Figure 2 shows an example FRAP study of protein mobility. The mobility of dEGFP-ER protein is demonstrated by the rapid fluorescence recovery after photobleaching in bleached OSERs. For the quantitative analysis, the half time and mobile fraction were derived from experimentally measured data by fitting the following monoexponential equation:
F(t) = Fpost + (Frec-Fpost)(1-e-t/τ)
where Fpost is the fluorescence signal after photobleaching, Frec is the maximum fluorescence recovery value that is reach after bleaching, t the time of registration and τ the time constant.
Please note the importance of acquiring images without saturated pixels that could alter the fluorescence recovery and, consequently, the protein mobility analysis. It is also essential to always normalize the fluorescence signal in the bleached ROI to the total fluorescence of the same cell in order to consider fluorescence intensity variations due to bleaching during image acquisition or small changes in the focus plane.
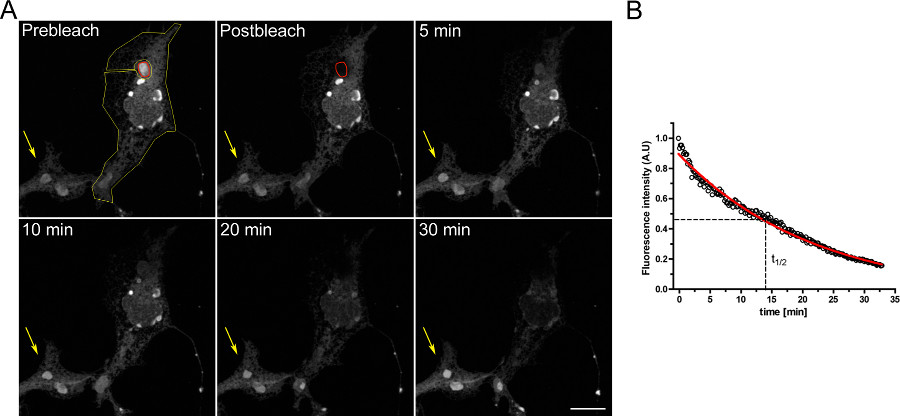
An example of a FLIP experiment to study the continuity between intracellular compartments is shown in Figure 3. OSERs are physically connected with the rest of the ER as demonstrated by the progressive emptying of the ER when the OSER domain is continuously bleached.
For a proper analysis, the acquisition of saturated pixels must be avoided (see above); furthermore, the acquisition parameters must be set up with laser powers as low as possible in order to avoid photobleaching due to image acquisition. For this reason it is strongly recommended to image an unbleached cell in the same field that will be used to normalize the fluorescence signal of the bleached cell.
All experiments have to be performed in the presence of cycloheximide, a translation inhibitor, in order to avoid any increase in the ER fluorescence signal (and consequently total fluorescence) due to protein biosynthesis.
Transmission electron microscopy demonstrated that the fluorescent aggregates observed in cultured cells transfected with dEGFP-ER represent patches of smooth and flattened ER cisternae that spatially organized themselves into well-defined 3D geometries classified on the basis of their patterns: linear or curved stacks (often associated with the nuclear envelope, not shown) (Figures 4A and B) that may be continuous with regions of sinusoidal ER (Figure 4A); the membranes in some regions are organized into lattices with a square or hexagonal symmetry (crystalloid ER, not shown). Adjacent cisternae are separated by a thin layer of slightly electron-dense cytoplasm about 11 nm thick that is continuous with the cytoplasm surrounding the aggregates.

Figure 1. Flow chart of the experimental procedure. The cultured cells are first transfected with jetPEI (see Protocol) in order to over-express the fluorescent fusion protein of interest. After 24 hr, the live transfected cells are visualized and FRAP and FLIP experiments are performed using a confocal microscope equipped with a controlled temperature CO2 incubator, and the recorded images are exported and analyzed using appropriate software (e.g. ImageJ). For the ultrastructural analysis, the transfected cells are fixed, pelleted and embedded in epon epoxy resin blocs. Ultrathin sections are obtained using a diamond knife, collected on copper grids, and observed under a transmission electron microscope. Click here to view larger figure.

Figure 2. FRAP experiment using COS-7 cells transiently transfected with dEGFP-ER. A) Two OSER structures (red ROIs) were bleached, and fluorescence recovery was recorded over time. Clear fluorescence recovery can be detected 1 min post-bleaching, and the signal further increases 4 min later (scale bar 10 μm). B): Quantitative analysis of the FRAP experiment showing the recovery half-time and the mobile fraction of the dEGFP-ER protein. Click here to view larger figure

Figure 3. FLIP experiment using COS-7 cells transiently transfected with dEGFP-ER. A) The continuous bleaching of an OSER (indicated by the red ROI) causes a progressive decrease in fluorescence in the rest of the ER and in other OSER structures within the same cell (indicated by the yellow ROI). The yellow arrow indicates a portion of an unbleached cell in which the fluorescence signal is constant over time. (Scale bar 10 μm). B) Quantitative analysis of the FLIP experiment. Click here to view larger figure

Figure 4. After fixation and embedding, cells expressing high levels of dEGFP-ER in which OSER structures could be detected by means of fluorescence optical microscopy were observed through a transmission electron microscope. A) Low magnification view of a portion of cytoplasm of a cell containing an OSER consisting of stacked cisternae and undulating sinusoidal membranes. Mitochondria (M) can be seen clustered around the OSER structures, whereas ribosomes decorate only the membranes of the outermost cisternae (arrowheads and inset). The 11 nm thick electron-dense space between the membranes is continuous with the cytoplasm (arrow and inset) (L = lysosomes/(auto-)phagosomes) (scale bar 1.5 μm; inset 0.25 μm). B) An OSER can be formed by lamellar ER: i.e. stacks of flattened ER cisternae that can be continuous or fragmented in their appearance in thin sections. Vesicles budding from the outermost cisternae of the stack can occasionally be observed (asterisk) (PM, plasma membrane) (Scale bar 150 nm).

{kind=link}