Community DNA Extraction from Bacterial Colonies
English
Share
Overview
Source: Laboratories of Dr. Ian Pepper and Dr. Charles Gerba – The University of Arizona
Demonstrating Author: Luisa Ikner
Traditional methods of analysis for microbial communities within soils have usually involved either cultural assays utilizing dilution and plating methodology on selective and differential media or direct count assays. Direct counts offer information about the total number of bacteria present, but give no information about the number or diversity of populations present within the community. Plate counts allow enumeration of total cultural or selected cultural populations, and hence provide information on the different populations present. However, since less than 1% of soil bacteria are readily culturable, cultural information offers only a piece of the picture. The actual fraction of the community that can be cultured depends on the medium chosen for cultural counts. Any single medium will select for the populations that are best suited to that particular medium.
In recent years, the advantages of studying community DNA extracted from soil samples have become apparent. This nonculture-based approach is thought to be more representative of the actual community present than culture-based approaches. In addition to providing information about the types of populations present, this approach can also provide information about their genetic potential. As with any technique, there are limitations to the data that can be obtained with DNA extraction. Therefore, many researchers now use DNA extraction in conjunction with direct and cultural counts to maximize the data obtained from an environmental sample.
Principles
DNA extraction from soil can be conducted in one of two ways (Table 1). In the in situ method, a combination of chemical-based and mechanical techniques is used. For this extraction, a mass of soil is combined with an equivalent volume of an extraction buffer. Glass beads are then added to the suspension along with a volume of detergent (sodium dodecyl sulfate, or SDS, is typically used), and the sample is blended to facilitate separation from soil particles followed by incubation at an elevated temperature to promote cell lysis. After centrifugation, the supernatant is subjected to further extraction and incubation steps in order to purify the DNA product.
Alternately, cells may first be fractionated (or separated) from the soil matrix prior to extraction of the genetic material. A mass of soil sample undergoes successive cycles of blending and slow centrifugation. The bead-beating step is eliminated here, however, in order to maintain intact cells, which are centrifuged to obtain a pellet. A lysozyme-based extraction is then performed in conjunction with incubation to disrupt the cell walls and liberate DNA for purification.
This manuscript and video will demonstrate the in situ method of DNA extraction from soil, as this procedure has been demonstrated to yield greater concentrations of DNA from soil samples relative to the cell fractionation method.
| Issue | Bacterial Fractionation | In Situ Lysis |
| Yield of DNA | 1-5 μg/g | 1-20 μg/g |
| Representative of community | Less representative because of cell sorption | More representative, unaffected cell sorption |
| Source of DNA recovered | Only bacteria | Mostly bacteria but also fungi and protozoa |
| Degree of DNA shearing | Less shearing | More shearing |
| Average size of DNA fragments | 50 kb | 25 kb |
| Degree of humic contamination | Less contaminated | More contaminated |
| Ease of methodology | Low, laborious | Faster, less labor-intensive |
Table 1. Comparison of bacterial fractionation and in situ lysis methodologies for the recovery of DNA from soil.
Procedure
1. Bacterial Community DNA Extraction
- To begin the procedure, weigh out 100 g of sieved soil. Add this to a polypropylene vessel, and add 100 mL of extraction buffer comprised of Tris buffer amended with EDTA to promote the release of bacteria from the soil matrix, then shake by hand.
- Next, weigh 100 g of glass beads, and add these to the mixing vessel. Agitate the sample for 5 min using a bead beating device or mechanical wrist-action shaker for 15 min. Add 10 mL 20% sodium dodecyl sulfate, or SDS, to the mixture, then agitate for an additional minute. Incubate at a high temperature of 60 – 65 °C for 60 min.
- Equally distribute the sample among separate 50-mL tubes, and centrifuge for 10 min at 6,000 x g. Transfer the supernatant from the tubes to a single sterile container. Next, repeat the extraction on the soil pellet as previously described, using a fresh volume of extraction buffer.
- Next, add the total volume of processed supernatant, approximately 200 mL, to a clean 50-mL tube filled to half volume with a solution of 30% polyethylene glycol and 1.6 M sodium chloride. Invert the bottles several times by hand to mix, and incubate at room temperature for 2 h. Centrifuge samples at 10,000 x g for 20 min to pellet the DNA.
- Remove the supernatant carefully from the centrifuge tube, leaving behind the partially purified nucleic acid pellet. Add 20 mL of TE Buffer and 1.5 mL of a 7.5 M potassium acetate solution to resuspend the pellet, then vortex. Place the suspension on ice for 5 min. Centrifuge at 16,000 x g for 30 min at 4 °C to precipitate proteins and polysaccharides.
- Next, add an RNAse and proteinase K to the sample, mix gently by hand, and let sit for moment. Add an equivalent volume of phenol:chloroform:isoamyl alcohol (ratio mixture of 25:24:1) to the suspension to be extracted, and mix gently by hand. Centrifuge the preparation for 10 min at 13,000 x g. Carefully remove the vessel from the centrifuge, and note the two layers.
- The bottom, heavier layer is comprised of the phenol:chloroform:isoamyl alcohol and extracted debris, and the top layer is the aqueous and contains the DNA. Place the aqueous phase into a sterile vessel, add an equivalent volume of isopropanol, and invert gently to initiate DNA precipitation. Incubate the suspension at room temperature for 2 h. Pellet the purified DNA by centrifugation at 16,000 x g for 30 min. Carefully remove the supernatant as the pelleted DNA may or may not be visible at the bottom of the vessel, and then resuspend in 1 mL of TE Buffer.
- Using a spectrophotometer or DNA/RNA quantification fluorimeter, measure the level of DNA extracted from the sample. The amount of DNA is estimated from the 260 nm reading. An absorbance reading of 1.0 is equivalent to 50 µg of DNA per mL of solution. If the concentration is too high for accurate readings, dilute the suspension 1 to 10, or 1 to 100 using molecular grade water.
- The purity of DNA is estimated from the ratio of the reading at 260 nm to that at 280 nm. A value > 1.7 indicates relatively pure DNA. The maximum theoretical value is 2.0.
Bacterial community DNA extraction is a process by which DNA is obtained from multiple bacterial species within a community during a single extraction procedure.
Traditional analyses of microbial communities in soil have usually involved cultural assays, utilizing dilution and plating methodology on different selective media. However, many bacteria grow poorly under laboratory conditions or on the specific growth media conditions selected, meaning they may be missed or severely underrepresented.
Recently, extracting community DNA from soil bacterial samples has allowed for a more comprehensive sampling of bacterial communities. This non-culture based approach is thought to be more representative of the actual communities present than traditional culture based methods.
This video will demonstrate a non-culture method of bacterial community DNA extraction, how to check the quality and quantity of extracted DNA, and explore how this DNA may be utilized to study bacterial diversity.
DNA extraction from soil can be conducted in one of two ways. In the fractionation method, cells are first separated from the soil matrix prior to extraction of the genetic material. The sample is then subjected to successive cycles of blending and slow centrifugation in order to collect intact cells in a pellet.
Lysozymes are then added to the cells, and the suspension is incubated. Lysozymes are enzymes that break down bacterial cell walls. Once the cell wall structure has been compromised, the DNA may then be liberated for purification. However, a second method of community DNA extraction, the in situ method, has been shown to yield greater DNA concentration.
Here, a mass of soil is combined with an equivalent volume of Tris-EDTA extraction buffer and glass beads, and mixed aggressively to facilitate separation of cells from the soil particles. A detergent is then added, generally sodium dodecyl sulfate, or SDS, and the sample is further blended to promote lysing of the cells and release of their contents, including DNA.
Incubation at a high temperature is then undertaken to lyse any remaining bacterial cells. Samples are centrifuged, and a polyethylene glycol extraction and incubation is performed on the supernatant in order to precipitate the DNA, which is then centrifuged into a pellet.
The DNA is resuspended in TE Buffer and potassium acetate in order to further wash the DNA of proteins and polysaccharides, then centrifugation is carried out to pellet these undesirable components. The aqueous supernatant containing the DNA is removed, and subjected to a phenol-chloroform extraction and isopropanol precipitation to further clean and concentrate the DNA. Following a room temperature incubation period of two hours, the DNA is centrifuged and resuspended in TE Buffer for storage until analysis.
Now that we are familiar with the concepts and processes behind bacterial community DNA extraction, let's take a look at how it is carried out in the laboratory.
To begin the procedure, weigh out 100 g of sieved soil. Add this to a polypropylene vessel, and add 100 mL of extraction buffer comprised of Tris buffer amended with EDTA to promote the release of bacteria from the soil matrix, then shake by hand.
Next, weigh 100 g of glass beads, and add these to the mixing vessel. Agitate the sample for 5 min using a bead beating device or mechanical wrist-action shaker. Add 10 mL 20% sodium dodecyl sulfate, or SDS, to the mixture, then agitate for an additional minute. Incubate at a high temperature for 60 min.
Equally distribute the sample among separate 50 mL tubes, and centrifuge for 10 min at 6,000 x g. Transfer the supernatant from the tubes to a single sterile container. Next, repeat the extraction on the soil pellet as previously described, using a fresh volume of extraction buffer.
Next, add the total volume of processed supernatant to a clean 50-mL tube filled to half volume with a solution of 30% polyethylene glycol and 1.6 M sodium chloride. Invert the bottles several times by hand to mix, and incubate at room temperature for 2 h. Centrifuge samples at 10,000 x g for 20 min to pellet the DNA.
Remove the supernatant carefully from the centrifuge tube, leaving behind the partially purified nucleic acid pellet. Add 20 mL of TE Buffer and 1.5 mL of a 7.5 M potassium acetate solution to resuspend the pellet, then vortex. Place the suspension on ice for 5 min. Centrifuge at 16,000 x g for 30 min at 4 °C to precipitate proteins and polysaccharides.
Next, add an RNAse and proteinase K to the sample, mix gently by hand, and let sit for moment. Add an equivalent volume of phenol:chloroform:isoamyl alcohol to the suspension to be extracted, and mix gently by hand. Centrifuge the preparation for 10 min at 13,000 x g. Carefully remove the vessel from the centrifuge, and note the two layers.
The bottom, heavier layer is comprised of the phenol:chloroform:isoamyl alcohol and extracted debris, and the top layer is the aqueous and contains the DNA. Place the aqueous phase into a sterile vessel, add an equivalent volume of isopropanol, and invert gently to initiate DNA precipitation. Incubate the suspension at room temperature for 2 h. Pellet the purified DNA by centrifugation at 16,000 x g for 30 min. Carefully remove the supernatant as the pelleted DNA may or may not be visible at the bottom of the vessel, and then resuspend in 1 mL of TE Buffer.
Using a spectrophotometer or DNA/RNA quantification fluorimeter, measure the level of DNA extracted from the sample. DNA/RNA fluorimeters will output DNA levels in units of nanograms per milliliter. If the concentration is too high for accurate readings, dilute the suspension 1 to 10, or 1 to 100 using molecular grade water.
Once DNA is obtained from a bacterial community, this can be utilized in a number of different ways. Some of those applications are explored here.
Spectrophotographic analyses of the DNA extracted from community DNA can provide an insight into the number of bacterial cells present in a given soil sample. The estimated quantity of DNA in ng per mL of solution can be related back to the total volume of DNA extracted in solution, to give the total amount of DNA per g of soil. Knowing the theoretical value of DNA per cell, the total number of cells per g of soil can be calculated.
For more targeted applications, DNA extracted from bacterial communities can be subjected to PCR to determine if a particular species is present within the community. For example, scientists may want to identify whether soil samples contain specific pathogens, such as Clostridium perfringens or Bacillus anthracis.
Finally, to obtain a more comprehensive understanding of the bacteria present in a community, DNA samples can be subjected to "omic" and bioinformatic characterization that allow for deeper analysis of the original bacteria within the sample. “Omics” describes a range of technologies that explore roles, relationships, and actions of molecules in organisms or communities. This includes the studies of genes and their function, or “Genomics”, and “Proteomics”, the study of proteins and their roles. For example, sequencing of 16S RNA from the samples can allow a metagenomic determination of specific species within the community, giving a more detailed estimate of diversity. This approach can give scientists a better understanding of the species makeup of a community, and what roles they may be undertaking.
You've just watched JoVE's introduction to bacterial community DNA extraction. You should now understand how to extract DNA from a bacterial community, how to check the quality of this DNA, and how this DNA can be used for investigations of bacterial community composition. Thanks for watching!
Applications and Summary
Community DNA from cultured colonies or extracted from soil can be subjected to bioinformatics and “omic” approaches that allow for characterization of the original bacteria within the sample. The omic approaches include metagenomics – determination of “who” is within the community via 16S rRNA sequencing. This gives an estimate of the diversity within the community.
The number of bacterial cells in the original soil sample can also be calculated. Community DNA is extracted from a soil and quantified by spectroscopic analyses. The estimated quantity of DNA measured as µg DNA per mL of solution is related back to the total volume of DNA extracted in solution to give a total amount of DNA per g of soil. By knowing the theoretical value of DNA per cell, the total number of cells per g of soil can be calculated.
Example
A soil has 0.12 µg DNA per g of soil
If each cell has 4 fg of DNA
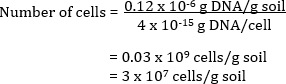
The extracted community DNA can be subjected to PCR analysis using specific primers to determine if a particular species is present within the community. Examples include specific bacterial pathogens such as Clostridium perfringens or Bacillus anthracis.
Transcript
Bacterial community DNA extraction is a process by which DNA is obtained from multiple bacterial species within a community during a single extraction procedure.
Traditional analyses of microbial communities in soil have usually involved cultural assays, utilizing dilution and plating methodology on different selective media. However, many bacteria grow poorly under laboratory conditions or on the specific growth media conditions selected, meaning they may be missed or severely underrepresented.
Recently, extracting community DNA from soil bacterial samples has allowed for a more comprehensive sampling of bacterial communities. This non-culture based approach is thought to be more representative of the actual communities present than traditional culture based methods.
This video will demonstrate a non-culture method of bacterial community DNA extraction, how to check the quality and quantity of extracted DNA, and explore how this DNA may be utilized to study bacterial diversity.
DNA extraction from soil can be conducted in one of two ways. In the fractionation method, cells are first separated from the soil matrix prior to extraction of the genetic material. The sample is then subjected to successive cycles of blending and slow centrifugation in order to collect intact cells in a pellet.
Lysozymes are then added to the cells, and the suspension is incubated. Lysozymes are enzymes that break down bacterial cell walls. Once the cell wall structure has been compromised, the DNA may then be liberated for purification. However, a second method of community DNA extraction, the in situ method, has been shown to yield greater DNA concentration.
Here, a mass of soil is combined with an equivalent volume of Tris-EDTA extraction buffer and glass beads, and mixed aggressively to facilitate separation of cells from the soil particles. A detergent is then added, generally sodium dodecyl sulfate, or SDS, and the sample is further blended to promote lysing of the cells and release of their contents, including DNA.
Incubation at a high temperature is then undertaken to lyse any remaining bacterial cells. Samples are centrifuged, and a polyethylene glycol extraction and incubation is performed on the supernatant in order to precipitate the DNA, which is then centrifuged into a pellet.
The DNA is resuspended in TE Buffer and potassium acetate in order to further wash the DNA of proteins and polysaccharides, then centrifugation is carried out to pellet these undesirable components. The aqueous supernatant containing the DNA is removed, and subjected to a phenol-chloroform extraction and isopropanol precipitation to further clean and concentrate the DNA. Following a room temperature incubation period of two hours, the DNA is centrifuged and resuspended in TE Buffer for storage until analysis.
Now that we are familiar with the concepts and processes behind bacterial community DNA extraction, let’s take a look at how it is carried out in the laboratory.
To begin the procedure, weigh out 100 g of sieved soil. Add this to a polypropylene vessel, and add 100 mL of extraction buffer comprised of Tris buffer amended with EDTA to promote the release of bacteria from the soil matrix, then shake by hand.
Next, weigh 100 g of glass beads, and add these to the mixing vessel. Agitate the sample for 5 min using a bead beating device or mechanical wrist-action shaker. Add 10 mL 20% sodium dodecyl sulfate, or SDS, to the mixture, then agitate for an additional minute. Incubate at a high temperature for 60 min.
Equally distribute the sample among separate 50 mL tubes, and centrifuge for 10 min at 6,000 x g. Transfer the supernatant from the tubes to a single sterile container. Next, repeat the extraction on the soil pellet as previously described, using a fresh volume of extraction buffer.
Next, add the total volume of processed supernatant to a clean 50-mL tube filled to half volume with a solution of 30% polyethylene glycol and 1.6 M sodium chloride. Invert the bottles several times by hand to mix, and incubate at room temperature for 2 h. Centrifuge samples at 10,000 x g for 20 min to pellet the DNA.
Remove the supernatant carefully from the centrifuge tube, leaving behind the partially purified nucleic acid pellet. Add 20 mL of TE Buffer and 1.5 mL of a 7.5 M potassium acetate solution to resuspend the pellet, then vortex. Place the suspension on ice for 5 min. Centrifuge at 16,000 x g for 30 min at 4 °C to precipitate proteins and polysaccharides.
Next, add an RNAse and proteinase K to the sample, mix gently by hand, and let sit for moment. Add an equivalent volume of phenol:chloroform:isoamyl alcohol to the suspension to be extracted, and mix gently by hand. Centrifuge the preparation for 10 min at 13,000 x g. Carefully remove the vessel from the centrifuge, and note the two layers.
The bottom, heavier layer is comprised of the phenol:chloroform:isoamyl alcohol and extracted debris, and the top layer is the aqueous and contains the DNA. Place the aqueous phase into a sterile vessel, add an equivalent volume of isopropanol, and invert gently to initiate DNA precipitation. Incubate the suspension at room temperature for 2 h. Pellet the purified DNA by centrifugation at 16,000 x g for 30 min. Carefully remove the supernatant as the pelleted DNA may or may not be visible at the bottom of the vessel, and then resuspend in 1 mL of TE Buffer.
Using a spectrophotometer or DNA/RNA quantification fluorimeter, measure the level of DNA extracted from the sample. DNA/RNA fluorimeters will output DNA levels in units of nanograms per milliliter. If the concentration is too high for accurate readings, dilute the suspension 1 to 10, or 1 to 100 using molecular grade water.
Once DNA is obtained from a bacterial community, this can be utilized in a number of different ways. Some of those applications are explored here.
Spectrophotographic analyses of the DNA extracted from community DNA can provide an insight into the number of bacterial cells present in a given soil sample. The estimated quantity of DNA in ng per mL of solution can be related back to the total volume of DNA extracted in solution, to give the total amount of DNA per g of soil. Knowing the theoretical value of DNA per cell, the total number of cells per g of soil can be calculated.
For more targeted applications, DNA extracted from bacterial communities can be subjected to PCR to determine if a particular species is present within the community. For example, scientists may want to identify whether soil samples contain specific pathogens, such as Clostridium perfringens or Bacillus anthracis.
Finally, to obtain a more comprehensive understanding of the bacteria present in a community, DNA samples can be subjected to “omic” and bioinformatic characterization that allow for deeper analysis of the original bacteria within the sample. “Omics” describes a range of technologies that explore roles, relationships, and actions of molecules in organisms or communities. This includes the studies of genes and their function, or “Genomics”, and “Proteomics”, the study of proteins and their roles. For example, sequencing of 16S RNA from the samples can allow a metagenomic determination of specific species within the community, giving a more detailed estimate of diversity. This approach can give scientists a better understanding of the species makeup of a community, and what roles they may be undertaking.
You’ve just watched JoVE’s introduction to bacterial community DNA extraction. You should now understand how to extract DNA from a bacterial community, how to check the quality of this DNA, and how this DNA can be used for investigations of bacterial community composition. Thanks for watching!