Methyl 4-(N-hydroxyacetamido)benzoate (1a) was synthesized in 92% isolated yield through a two-step procedure (i.e., reducing methyl 4-nitrobenzoate with hydrazine using 5% Rh/C as a catalyst to form methyl 4-(N-hydroxyamino)benzoate, followed by acetyl protection of the resulting hydroxylamine). O-Trifluoromethylation of 1a with Togni reagent II in the presence of catalytic amount of cesium carbonate (Cs2CO3) in chloroform at RT afforded the desired 4-(N-(trifluoromethoxy)acetamido)benzoate (2a) in 95% isolated yield. This compound underwent thermally induced OCF3-migration in MeNO2 at 120 °C to give the desired methyl 4-acetamido-3-(trifluoromethoxy)benzoate (3a) in 85% isolated yield.
The 1H, 13C, and 19F NMR spectrum of the final product 3a are depicted in Figure 1, Figure 2, and Figure 3, respectively. A distinguish quartet peak at 120.6 ppm with a large coupling constant (258.9 Hz) in 13C NMR spectra corresponds to the CF3 carbon. When the OCF3-migration takes place, a sharp change in the 19F NMR from -65 ppm (2a) to -58.1ppm (3a) is observed. The detail characterization data of 3a is reported as follow: Rf = 0.51 (hexanes/EtOAc 4:1 (v/v)). NMR Spectroscopy: 1H NMR (700 MHz, CDCl3, 25 °C, δ): 8.56 (d, J = 8.6 Hz, 1H), 7.97 (d, J = 8.6 Hz, 1H), 7.93 (s, 1H), 7.56 (br. s, 1H), 3.92 (s, 3H), 2.27 (s, 3H). 13C NMR (175 MHz, CDCl3, 25 °C, δ): 168.5, 165.6, 137.2, 134.7, 129.3, 125.8, 121.5, 120.8, 120.6 (q, J = 258.9 Hz), 52.5, 25.2. 19F NMR (376 MHz, CDCl3, 25 °C, δ): -58.1 (s). Mass Spectrometry: HRMS (ESI-TOF) (m/z): calcd for C11H11NO4F3 ([M + H]+) 278.0640, found 278.0643.
This protocol is general and applicable to a wide array of aromatic compounds (Table 1). The reaction tolerates a broad spectrum of functional groups including ester (3a, 3d), ketone (3b), nitrile (3c), ethers (3e, 3m), halogens (3g – 3l), CF3 group (3m, 3n), amide (3o) and heterocycle substituent (3o). The halogen substituents, especially Br and I, are particularly useful because they provide synthetic handles for further functionalization. In addition, high levels of ortho– over para-selectivity are observed (3f, 3k – 3l). In the presence of two non-identical ortho positions, low levels of regiocontrol are obtained (3d, 3e, 3k, 3m). Furthermore, the reaction temperature for the OCF3-migration step depends on the electronic nature of arenes. Generally, more electron deficient arenes require higher reaction temperature.

Figure 1. 1H NMR spectrum of 3a. Chemical shift and relative integration of characteristic protons are labeled. Please click here to view a larger version of this figure.

Figure 2. 13C NMR spectrum of 3a. Chemical shift of characteristic carbons is labeled. Please click here to view a larger version of this figure.

Figure 3. 19F NMR spectrum of 3a. Chemical shift of characteristic fluorine is labeled using trifluorotoluene (-63.3 ppm) as internal reference. Please click here to view a larger version of this figure.
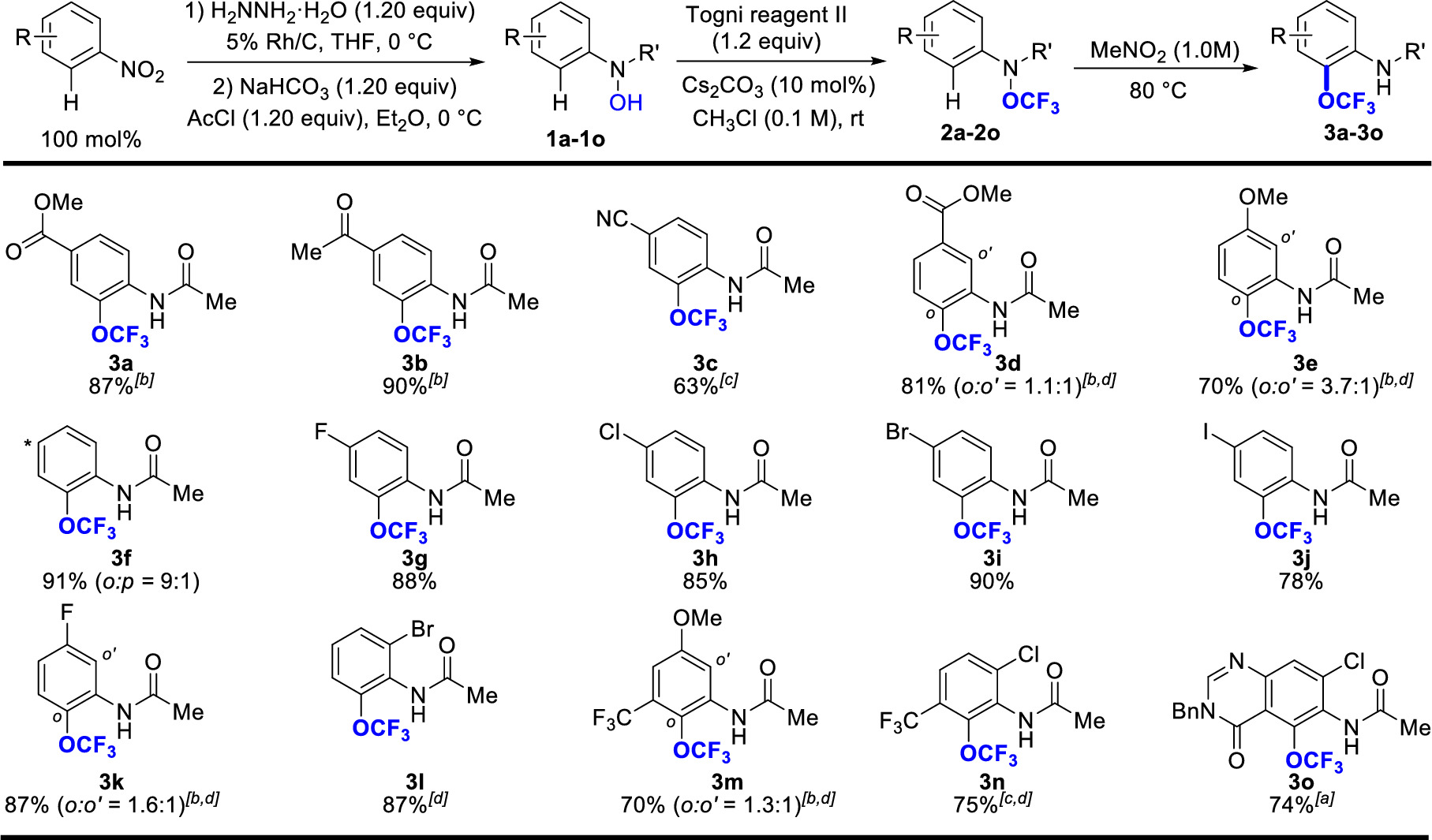
Table 1. Selected examples of trifluoromethoxylation of arenes. Reaction time: 11-48 hr. Cited yields and isomeric ratios are for OCF3-migration step (from 2 to 3) and of isolated material by flash column chromatography. [a] 50 °C. [b] 120 °C. [c] 140 °C. [d] Less than 5% para-product was detected. THF = Tetrahydrofuran; AcCl = acetyl chloride. Please click here to view a larger version of this table.
