade para o ensaio do micronucleus.
Caracterização da linha de base do modelo spheroid hepg2 hepg2 3D
Antes de qualquer avaliação toxicológica in vitro, é importante verificar se os esferoides HepG2 3D formaram-se adequadamente antes de realizar a transferência de agarose ou tratamento químico/ENM. Os spheróides hepG2 produzidos usando o método de queda de suspensão geralmente levam de 2 a 3 dias após a semeadura (4000 células/esferoides) para formar esferoides compactos e esféricos com diâmetro médio de 495,52 μm W x 482,69 μm H, como mostrado na Figura 5A-5C. Os spheróides hepG2 que se formaram corretamente e são aceitáveis para serem usados para avaliação toxicológica in vitro devem ter uma estrutura compacta e esférica com uma superfície lisa e sem projeções visuais. A Figura 5 fornece exemplos de boa qualidade(Figura 5D-F) e um esferoides de má qualidade(Figura 5G-I). Este último deve ser descartado. Normalmente, 90-95% dos esferoides formados por placa se formarão corretamente e serão viáveis para novas experimentações.

Figura 5: Imagens de microscopia leve que exibem a morfologia natural dos esferoidesHepG2 formadas através do método de queda de suspensão . (A-C) mostrar dia 2 e(D-I) Dia 4 HepG2 hefóides pós semeadura. (D-F) são exemplos de spheroids HepG2 de boa qualidade, enquanto(G-I) mostra esferoides mal formados. Todas as imagens foram tiradas em um objetivo X20 usando um microscópio. A barra de escala representa 20 μm. Clique aqui para ver uma versão maior desta figura.
Para confirmar ainda mais a viabilidade esferoide HepG2, um ensaio colorido básico do Bromocresol Green Albumin (BCG) ou Urea Assay pode ser realizado para avaliar sua funcionalidade semelhante ao fígado. A funcionalidade semelhante ao fígado foi avaliada de acordo com a viabilidade usando o Ensaio de Exclusão Azul trypan durante um período de cultura de 14 dias para determinar a longevidade do modelo esferoide hepático e estabelecer se poderia suportar a avaliação de risco baseada em ENM/química a longo prazo(Figura 6). A concentração de albumina permaneceu consistente durante o período cultural. A produção de ureia apresenta um aumento na concentração de ureia produzida por esferoide ao longo de uma semana na cultura antes de atingir um patamar no dia 7. É importante notar que os níveis de albumina e ureia produzidos nos esferoides HepG2 3D são substancialmente maiores do que os observados na mesma linha celular cultivada em formato 2D. De fato, as culturas 2D das células HepG2, os níveis de pico de albumina e ureia foram de 0,001 mg/mL e 0,010 ng/μL, respectivamente. Além disso, em trabalhos anteriores publicados por Shah et al. utilizando um sistema esferoide HepG2 quase idêntico, os autores destacam uma notável melhora na atividade metabólica (CYP1A1 e CYP1A2) nos sistemas de modelos in vitro 3D HepG2 quando comparados com as células HepG2 cultivadas em 2D5.

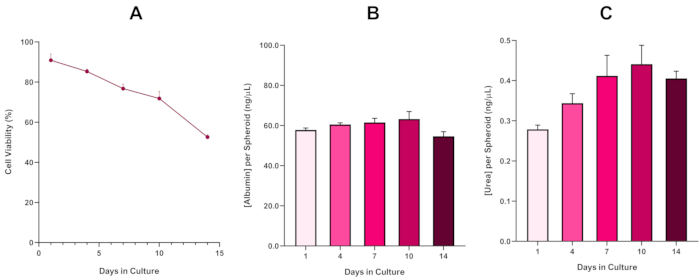
Figura 6: Dados de caracterização da linha de base de 14 dias para esferoides hepG2 de fígado. Após a transferência da queda de suspensão, (A) destaca a viabilidade do modelo esferoide HepG2 durante um período de 14 dias, enquanto(B) e (C) destacam a funcionalidade de albumina e ureia semelhante ao fígado, respectivamente. Dados médios ± SEM apresentados, n = 4. Clique aqui para ver uma versão maior desta figura.
Com o desenvolvimento inevitável de um núcleo necrosado, uma limitação conhecida das culturas esferoides hepáticos 3D, a viabilidade deste modelo baseado em HepG2 teve que ser estabelecida para demonstrar que era capaz de sustentar regimes de exposição a longo prazo (5-10 dias) mantendo a capacidade proliferativa necessária para suportar o ensaio micronucleus5. De fato, este modelo de spheroid hepático 3D tem sido mostrado para manter >70% de viabilidade ao longo de 10 dias na cultura. Com base nisso e em conjunto com a funcionalidade sustentada do fígado observada ao longo do período de cultura de ≥ 14 dias, este modelo de spheroid hepático 3D pode, assim, suportar regimes de exposição enm a longo prazo e repetidos até 10 dias de duração (ou seja, antes que a viabilidade dos esferoides caia abaixo de 70%). Para referência, é aconselhável que os níveis de albumina para spheróides HepG2 semeados em 4000 células/esferoides devem ser ≥20,0 ng/μL enquanto a produção de ureia deve ser ≥0,25 ng/μL antes de realizar uma avaliação toxicológica in vitro com este modelo.
Avaliação genotoxicidade de nanomateriais projetados
Para avaliação da genotoxicidade, o ensaio micronucleus foi utilizado para determinar a presença de micronuclei após exposições enm agudas (24 h) e de longo prazo (120 h). Aflatoxina B1 é um conhecido carcinógeno hepático16,17 e é um controle positivo recomendado para o ensaio de micronucleus. Experimentos de otimização mostraram que 0,1 μM de AlfatoxinA B1 induz uma resposta genotoxica positiva (≥2,0 vezes) em esferoides hepáticos hepg2 3D e, portanto, é usada em todos os ensaios micronucleus realizados com este modelo. Para garantir a validade dos resultados do ensaio de micronuclear usando o modelo spheroid HepG2, a frequência de micronucleus de fundo para células HepG2 utilizadas neste modelo in vitro 3D deve estar dentro de uma faixa de 0,6% a 1,2%. Como resultado, a AlfatoxinA B1 deve induzir uma resposta genotoxica de pelo menos duas vezes maior do que a observada com o controle negativo; assim, 0,1 μM de Alfatoxina B1 deve induzir uma frequência micronuclei entre 1,5% e 3,0%. Usando esses parâmetros de controle, a genotoxicidade associada da ENM in vitro pode então ser avaliada de forma confiável. Com base na Diretriz de Teste da OCDE 487, é importante notar que, ao testar um ENM ou químico, as concentrações selecionadas não devem induzir mais de 55% ± citotoxicidade de 5% (indicada por uma redução dos valores de CPBI ou RVCC em relação ao controle negativo)11. A Figura 7 ilustra os dados gerados quando a AflatoxinA B1 e dois ENMs (dióxido de titânio (TiO2) e lasco (Ag)) foram avaliados após exposições agudas e de longo prazo nos esferoides HepG2, e o potencial genotóxico subsequente foi analisado usando o ensaio micronucleus. Ambos os ENMs avaliados foram testados em um regime noncytotóxico, de baixa dose de 5,00 μg/mL sobre uma exposição aguda (24 h) e regime de exposição a longo prazo (120 h). Uma tendência semelhante para genotoxicidade em ambos os ENMs TiO2 e Ag podem ser observadas, pela qual a resposta elevada de genotoxicidade que resultou após a exposição de 24 horas não ficou evidente após uma exposição de longo prazo de 5 dias. Isso foi apesar da genotoxicidade sustentada induzida pelo controle positivo da Aflatoxin B1 em ambos os pontos de tempo.

Figura 7: Avaliação genotoxicidade após a exposiçãotio 2 e ag ENM em spheroids hepG2. Avaliação genotoxicidade (frequência de micronucleus) utilizando o posto de ensaio micronucleus(A) aguda (24 horas) e(B)exposição a longo prazo (120 horas) a 5,00 μg/mL de TiO2 e Ag ENM. O controle negativo é apenas uma mídia, enquanto o controle positivo é de 0,1 μM de Aflatoxin B1. Os dados médios (n=2) apresentaram ± SD. Significância indicada em relação ao controle negativo: * = p≤ 0,05. Clique aqui para ver uma versão maior desta figura.
Discussion
As aplicações para modelos hepáticos 3D variam consideravelmente dependendo do ponto final bioquímico específico ou da via de desfecho adverso que está sendo alvo. Cada modelo tem seus benefícios e limitações, desde a variação interdonor nos modelos primários de hepatócito humano (PHH) até a redução da atividade citocromática p450 em modelos baseados em linha celular, mas todos são valiosos por si só6,12,18,19. Ao avaliar a genotoxicidade, há limitações na compatibilidade dos modelos com pontos finais aprovados pela regulamentação, como o ensaio de micronuclear in vitro, pois a proliferação ativa é necessária. Isso é necessário, pois a avaliação da genotoxicidade requer a quantificação de danos fixos de DNA a serem avaliados na divisão pós-celular quando há oportunidade de reparação de DNA para corrigir lesões transitórias. Infelizmente, hepatocitos altamente diferenciados (ou seja, esferoides baseados em HepaRG) ou microtissues PHH, que são considerados como apresentar as características hepáticas mais fisiologicamente relevantes formam modelos estáticos (não proliferativos)12,19,20. Como resultado, o modelo esferoide HepG2 3D apresentado aqui fornece um modelo adequado e alternativo capaz de suportar testes de genotoxicidade. Os esferoides baseados em linha celular HepG2 têm células divisórias suficientes na superfície externa dos esferoides, mantendo características básicas semelhantes ao fígado, como a produção de albumina e ureia e alguma atividade CYP4505,12,19. Principalmente este modelo hepático in vitro foi desenvolvido para complementar o ensaio micronucleus, pois este é um dos dois ensaios in vitro recomendados na bateria para testes de genotoxicidade8,10,11,21. No entanto, o modelo pode ser facilmente aplicado às tecnologias de análise de sequenciamento de DNA e expressão genética (RNA), enquanto tem o potencial de ser ainda mais adaptado e utilizado para outros pontos finais de dano de DNA, como o ensaio do cometa. No entanto, é importante considerar o papel que a interferência do ENM desempenha em algumas análises de ponto final. Por exemplo, análises baseadas em citometria de fluxo podem não ser adequadas para avaliação de genotoxicidade enm especificamente devido à interferência de partículas22.
Um fator limitante dos modelos esferoides que se submetem ativamente à divisão celular é o seu tamanho. A otimização da densidade de semeadura é fundamental, pois é preciso haver células suficientes que permitam que o modelo continue a proliferar; mas não muito alto um número de células, o que resulta no esferoide se tornando excessivamente compacto, levando a um núcleo necrosado aumentado. Acredita-se que a causa dessa necrose seja a restrição de oxigênio e difusão de nutrientes, já que o limite dessa difusão é de aproximadamente 100 – 150 μm de tecido23,24. No entanto, isso depende do tipo celular, número celular, interações de andaimes e condições de cultura25. Desde então, foi demonstrado que aproximadamente 700 μm de diâmetro é o limite para evitar o início prematuro da necrose no centro de esferoides C3A, semeando 4000 células HepG2 por esferoide garante que o diâmetro do modelo no momento da exposição seja ≤500 μm26. Além disso, Shah et al. estabeleceram que as células HepG2 semeadas acima de 5000 células por esferoide apresentaram uma redução de 25% na viabilidade após 7 dias de cultura, o que poderia pertencer ao diâmetro médio de 680 μm e disponibilidade limitada de nutrientes em uma queda de 20 μL de suspensão5. Para superar isso, o modelo concebido no presente protocolo passa por um passo crítico onde a queda de enforcamento é transferida para poços revestidos de agarose após a formação inicial do esferoide. Isso garante um maior volume de cultura média está presente para sustentar o número crescente de células dentro dos esferoides. Como resultado, o modelo esferoide HepG2 permanece mais de 70% viável após 10 dias de cultura e pode ser utilizado para avaliação de risco de longo prazo in vitro.
Embora o modelo esferoide HepG2 possa suportar regimes de exposição aguda e de longo prazo, o meio de cultura celular refrescante durante períodos de cultura prolongado é restrito para este modelo, pois a substituição completa do meio não é aconselhada devido à perda potencial dos esferoides. Presume-se que, com as exposições enm, a tendência de dispersões homogêneas de ENM para aglomeração e sedimentos é alta. No entanto, é notável que a taxa em que um sedimento ENM pode variar dependendo dos parâmetros de partículas (por exemplo, tamanho, forma e densidade) e pode ser determinada teoricamente usando o modelo de sedimentação in vitro, difusão e dosimetria (ISDD), ou seus derivados recentes, muitas vezes referidos quando se aproxima da exposição ENM (suspensão) se aproxima27,28. Com isso é mente, supõe-se que se apenas 50% do meio de cultura celular for cuidadosamente removido da superfície da cultura celular, a interrupção e posterior remoção da dose ENM deve, em teoria, ser mínima. No entanto, com a moção browniana em jogo, isso pode não ser estritamente o caso e mais trabalhos na deposição e sedimentação de cada ENM em particular a ser testado devem ser realizados para garantir que a dosimetria correta seja mantida ao longo dos regimes de exposição a longo prazo27. Principalmente, esta é uma limitação potencial a ser considerada ao realizar regimes repetidos de dosagem, pois isso poderia ser fundamental para a concentração final e acumulada. Exposições baseadas em produtos químicos, por outro lado, embora não sem suas próprias limitações a considerar, oferecem uma abordagem mais simplista na forma de que as substâncias químicas tendem a permanecer em solução e, portanto, uma substituição direta da concentração química original, além da concentração recém-adicionada, garante que qualquer produto químico perdido durante o refresco da mídia seja substituído emconformidade com 29. Aplicações futuras incluiriam avaliar a adequação do modelo para regimes de exposição repetida em períodos de cultura de longo prazo, uma vez que estratégias de dosagem repetidas são crucialmente importantes para avaliar a capacidade de um determinado sistema de órgãos de amenizar ou superar os efeitos adversos, se houver, induzidos pela bioaccumulação de substância abiótica.
Em conclusão, este modelo hepático in vitro 3D tem a capacidade de ser utilizado para avaliar uma série de cenários de exposição realista, proporcionando assim uma futura abordagem in vitro para melhor suportar tanto a AVALIAÇÃO DE ENM quanto a avaliação de riscos químicos de forma rotineira e de fácil acesso.
Disclosures
Os autores não têm nada a revelar.
Acknowledgments
Os autores gostariam de reconhecer que esta pesquisa recebeu financiamento do programa de pesquisa e inovação Horizon 2020 da União Europeia para o projeto PATROLS, sob o acordo de subvenção nº 760813
Materials
| Name | Company | Catalog Number | Comments |
| Aflotoxin B1 | Sigma Aldrich, UK | A6636-5MG | |
| Agarose | Sigma Aldrich, UK | A9539-50G | |
| Autoclave Tape | |||
| BCG Albumin Assay | Sigma Aldrich, UK | MAK124 | |
| Bovine Serum Albumin Powder | Sigma Aldrich, UK | A9418 | |
| Cell Freezing Aid | Thermo Fisher Scientific, UK | 5100-0001 - Mr Frosty | |
| Centrifuge | Eppendorf | 5810 R | |
| Cytochalasin B | Merck, UK | 250233 | |
| Cytology Metal Clips | |||
| Cytospin 4 Centrifuge | ThermoFisher Scientific, UK | CM00730202 | |
| DMEM with 4.5g/L D-Glucose, L-Glutamine | GIBCO, Paisley, UK | 41965-039 | |
| DMEM, phenol-red free with 4.5g/L D-Glucose, L-Glutamine with Hepes | GIBCO, Paisley, UK | 21063-029 | |
| DPX Mounting Medium | FisherScientific, UK | D/5330/05 | |
| Ethanol | FisherScientific, UK | 10048291 | |
| FBS | GIBCO, Paisley, UK | 10270-106 | |
| Filter Cards for Shandon Cytospin | FisherScientific, UK | 15995742 | |
| Frosted Glass Slides | ThermoFisher Scientific, UK | ||
| Giemsa's Stain Improved R66 Solution, Gurr | VWR Chemicals, UK | MFCD00081642 | |
| Glass Coverslips (24 x 60) | Deckglaser, VWR | ECN631-1575 | |
| Haemocytometer and Coverslip | |||
| Immersion Oil for Microscope | Zeiss, UK | 518F, ISO8034 | |
| Laminar Class II Tissue Culture Hood | Scanlaf Mars | ||
| Light Microscope | Zeiss, UK | Axiovert 40C | |
| Liquid Nitrogen | |||
| Methanol | FisherScientific, UK | 10284580 | |
| Microwave | |||
| Non-Filtered, Sterile 200µl and 1000µl Pipette tips | Greiner-Bio-One, UK | ||
| NuncMicroWell 96-Well Microplates | ThermoFisher Scientific, Denmark | 167008 | |
| P1000 and P200 micropipettes | |||
| P300 and P50 multi-channel pipettes | |||
| PBS pH 7.4 1X, MgCl2 and CaCl2 Free | GIBCO, Paisley, UK | 14190-094 | |
| Pen/Strep | GIBCO, Paisley, UK | 15140-122, Penicillin/Strepmyocin 100X or 10,000U/ml | |
| Phosphatase Buffer Tablets | GIBCO, Paisley, UK | 10582-013 | |
| Pipette Boy | |||
| Simport Scientific CytoSep Funnels for Shandon Cytospin 4 Centrifuges | FisherScientific, UK | 11690581 | |
| Sonifier SFX 550 240V CE 1/2" - Probe | Branson, USA | 101-063-971R | |
| T-25 and T-75 Tissue Culture Flask | Greiner-Bio-One, UK | T-25 (690175) and T-75 (660175) | |
| Trypan Blue Solution | Sigma Aldrich, UK | T8154-100mL | |
| Urea Assay Kit | Sigma Aldrich, UK | MAK006 | |
| Virkon Disinfectant | DuPont, UK | Rely+On Virkon | |
| Water Bath (37?C) | Grant JBNova 18 | ||
| Weighing Balance | |||
| Xylene | FisherScientific, UK | 10588070 | |
| 0.05% Trypsin-EDTA | GIBCO, Paisley, UK | 5300-054 | |
| 0.2mL and 1.0mL Eppendorf Tubes | Greiner-Bio-One, UK | ||
| 0.45µm Filter Unit | Millex HA, MF-Millipore, UK | SLHA033SS | |
| 1.0mL Syringe | BD Plastipak, FisherScientific, UK | 300185 | |
| 20mL LS Scintillation Glass Vials, 22-400 Foil Lined PP Caps | DWK Life Sciences GmbH, Germany | WHEA986581 | |
| 37?C and 5% CO2 ISO Class 5 Hepa Filter Incubator | NUAIRE DHD Autoflow | ||
| 3mL Pasteur Pipette | Greiner-Bio-One, UK | ||
| 50mL Conical Falcon Tubes | Greiner-Bio-One, UK | ||
| 50mL or 100mL Glass Bottles | |||
| 50mL Skirted Falcon Tubes | Greiner-Bio-One, UK | ||
| 5mL, 10mL and 25mL Pipettes | Greiner-Bio-One, UK | ||
| 9.4cm Square, Petri Dish | Greiner-Bio-One, UK | 688161 |
References
- Geiser, M., Kreyling, W. G. Deposition and biokinetics of inhaled nanoparticles. Particle and Fibre Toxicology. 7, 2 (2010).
- Modrzynska, J. Toxicological effects of nanoparticle deposition in the liver. Kgs. Lyngby, Denmark: Technical University of Denmark. , (2018).
- Elje, E., et al. The comet assay applied to HepG2 liver spheroids. Mutation Research/Genetic Toxicology and Environmental Mutagenesis. 845, 403033 (2019).
- Breslin, S., O'Driscoll, L. Three-dimensional cell culture: the missing link in drug discovery. Drug Discovery Today. 18, 240-249 (2013).
- Shah, U. -K., et al. A three-dimensional in vitro HepG2 cells liver spheroid model for genotoxicity studies. Mutation Research/Genetic Toxicology and Environmental Mutagenesis. 825, 51-58 (2018).
- Lauschke, V. M., Hendriks, D. F. G., Bell, C. C., Andersson, T. B., Ingelman-Sundberg, M. Novel 3D Culture Systems for Studies of Human Liver Function and Assessments of the Hepatotoxicity of Drugs and Drug Candidates. Chemical Research in Toxicology. 29, 1936-1955 (2016).
- van Grunsven, L. A. 3D in vitro models of liver fibrosis. Advanced Drug Delivery Reviews. 121, 133-146 (2017).
- Corvi, R., Madia, F. In vitro genotoxicity testing - can the performance be enhanced. Food and Chemical Toxicology. 106, 600-608 (2017).
- Doak, S. H., Manshian, B., Jenkins, G. J. S., Singh, N. In vitro genotoxicity testing strategy for nanomaterials and the adaptation of current OECD guidelines. Mutation Research/Genetic Toxicology and Environmental Mutagenesis. 745, 104-111 (2012).
- Fenech, M. Cytokinesis-block micronucleus cytome assay. Nature Protocols. 2, 1084-1104 (2007).
- OECD. OECD Guidelines. Test 489: In vivo Mammalian Alkaline Comet Assay. , (2016).
- Gerets, H. H. J., et al. Characterization of primary human hepatocytes, HepG2 cells, and HepaRG cells at the mRNA level and CYP activity in response to inducers and their predictivity for the detection of human hepatotoxins. Cell Biology and Toxicology. 28, 69-87 (2012).
- Sison-Young, R. L., et al. A multicenter assessment of single-cell models aligned to standard measures of cell health for prediction of acute hepatotoxicity. Archives of Toxicology. 91, 1385-1400 (2017).
- European Guidelines 2019. European Agency for Safety and Health at Work. , Available from: https://osha.europa.eu/en/safety-and-health-legislation/european-guidelines (2019).
- Jensen, K. A. The NANOGENOTOX Dispersion Protocol for NANoREG. European Union Grant Agreement n° 2009. 21, 01 (2014).
- Marchese, S., et al. Aflatoxin B1 and M1: Biological Properties and Their Involvement in Cancer Development. Toxins. 10, 214 (2018).
- Rushing, B. R., Selim, M. I. Aflatoxin B1: A review on metabolism, toxicity, occurrence in food, occupational exposure, and detoxification methods. Food and Chemical Toxicology. 124, 81-100 (2019).
- Kermanizadeh, A., Brown, D. M., Moritz, W., Stone, V. The importance of inter-individual Kupffer cell variability in the governance of hepatic toxicity in a 3D primary human liver microtissue model. Scientific Reports. 9, 7295 (2019).
- Berger, B., et al. Comparison of Liver Cell Models Using the Basel Phenotyping Cocktail. Frontiers in Pharmacology. 7, 443 (2016).
- Ramaiahgari, S. C., et al. A 3D in vitro model of differentiated HepG2 cell spheroids with improved liver-like properties for repeated dose high-throughput toxicity studies. Archives of Toxicology. , (2014).
- Li, Y., et al. Factors affecting the in vitro micronucleus assay for evaluation of nanomaterials. Mutagenesis. 32 (1), 151-159 (2016).
- Kirkland, D., Reeve, L., Gatehouse, D., Vanparys, P. A core in vitro genotoxicity battery comprising the Ames test plus the in vitro micronucleus test is sufficient to detect rodent carcinogens and in vivo genotoxins. Mutation Research/Genetic Toxicology and Environmental Mutagenesis. 721 (1), 27-73 (2011).
- Curcio, E., et al. Mass transfer and metabolic reactions in hepatocyte spheroids cultured in rotating wall gas-permeable membrane system. Biomaterials. 28, 5487-5497 (2007).
- Glicklis, R., Merchuk, J. C., Cohen, S. Modeling mass transfer in hepatocyte spheroids via cell viability, spheroid size, and hepatocellular functions. Biotechnology and Bioengineering. 86, 672-680 (2004).
- Asthana, A., Kisaalita, W. S. Microtissue size and hypoxia in HTS with 3D cultures. Drug Discovery Today. 17, 810-817 (2012).
- Gaskell, H., et al. Characterization of a functional C3A liver spheroid model. Toxicology Research. 5, 1053-1065 (2016).
- Cho, E. C., Zhang, Q., Xia, Y. The effect of sedimentation and diffusion on cellular uptake of gold nanoparticles. Nature Nanotechnology. 6, 385-391 (2011).
- Hinderliter, P. M., et al. ISDD: A computational model of particle sedimentation, diffusion and target cell dosimetry for in vitro toxicity studies. Particle and Fiber Toxicology. 7, 36 (2010).
- Kramer, N. I., di Consiglio, E., Blaauboer, B. J., Testai, E. Biokinetics in repeated-dosing in vitro drug toxicity studies. Toxicology in vitro. 30, 217-224 (2015).
Tags
Bioengenharia Edição 160 Modelos in vitro de fígado nanomateriais avaliação de riscos exposição a longo prazo nano (geno)toxicologia dano de DNAErratum
Formal Correction: Erratum: Advanced 3D Liver Models for In vitro Genotoxicity Testing Following Long-Term Nanomaterial Exposure
Posted by JoVE Editors on 01/26/2021.
Citeable Link.
An erratum was issued for: Advanced 3D Liver Models for In vitro Genotoxicity Testing Following Long-Term Nanomaterial Exposure. The Representative Results section was updated.
Figure 6 in the Representative Results section was updated from:
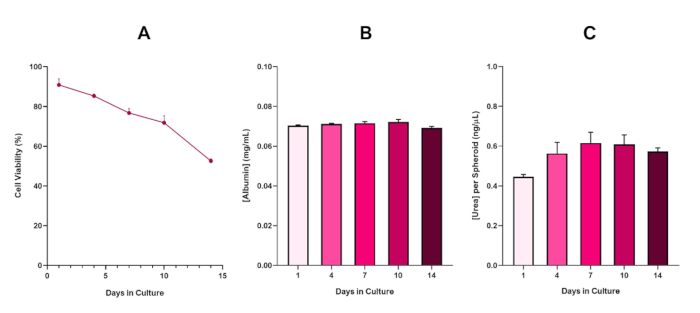
to:
The fourth paragraph in the Representative Results section was updated from:
With the inevitable development of a necrotic core, a known limitation of 3D liver spheroid cultures, the viability of this HepG2 based model had to be established to demonstrate it was able to sustain long-term (5-10 day) exposure regimes whilst maintaining the proliferative capability required to support the micronucleus assay5. Indeed, this 3D liver spheroid model has been shown to retain >70% viability over 10 days in culture. Based on this and in conjunction with the sustained liver-like functionality observed over the ≥14 day culture period, this 3D liver spheroid model can thus support long-term, repeated ENM exposure regimes up to 10 days long (i.e., before viability of the spheroids drop below 70%). For reference, it is advised that albumin levels for HepG2 spheroids seeded at 4000 cells/spheroid should be ≥0.06 mg/mL whilst urea production should be ≥0.4 ng/µL before conducting an in vitro toxicological assessment with this model.
to:
With the inevitable development of a necrotic core, a known limitation of 3D liver spheroid cultures, the viability of this HepG2 based model had to be established to demonstrate it was able to sustain long-term (5-10 day) exposure regimes whilst maintaining the proliferative capability required to support the micronucleus assay5. Indeed, this 3D liver spheroid model has been shown to retain >70% viability over 10 days in culture. Based on this and in conjunction with the sustained liver-like functionality observed over the ≥14 day culture period, this 3D liver spheroid model can thus support long-term, repeated ENM exposure regimes up to 10 days long (i.e., before viability of the spheroids drop below 70%). For reference, it is advised that albumin levels for HepG2 spheroids seeded at 4000 cells/spheroid should be ≥50.0 ng/μL whilst urea production should be ≥0.25 ng/µL before conducting an in vitro toxicological assessment with this model.
