Fonte: Alexander S. Gold1, Tonya M. Colpitts1
1 Departamento de Microbiologia, Escola de Medicina da Universidade de Boston, Laboratórios Nacionais de Doenças de Infecções Emergentes, Boston, MA
Transdução é uma forma de troca genética entre bactérias que utilizam bacteriófagos, ou phages, uma classe de vírus que infecta exclusivamente organismos procarióticos. Esta forma de transferência de DNA, de uma bactéria para outra por meio de um phage, foi descoberta em 1951 por Norton Zinder e Joshua Ledererg, que chamou o processo de “transdução” (1). Bacteriphages foram descobertos pela primeira vez em 1915 pelo bacteriologista britânico Frederick Twort, então descoberto independentemente novamente em 1917 pelo microbiologista franco-canadense Felix d’Herelle (2). Desde então, a estrutura e a função desses phages têm sido amplamente caracterizadas (3), dividindo essas pragas em duas classes. A primeira dessas classes são as fábulas líticas que, após a infecção, se multiplicam dentro da bactéria hospedeira, interrompendo o metabolismo bacteriano, lisendo a célula e liberando phage progênero (4). Como resultado dessa atividade antibacteriana e da crescente prevalência de bactérias resistentes a antibióticos, esses frascos líticos têm se mostrado recentemente úteis como um tratamento substituto para antibióticos. A segunda dessas classes são as fálvias lisogênicas que podem se multiplicar dentro do hospedeiro através do ciclo lítico ou entrar em um estado quiescente no qual seu genoma é integrado ao hospedeiro (Figura 1), um processo conhecido como lisogenia, com a capacidade de a produção de phage ser induzida em múltiplas gerações posteriores (4).
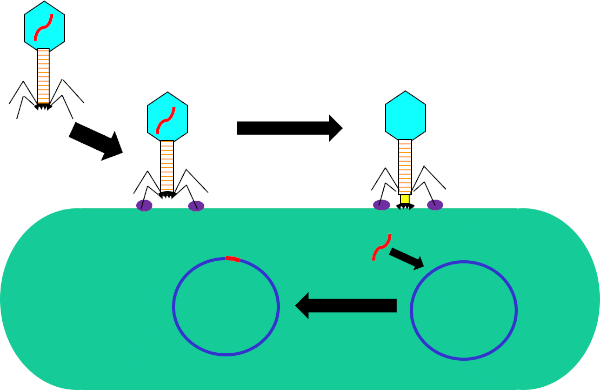
Figura 1: Infecção de célula hospedeira por bacteriófago. Adsorção pela phage para a parede celular bacteriana através de interações entre as fibras traseiras e receptor (roxo). Uma vez na superfície celular, o phage é irreversivelmente ligado à célula bacteriana usando a placa base (preta) que é movida para a parede celular pela baia contraltil (amarelo). O genoma phage (vermelho) então entra na célula e se integra ao genoma das células hospedeiras.
Embora a transdução bacteriana seja um processo natural, usando tecnologia moderna foi manipulado para a transferência de genes para bactérias no ambiente laboratorial. Ao inserir genes de interesse no genoma de uma praga lisogênica, como a praga, é capaz de transferir esses genes para os genomas das bactérias e, consequentemente, expressá-los dentro dessas células. Enquanto outros métodos de transferência genética, como a transformação, usam um plasmídeo para transferência e expressão genética, a inserção do genoma phage na da bactéria receptora não só tem o potencial de conferir novos traços a esta bactéria, mas também permite que mutações que ocorram naturalmente e outros fatores do ambiente celular alterem a função do gene transferido.
Em comparação com outros métodos de transferência de genes horizontais, como a conjugação, a transdução é bastante flexível nos critérios necessários para as células doadoras e receptoras. Qualquer elemento genético que possa caber dentro do genoma da praga que está sendo usada pode ser transferido de qualquer cepa de bactérias doadoras para qualquer cepa de bactérias receptoras, desde que ambas sejam permissivas ao phage, exigindo a expressão de receptores phage necessários nas superfícies celulares. Uma vez que este gene é removido do genoma do doador e embalado para o phage, ele pode ser transferido para o receptor. Após a transdução, é necessário selecionar para o receptor bactérias que contenham o gene de interesse devem ser selecionados. Isso poderia ser feito pelo uso de um marcador genético, como uma marca BANDEIRA ou polyhistidina-tag, para marcar o gene de interesse, ou a função intrínseca do gene, no caso de genes que codificam para resistência a antibióticos. Além disso, o PCR poderia ser usado para confirmar ainda mais a transdução bem sucedida. Usando primers para uma região dentro do gene de interesse e comparando sinal a um controle positivo, bactérias que têm o gene de interesse, e um controle negativo, bactérias que passaram pelos mesmos passos da reação de transdução sem phage. Embora a transdução bacteriana seja uma ferramenta útil na biologia molecular, ela tem e continua a desempenhar um papel importante na evolução das bactérias, particularmente no que diz respeito ao recente aumento da resistência a antibióticos.
Neste experimento, a transdução bacteriana foi utilizada para transferir a codificação genética para resistência à ampicilina antibiótico da cepa W3110 de E.coli para a cepa J53 através do bacteriófago P1 (5). Este experimento consistia em dois passos principais. Primeiro, a preparação do phage P1 contendo o gene de resistência à ampicilina da cepa do doador. Em segundo lugar, a transferência deste gene para a cepa receptora por transdução com a phage P1 (Figura 1). Uma vez realizada, a transferência bem sucedida do gene de resistência à ampicilina poderia ser determinada por qPCR (Figura 2). Se a transdução fosse bem sucedida, a cepa J53 de E. coli seria resistente à ampicilina, e o gene conferenciando essa resistência detectável por qPCR. Se não tivesse sucesso, não haveria detecção do gene de resistência à ampicilina e a ampicillina ainda funcionaria como um antibiótico eficaz contra a cepa J53.
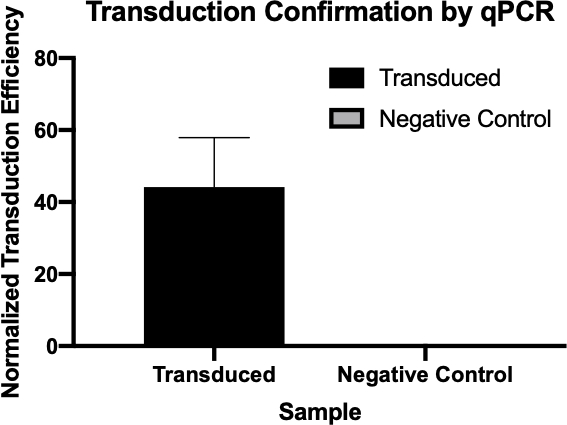
Figura 2: A confirmação da transdução bem sucedida por qPCR. Comparando os valores Cq detectados para o gene de interesse da reação de transdução e a reação de controle negativo, e normalizando esses valores em relação a um gene de limpeza, foi possível confirmar que a transdução bacteriana foi bem sucedida.