Synaptic connectivity is evident by stimulating the presynaptic neuron to fire an action potential by passing a depolarizing current pulse (typically 20-50 pA for 20 msec) via the recording electrode. The postsynaptic current trace is then examined for the presence of a monosynaptic EPSC evoked at short (<5 msec) and consistent latencies after the peak of the presynaptic action potential (Figure 3A). In most experiments multiple postsynaptic neurons are tested before a synaptically-connected pair can be obtained. Overall, ~1/3 of presynaptic CA3 cells are monosynaptically coupled to the postsynaptic CA3 cell by active synaptic connections (Figure 3A), and approximately 20% of CA3 neurons are connected by all-silent synaptic connections6,8.
The amplitude of baseline AMPA receptor mediated currents is variable from trial to trial within a paired recording, and also between independent paired recordings (Figure 3B)6,8. Average AMPAR EPSC amplitude ranged from <10 pA to >800 pA, and likely arises from differences in the number of functional synapses between paired recordings. Failure rates have also been observed to vary significantly between paired recordings6,7, ranging from 100% AMPAR EPSC failure rates at silent synapses, to 0-95% failure rates in pairs connected by active synapses6,7. Within individual paired recordings, synaptic failures occurred, especially in recordings with smaller AMPAR EPSC amplitudes. The variability in AMPAR EPSC amplitude is evident from trial to trial in the raw data (Figure 3B) is likely due to fluctuation in quantal number released from trial to trial, as occurs at other synapses6.
Dual whole cell recordings also provide direct access to both the pre- and the postsynaptic neuronal cytoplasm, enabling pharmacological manipulation of either/both the presynaptic and postsynaptic cells via the recording electrodes6. Typically, presynaptic pharmacological manipulations are very rapid (within 10 min). This is not limited to small molecules (e.g., BAPTA), as fluorescently labeled dextrans can access presynaptic terminals ≤200 μm away from the recording electrode. Therefore analysis similar to that performed at the squid giant synapse25 could be extended to studies of synaptic vesicle release machinery in the hippocampal slice. The position of the specific synapses measured in the paired recordings are not identified in the recording process. Therefore the proximal versus distal sites of synapses cannot be assessed as readily as local stimulation with extracellular stimulating electrodes or focal glutamate application. However, dye filling of each neuron during the paired recording can enable morphological reconstruction of axonal arbors and potential synaptic sites6.
LTP and LTD are reliably induced at CA3-CA3 synapses in this culture system (Figure 3C), and both forms of plasticity last for the duration of whole cell recordings (over 2 hr). The LTP and LTD induction paradigms utilized are identical to those performed in acute slices and LTP and LTD exhibit the properties of NMDAR-dependence, associativity and pathway independence, and therefore this combination of culture system and paired recordings provides a valuable model system to examine synaptic plasticity7.
Polysynaptic inhibitory events are frequently observed in paired recordings between CA3 pyramidal cell pairs (Figure 3D). We do not pharmacologically block this GABAergic inhibition as this produces highly disruptive hyperactivity. However, due to the longer latency of these polysynaptic inhibitory events, they generally do not prevent measurement of the monosynaptic excitatory current. If the polysynaptic inhibitory events were observed to interfere with the monosynaptic current, obscuring the peak of the monosynaptic current, these pairs need to be excluded from the analysis. Polysynaptic excitatory connections are also sometimes observed, but are significantly less common and also are excluded from analysis. Rarely, a monosynaptic inhibitory synaptic current is obtained due to the presynaptic neuron being an inhibitory interneuron. This is readily identified by the lack of accommodation of action potential firing in response to longer (1 sec) current injection. This is not possible in postsynaptic neurons in which the internal solution is cesium gluconate-based. However, as neurons are identified visually prior to recording, in our experience this occurs <1% of the time. Interneurons within organotypic hippocampal slices are readily identified visually by their non-pyramidal cell body, especially in the stratum radiatum and oriens. Therefore this preparation also enables recordings of interneuron excitatory and inhibitory currents. However, very little is known about the inhibitory networks in organotypic slices, and whether they maintain connectivity similar to that in the brain is not clear.
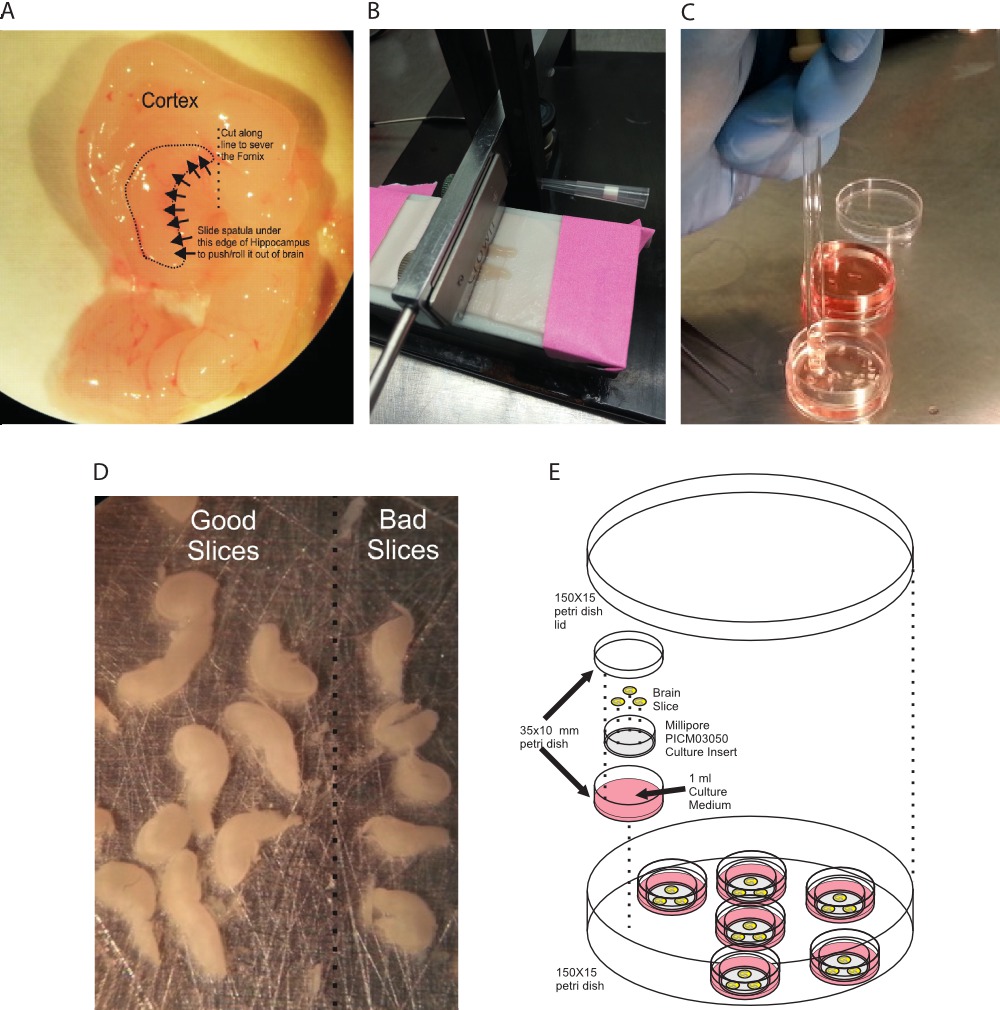
Figure 1. Preparation of organotypic hippocampal slice cultures. (A) The hippocampus, in situ, outlined by the dotted line. To remove the hippocampus, the connections to the fornix are severed and the hippocampus gently rolled out of the brain. (B) Hippocampi are positioned on the stage of the slicer on top of filter paper. (C) Hippocampal slices are transferred via the wide bore of a Pasteur pipette to prevent damage. (D) Examples of ‘good’ versus ‘bad’ slices. (E) Cartoon of slice setup on membranes. Please click here to view a larger version of this figure.

Figure 2. Obtaining paired whole cell recordings from hippocampal CA3 neurons. Sequential images of attaining a paired recording, showing optimal electrode and neuronal positioning. (A) After obtaining the first whole cell recording the microscope is moved 10-200 mm in the x-axis so that the established recording (arrow) is located at the edge of the area visible on the monitor. Scale bar: 25 μm. (B) To enable room for the second electrode, the lens of the microscope is then raised ~5-10 mm, ensuring that the ACSF still maintains contact with the lens. To ensure the second electrode does not bump the other recording electrode, the second electrode is moved in the x-axis until it is directly under the light path through the lens but still significantly above the established recording electrode. (C) Established paired recording showing both electrodes (arrows) in the same focal plane. Scale bar: 25 μm. (D) Paired recording from adjacent CA3 neurons in a transgenic animal, showing the EGFP-positive neurons on the RHS. Scale bar: 20 μm. Please click here to view a larger version of this figure.
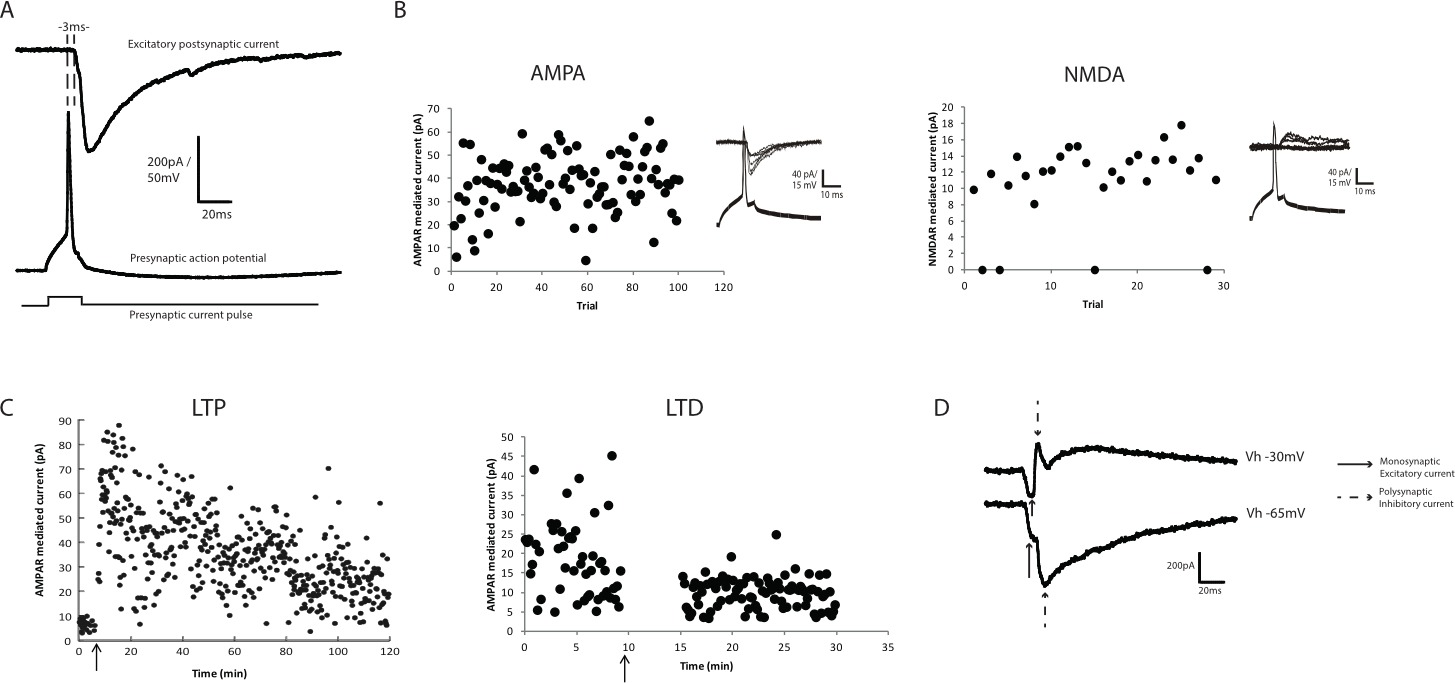
Figure 3. Synaptic transmission and plasticity between CA3-CA3 pyramidal cell pairs. (A) Example of pre- and postsynaptic traces from a paired recording between two CA3 pyramidal neurons. In response to a 20 sec current pulse to induce presynaptic action potential firing, the postsynaptic AMPAR-mediated current (recorded at -65 mV) occurs in direct response to the action potential. (B) Left: The amplitude of AMPAR-mediated EPSCs varies not only between paired recordings, but also from trial-to-trial within a paired recording. Each trial represents the amplitude of the EPSC measured at 0.1 Hz. In this example, AMPAR EPSC amplitude fluctuates from 5 pA to >60 pA. Right: NMDAR EPSC amplitudes measured at +40 mV in the presence of 10 μM CNQX. NMDAR EPSC amplitudes are typically significantly smaller in amplitude that AMPAR EPSCs, but still show significant variability. NMDAR EPSC amplitude typically averages between 10-20 pA in paired recordings between CA3 pyramidal cells, making failures readily identifiable. (C) Left: CA3 pyramidal cell pairs express LTP that persists for the length of the paired recording. LTP is readily evident by an increase in the amplitude of AMPAR EPSCs. Significant trial-to-trial variability in the AMPAR EPSC amplitude remains after the induction of LTP. Right: Expression of long-term depression (LTD) between CA3 pyramidal neurons. Note the decrease in average amplitude of the AMPAR EPSC and concomitant decrease in the trial to trial amplitude variability. (D) Example of postsynaptic polysynaptic inhibitory currents, which occur at a longer latency compared to the monosynaptic AMPAR EPSC. When the postsynaptic neuron is voltage clamped at -65 mV, the polysynaptic currents are visible as a second peak at a latency of >5 msec after the peak of the presynaptic action potential. Holding the postsynaptic neuron at -30 mV confirms that the polysynaptic current is inhibitory due to the reversal of the current direction. Please click here to view a larger version of this figure.

Table 1. Potential problems arising in paired recordings and preparation of organotypic slice cultures. This lists common potential problems that occur with paired whole cell recordings in organotypic slice cultures. In addition, we also advise that significant time is required to be spent ‘driving’ the electrophysiology setup for paired recordings, as most problems are related to movement disruptions that can readily be visualized and rectified.
