

Summary
Hier präsentieren wir ein Protokoll Single, SNARE-vermittelte Fusion Ereignisse zwischen Liposomen und unterstützt Bilayer in mikrofluidischen Kanälen unter Verwendung von polarisierten TIRFM, mit Einzelmolekülempfindlichkeit und ~ 15 ms Zeitauflösung zu erfassen. Lipid und lösliche Ladung Freisetzung können gleichzeitig erfasst werden. Liposomgröße, Lipid-Diffusität und Fusion Poreneigenschaften gemessen werden.
Abstract
In der allgegenwärtigen Prozess der Membranfusion die Eröffnung einer Fusionspore gründet die erste Verbindung zwischen zwei ehemals getrennten Fächern. Während Neurotransmitter oder Hormonfreisetzung über Exozytose, kann die Fusionspore vorübergehend öffnen und schließen sich immer wieder, die Regulierung Ladung Freisetzungskinetik. Pore Dynamik bestimmen auch die Art der Vesikelrecycling; irreversible Wiederverschließbarkeit Ergebnisse in transient, "kiss-and-run" Fusion, während Dilatation führt zu Vollfusion. Um besser zu verstehen , welche Faktoren bestimmen Poren Dynamik entwickelten wir einen Test der Membranfusion unter Verwendung von polarisierten Totalreflexions - Fluoreszenz (TIRF) Mikroskopie mit Einzelmolekülempfindlichkeit und ~ 15 ms Zeitauflösung in einem biochemisch gut definierten in vitro - System zu überwachen. Fusion von fluoreszenzmarkierten kleine unilamellare Vesikel enthalten, v-SNARE-Proteine (v-SUVs) mit einem planaren Bilayer Lager T-SNAREs, auf einem weichen Polymer Kissen unterstützt (t-SBL, t-supported bilayer), Wird überwacht. Der Test verwendet mikrofluidischen Strömungskanäle, die nur minimale Probenverbrauch gewährleisten, während eine konstante Dichte von SUVs liefern. Ausnutzen der schnellen Signalverstärkung bei der Übertragung von Lipid-Etiketten aus dem SUV auf die SBL bei der Fusion, die Kinetik der Lipid-Farbstoffübertragung wird überwacht. Die Empfindlichkeit der TIRF-Mikroskopie ermöglicht es einzelnen fluoreszierenden Lipid-Etiketten-Tracking, von dem Lipid Diffusität und SUV-Größe kann für jede Fusion Veranstaltung abgeleitet werden. Lipid Farbstofffreisetzungszeiten können viel länger sein als für die ungehinderte Passage erwartet durch permanent offenen Poren. Mit Hilfe eines Modells, das Verzögerung der Lipid-Release übernimmt aufgrund flackert Poren, eine Pore "Offenheit", den Anteil der Zeit, die Poren bei der Fusion offen bleibt, kann abgeschätzt werden. Ein lösliches Marker kann in den SUVs für die gleichzeitige Überwachung von Lipid und lösliche Ladung Freisetzung verkapselt werden. Solche Messungen zeigen einige Poren nach dem Verlust einer Fraktion des löslichen Ladung wieder verschließen kann.
Introduction
Die Membranfusion ist ein universeller biologischer Prozess , die für intrazellulären Transport von Lipiden und Proteinen, Sekretion, Befruchtung, Entwicklung und umhüllte Virus den Eintritt in die Wirtsorganismen 1-3. Für die meisten intrazellulären Fusionsreaktionen einschließlich der Freisetzung von Hormonen und Neurotransmittern über Exozytose, die Energie zwei Lipid - Doppelschichten zu verschmelzen wird durch die Bildung eines Vier-Helix - Bündel von cognaten löslichen N-Ethylmaleimid-sensitive Faktor Attachment - Protein - Rezeptor (SNARE) Proteine zur Verfügung gestellt, verankert in das Vesikel (v-SNARE) und die Zielmembran (t-SNARE) 4. Synaptischen Vesikel Exocytose ist die streng reguliert Fusionsreaktion und tritt innerhalb einer Millisekunde nach der Ankunft eines Aktionspotentials 1,4,5. Die Fusionspore, die erste Verbindung zwischen den beiden Verschmelzen Fächer, flackern offen und geschlossen mehrere Male vor der Wiederverschließung oder irreversibel 5-7 erweitert. Die bisherigen Ergebnissein transient, "Kiss & run" Fusion, während Letzteres führt zur vollständigen Fusion. Faktoren , die die Balance zwischen diesen beiden Modi der Fusion regeln und Mechanismen Poren Flimmern Regulierung sind nicht gut 5,8 verstanden.
SNARE-Proteine sind für die Exocytose erforderlich; synaptischen Vesikel Fusion 9 , bei der Spaltung von SNARE durch Neurotoxine abgeschafft. Bulk - Fusionsexperimente mit kleinen unilamellaren Vesikeln (SUVs) haben gezeigt , dass SNAREs sind nicht nur erforderlich, sondern auch eine ausreichende Membranfusion 10 zu fahren. In diesem bulk Assay mit v-SNARE rekonstituiert SUVs (v-SUV) wurden mit fluoreszierenden Phospholipide (N dotiertem - (7-nitro-2-1,3-benzoxadiazol-4-yl) -phosphoethanolamine (NBD-PE) und ( N -. (Lissamins Rhodamin B sulfonyl) -phosphoethanolamine (LR-PE) und gemischt mit nicht - markierten Vesikeln t-SNAREs (t-SUV) Zunächst wird die Fluoreszenz von NBD-PE in v-SUVs wird abgeschreckt durch Förster - Resonanzenergietransfer (FRET enthaltend ) zu LR-PE. Da LaborELED v-SUVs Sicherung mit unmarkiertem t-SUVs, das Fluorophor Oberflächendichte in der nun kombinierten Membran reduziert wird und die sich ergebende Erhöhung der NBD-PE - Fluoreszenz berichtet , um das Ausmaß der Lipid - 10 gemischt wird . Da der Groß Assay einfach einzurichten und zu analysieren ist, wurde es allgemein zu untersuchen Mechanismen der SNARE-vermittelte Fusion 10-14 verwendet. Allerdings hat es einige Einschränkungen, wie niedrige Empfindlichkeit und schlechte Zeitauflösung. Am wichtigsten ist, als Ensemble-Messung, mittelt Ergebnisse aller Ereignisse zwischen docking und Fusion sowie Detektion von hemifusion Zwischenprodukte erschwert Diskriminierung.
In den letzten zehn Jahren mehrere Gruppen, darunter auch unsere, haben neue Assays entwickelt , um Fusionsereignisse an der einzelnen Vesikel Ebene 15-27 überwachen. Ha und Kollegen verwendeten V-SUVs auf eine Oberfläche gebunden und überwacht ihre Fusion mit kostenlosen T-SUVs 18,19. Lipidmischung wurde unter Verwendung von FRET zwischen einem Paar von Lipid-gebundenen Fluorophoren überwacht emin der v- und T-SUVs bett jeweils 18 Totalreflexions - Fluoreszenz (TIRF) Mikroskopie. Später benutzte Brunger Labor eine einzige Lipid-Label - Spezies zusammen mit einem Inhalt Marker für die simultane Detektion von Lipid und Inhalt 20,28 mischen. Sowohl das Lipid und die Inhalte Marker wurden bei hohen, selbstlöschenden Konzentrationen enthalten sind; Fusion mit unmarkierten SUVs führte Fluoreszenz Dequenching 20,28.
Andere haben fusioniert v-SUVs auf planaren Doppel Rekonstitution mit t-SNAREs 15-17,21-27,29. Die planare Geometrie des Targets (t-SNARE enthält) bilayer besser imitiert die physiologische Fusionsprozess von kleinen, stark gekrümmten Vesikel mit einer flachen Plasmamembran. Die Steinem Gruppe poren Spanning Membranen rekonstituiert mit t-SNAREs, suspendiert über ein poröses Siliciumnitridsubstrats und detektiert Fusion mit einzelnen V-SUVs mit der konfokalen Laser - Scanning - Mikroskopie 23 eingesetzt. Andere fgebrauchte v-SUVs zu planar bilayers mit t-SNAREs rekonstituiert, auf einem Glassubstrat 15-17,21,22,24-27,29 unterstützt. Der große Vorteil bei der Verwendung unterstützt Bilayer (SBLS) ist, dass die TIRF-Mikroskopie verwendet werden können, Docking und Fusionsereignisse mit hervorragenden Signal-Rausch-Verhältnis und ohne Interferenz von freiem V-SUVs zu erkennen, obwohl auch Einzelereignis Mikrofluidik mit Auflösung bietet mit Standard Fernfeld - Epifluoreszenzmikroskopie 24.
Ein wichtiges Anliegen ist, ob und wie Substrat-Wechselwirkungen Bilayer Bilayer Qualität und den Fusionsprozess unterstützt Affekt. Frühe Arbeiten gemacht Verwendung von planaren SBLS , die auf einem Glas- oder Quarzsubstrat 15-17 direkt unterstützt wurden. Diese SBLS wurden durch Adsorption hergestellt, bersten, Aufstreichen und Fusion von T-SUV Membranen auf dem Substrat. Es wurde jedoch bald erkannt, dass ein Schlüssel t-SNARE-Komponente weggelassen, SNAP25 aus SBLS auf diese Weise hergestellten führte V-SUV Andocken und Fusion Kinetik indistinguisHable von denen 17 mit der kompletten t-SNAREs erhalten. Da SNAP25 absolut für die Fusion in vivo 30,31 erforderlich ist, wurde die physiologische Relevanz dieser frühen Versuche in Frage gestellt. Tamm Gruppe überwanden diese Herausforderung durch eine bessere Verwendung von 21 unterstützten Doppelschichtbildung gesteuert. Es verwendete Langmuir-Blodgett - Abscheidung für die proteinfreie erste Beilage des SBL, gefolgt von der Fusion von dieser Monoschicht mit t-SUVs 21. Dies resultierte in SNAP25 abhängigen Fusion.
Potentielle Artefakte mit einer Doppelschicht direkt unterstützt auf einem Glassubstrat ohne die Notwendigkeit zu verwenden , Langmuir-Blodgett - Verfahren, zu vermeiden, Karatekin et al. Eingeführt einem weichen, hydratisierten Poly (ethylenglycol) (PEG) Polsters zwischen der Doppelschicht und dem Substrat 24. Diese Änderung führte auch in SNAP25 abhängigen Fusion 24. Doppelschichten auf einer Schicht weichen Polymer abgefedert war bekannt Trans besser zu bewahrenProtein Mobilität und Funktion 32, und hatte in der Fusionsstudien mit Viren 33 verwendet. Zusätzlich PEGylierten bilayers scheinen gewisse Fähigkeit zur Selbstheilung und sind sehr robust 34,35 zu halten. Zunächst wird eine Fraktion des handelsüblichen, Lipid-gebundenen PEG-Ketten sind in der t-SUV-Membran enthalten. Wenn diese t-SUVs bersten und eine planare Doppelschicht auf einem Glassubstrat bildet, erstreckt sich ein PEG brush beide Blättchen der planaren Bilayer. Weil planaren Doppelschichtbildung durch Anhaften der PEG-Ketten umgebenden t-SUVs auf die hydrophile Glasoberfläche, Liposom Berst- und planaren Doppelschichtbildung sind relativ unempfindlich gegenüber der Lipidzusammensetzung eingesetzt angetrieben wird. Wenn jedoch große Mengen an Cholesterin enthalten sind, die Kohäsionseigenschaften der SUVs zu erhöhen, können die SUVs nicht spontan platzen. Wenn dies der Fall ist, kann osmotischen Schock oder zweiwertige Ionen eingesetzt werden 25 planar Doppelschichtbildung zu helfen.
Wie oben erwähnt, in diesem apsatz eine PEG-Bürste auf beiden Seiten des ebenen umfasst, supported bilayer. Die Bürste den mikrofluidischen Strömungskanal zugewandt hilft unspezifischen Adhäsion von eingehenden v-SUVs zu verhindern, die sich auch in der Regel mit einer PEG-Schicht bedeckt. Die Bildung von v- und T-SNARE - Komplexen geht von der Membran-distalen N-Termini und verläuft in Stufen in Richtung der Membran-proximalen Domänen 36. Für die V-SUVs mit dem t-SBL, die v- und t-SNARE N-Termini Notwendigkeit vorzustehen über den PEG Bürsten zu interagieren, was der Fall unter den Bedingungen des Assays zu sein scheint. Bürstenhöhe können an andere Proteine als SNAREs studieren angepasst werden , indem die Dichte von PEGyliertem Lipiden variierende und die PEG - Kettenlänge 37,38. Ein weiterer Vorteil der PEG - Bürsten die proximalen Oberflächen der Fusionieren Bilayer bedeckt , ist , dass sie die überfüllten Umgebung von biologischen Membranen imitieren , die mit 30.000-40.000 integrale Membranproteine pro Quadratmikrometer 39 verpackt werden. Genau wie die PEG-Ketten in diesem Assay die repulsive Proteinschicht, die biologischen Membranen muss gedrückt werden, zur Seite für den Kontakt zwischen den beiden Phospholipiddoppelschichten zu ermöglichen für die Fusion auftritt.
Mikrofluidischen Strömungskanäle werden in diesem Test verwendet, da sie einzigartige Vorteile bieten. Erstens ermöglicht mikrofluidischen Strömungs einheitlichere Abscheidung von T-SUVs zu verbreiten und Sicherung der t-SBL zu bilden. Zweitens ist die kleine Kanalvolumen (<1 & mgr; l) minimiert Probenverbrauch. Drittens benötigt die kleinen Volumina ermöglichen das gesamte Experiment unter konstanten Fluss durchgeführt werden. Fluss entfernt schwach, vermutlich nicht spezifisch haftete v-SUVs von der SBL 16. Es unterhält auch eine konstante Dichte von v-SUVs über dem t-SBL, kinetische Analyse zu vereinfachen 17. Schließlich werden dockt Vesikel aus kostenlos sind durch die Strömung 25 durch leicht zu unterscheiden. Viertens mehrere mikrofluidische Kanäle auf dem gleichen Deckglas verwendet werden, die jeweils einen anderen Zustand Sondieren. Dies ermöglicht den Vergleich der Bedingungen during der gleichen Versuchslauf. Ein ähnlicher Ansatz wurde von der van Oijen Gruppe verwendet Fusion zwischen Influenza - Virus zu studieren und gepolsterten SBLS 33.
In TIRF Mikroskopie, das exponentielle Abklingen des evaneszenten Feldes (mit einer Zerfallskonstante ~ 100 nm) Fluoreszenzanregung beschränkt auf diejenigen Moleküle, die in unmittelbarer Nähe der Glaspufferschnittstelle sind. Dies minimiert Beitrag von fluoreszierenden Molekülen, die weiter entfernt sind, erhöht das Signal-zu-Rauschverhältnis und erlaubt Einzelmolekülempfindlichkeit mit Rahmen Belichtungszeiten von 10-40 msec. Das abklingende Feld führt auch zu einem Signalanstieg beim Schmelzen: als markierter Lipide Transfer vom SUV in die SBL, finden sie sich im Durchschnitt in einer stärkeren Erregungsfeld. Dieser Anstieg in der Fluoreszenz ist für größere Liposomen stärker.
Wenn polarisiertes Licht verwendet wird, das abklingende Feld zu erzeugen, tragen zusätzliche Effekte zu Veränderungen in der Fluoreszenz auf transfer von Etiketten aus dem SUV in die SBL. Einige Lipid-Farbstoffe haben einen Übergangsdipolmomente orientiert mit einem bevorzugten mittleren Winkel in Bezug auf die Doppelschicht in die sie eingebettet sind. Dies erzeugt eine Differenz in der Menge an Fluoreszenz , die durch die Fluorophore emittiert wird, wenn sie in der gegenüber dem SUV SBL sind, da die polarisierten Strahl Farbstoffe in den beiden Membranen unterschiedlich erregt wird. Für erstere wird der Anregungsstrahl mit Übergangsdipole interagieren um den sphärischen SUV orientiert, während für letztere Dipolorientierungen wird durch die flache SBL Geometrie beschränkt werden. Wenn beispielsweise s-polarisierte einfallende Licht (polarisiertes normal zur Einfallsebene) verwendet wird, ist Erregungs effizienter , wenn der Farbstoff in der SBL ist als im SUV für eine Lipidfarbstoff Übergangsdipolmomente parallel zu der Membran 29,40 (wie die von Dil oder DiD 41-43). Ein SUV mit einem solchen Fluorophor dotierten erscheint dunkler , wenn es Docks auf die SBL (Bild 7, Representat ive Ergebnisse). Als Fusions Pore öffnet und verbindet die SUV und SBL Membranen diffundieren Fluoreszenzsonden in das SBL und werden eher durch den s-polarisierten abklingende Feld 25,27,29 angeregt werden. Folglich erhöht sich das Fluoreszenzsignal um die Fusionsstelle integriert scharf während der Farbstoffübertragung von dem SUV in die SBL 27 (Abbildung 3 und Abbildung 7). Ein zusätzlicher Faktor, der auf Signaländerungen trägt, die Fusions begleiten wird Dequenching von Fluoreszenzmarkierungen, wie sie verdünnt werden, wenn sie in die SBL übertragen. Der Beitrag der Dequenching ist üblicherweise gering im Vergleich zu abklingende Feld Zerfalls und Polarisationseffekte in dem Assay hier beschrieben, da nur ein kleiner Bruchteil (  ) Der Lipide sind markiert.
) Der Lipide sind markiert.
Der Signalanstieg bei Fusion kann ausgenutzt werden, um Fusionsporeneigenschaften ableiten, indem sie die Zeit zu vergleichen,1 "src =" / files / ftp_upload / 54349 / 54349eq2.jpg "/>, die für ein Lipid durch eine Pore zu entkommen, die frei durchlässig Lipide auf die tatsächliche Release-Zeit ist,  . Wenn die beiden Zeitskalen vergleichbar sind, würde geschlossen werden, daß die Poren wenig Widerstand gegen Lipidfluss darstellt. Wenn jedoch die tatsächliche Auslösezeit wesentlich länger als die Zeit für die Diffusion begrenzte Freisetzung ist, wäre dies ein Verfahren zeigen, wie Poren Flackern, Lipid-Freisetzung zu verzögern. Die diffusionsbegrenzten Freigabezeit,
. Wenn die beiden Zeitskalen vergleichbar sind, würde geschlossen werden, daß die Poren wenig Widerstand gegen Lipidfluss darstellt. Wenn jedoch die tatsächliche Auslösezeit wesentlich länger als die Zeit für die Diffusion begrenzte Freisetzung ist, wäre dies ein Verfahren zeigen, wie Poren Flackern, Lipid-Freisetzung zu verzögern. Die diffusionsbegrenzten Freigabezeit,  , Hängt von der Größe des Schmelz Liposom und Lipid-Diffusionsvermögen; seine Schätzung erfordert diese beiden Parameter zu quantifizieren. Die Einzelmolekülempfindlichkeit des Assays ermöglicht Lipid Diffusität durch die Verfolgung mehrerer Einzellipid Fluorophore nach ihrer Freisetzung in die SBL für jedes Fusionsereignis 26 gemessen werden. Die Größe jedes Verschmelzen Vesikel27 durch die Kombination von (i) geschätzt werden , um die Intensität eines einzelnen Lipid Farbstoff kann, (ii) die Änderung der Gesamtfluoreszenz um eine Andockstelle nachdem alle Fluorophore in die SBL beim Schmelzen übertragen werden, (iii) die bekannte Markierungsdichte von SUV Lipide, und (iv) die Fläche pro Lipid. Für viele Fusionsereignisse wurden die tatsächlichen Lipid Freisetzungszeiten gefunden viel langsamer als 27 durch diffusionskontrollierte Freisetzung erwartet, wie zuvor einheitliche Größe SUV 44 unter der Annahme festgestellt wurde. Unter der Annahme , Verzögerung der Lipid - Freisetzung durch flackert Poren, ein quantitatives Modell ermöglicht Schätzung von "Poren Offenheit", der Anteil der Zeit bleibt die Pore offen bei der Fusion 27.
, Hängt von der Größe des Schmelz Liposom und Lipid-Diffusionsvermögen; seine Schätzung erfordert diese beiden Parameter zu quantifizieren. Die Einzelmolekülempfindlichkeit des Assays ermöglicht Lipid Diffusität durch die Verfolgung mehrerer Einzellipid Fluorophore nach ihrer Freisetzung in die SBL für jedes Fusionsereignis 26 gemessen werden. Die Größe jedes Verschmelzen Vesikel27 durch die Kombination von (i) geschätzt werden , um die Intensität eines einzelnen Lipid Farbstoff kann, (ii) die Änderung der Gesamtfluoreszenz um eine Andockstelle nachdem alle Fluorophore in die SBL beim Schmelzen übertragen werden, (iii) die bekannte Markierungsdichte von SUV Lipide, und (iv) die Fläche pro Lipid. Für viele Fusionsereignisse wurden die tatsächlichen Lipid Freisetzungszeiten gefunden viel langsamer als 27 durch diffusionskontrollierte Freisetzung erwartet, wie zuvor einheitliche Größe SUV 44 unter der Annahme festgestellt wurde. Unter der Annahme , Verzögerung der Lipid - Freisetzung durch flackert Poren, ein quantitatives Modell ermöglicht Schätzung von "Poren Offenheit", der Anteil der Zeit bleibt die Pore offen bei der Fusion 27.
Wann immer praktisch, ist es wichtig, Fusionsmechanismen zu testen, unter Verwendung von sowohl Lipid und löslichen Inhalt Etiketten. Zum Beispiel könnte Lipid-Freisetzung durch andere Prozesse als die Poren Flackern, wie die Beschränkung der Lipid-Diffusion durch die SNARE-Proteine, die Surround-t verzögert werdener Pore. Wenn dies der Fall wäre, dann loslassen Inhalts würde Freisetzung von Lipid Etiketten voran, vorausgesetzt die Poren groß genug ist, Durchgang von löslichen Sonden zu ermöglichen. Ein grundlegender Fehler im Ansatz könnte in der Annahme sein, dass die Übertragung von markierten Lipide an die SBL einer engen Fusionspore erfolgt durch die SBL zu einem Vesikel anschließen, der seine Pre-fusion Form weitgehend erhalten hat. Lipid - Transfer in die SBL könnte auch mit einer begleitenden von einer schnellen Erweiterung der Fusionspore führen, extrem schnellen Zusammenbruch des SUV in die SBL - Membran, wie zuvor auf den Lipidfreisetzungsdaten allein 29 basierend vorschlug. Überwachung sowohl Lipid und Inhalt freigeben gleichzeitig wurde festgestellt , dass viele Poren wieder verschlossen , nachdem alle Etiketten ihre Lipid zu veröffentlichen, aber 27 einige ihrer löslichen Ladung erhalten. Dies zeigt, dass zumindest einige Liposomen in die SBL nicht nach dem Verschmelzen kollabieren, und daß die Lipidfarbstoffübertragung in die SBL Fusions Pore erfolgt durch. Zudem lIPID und Inhalt Release aufgetreten 27 gleichzeitig, ist es unwahrscheinlich , dass Verzögerung der Lipid - Release machen zurückzuführen war durch die SNARE - Proteine von Lipiddiffusion Behinderung 45 der Pore umgibt.
Ein SUV-SBL Fusionsprotokoll , die nicht löslichen Anteile Release überwachen tat , war zuvor von Karatekin und Rothman 25 veröffentlicht. Hier neuere Entwicklungen enthalten sind, nämlich die gleichzeitige Überwachung von Lipid und Inhalte freigeben und die Schätzung von SUV, Lipid und Fusionspore - Eigenschaften 27. Das Protokoll beginnt mit Anweisungen für die Mikrofluidik - Zellen vorbereitet, hergestellt durch ein Poly (dimethylsiloxan) Bindung (PDMS) Elastomerblock enthält Rillen mit einem Deckglas 25. Als nächstes Herstellung von v-SUVs mit sowohl Lipid und Inhalt Marker erläutert. Abschnitte 4 und 5 enthalten Anweisungen für die Mikrofluidik - Zellen zusammenbauen, die SBLS in situ Bildung und Überprüfung auf Defekte und Flüssigkeit, die Einführung von vSUVS in den Flusszellen und die Detektion von Fusionsereignissen. Abschnitt 6 enthält Anweisungen für die Datenanalyse.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. Herstellung eines PDMS-Block der mikrofluidischen Kanal zu bilden

Abbildung 1. Mikrofertigung von Flusszelle Vorlage und PDMS Satzvorbereitung. (A) Entwurf eines Vier-Kanal - Durchflusszelle , die auf einem 24 x 60 mm Deckglas passt (unten). Sechs identische Entwürfe sind so angeordnet, auf ein 10 cm Silizium-Wafer passen (oben). (B) ausschneiden Block von etwa 5-8 mm dicken PDMS auf ein Loch Puncher. (C) Insertion von Schlauch in das gestanzte Loch eine Pinzette. Bitte hier klicken , um eine größere Version dieser Figur zu sehen.
- Besorgen Sie sich eine Durchflusszelle Schablone wie die in 1A. Typische Kanäle sind 0,3-1 mm breit, 70 bis 100 & mgr; m hoch und 1-2 cm lang. Fabrizieren eine Vorlage usStandard Photolithographietechniken ing in einem Reinraum - Anlage 25 oder bestellen , wenn Reinraum- Zugang ist nicht verfügbar. Alternativ Maschine eine Vorlage mit größeren Abmessungen aus einem geeigneten Material.
Hinweis: Reinraum- Mitarbeiter können trainieren und führen unerfahrene Benutzer (Zugang zu einem Reinraum - Anlage ist immer auf Benutzer beschränkt mit der richtigen Ausbildung) bei der Gestaltung und Anordnung einer Maske, Waferreinigung und Photolithographie. Sobald eine Vorlage erhalten wird, kann es immer wieder außerhalb des Reinraums verwendet werden, sofern es in einer geschlossenen Schale und Pflege Staub auszuschließen gehalten wird genommen. - Bereiten Sie eine Mischung von ~ 100 ml PDMS (Silikon-Elastomer-Basis) und ~ 10 ml Vernetzer (Härter) in einem Einweg-Plastikbecher. Brechen Sie die Spitze eines Einweg-Pipetten die zähflüssige PDMS leichter zu handhaben. Wiegen Sie die PDMS in den Plastikbecher als Pipettieren nicht korrekt ist.
- Rühren Sie die Mischung gut. Entfernen Sie die vielen Luftblasen durch Entgasen in einem Vakuumexsikkator (ca. 20min). Da die Tasse unter Vakuum gesetzt wird, wachsen die Blasen werden zunächst in der Größe stark das Volumen der Mischung zu erhöhen. Steuern Sie das Vakuum, das angewendet wird, und stellen Sie sicher, dass die Schale tief genug ist, ein Spill zu vermeiden.
- Gießen Sie einen großen Tropfen des entgasten PDMS-Mischung in eine Glaspetrischale (150 mm x 20 mm) und drücken Sie die Scheibe auf den PDMS mit der Vorlage nach oben zeigt. Dies vermeidet Blasen unter dem Wafer eingeschlossen werden. Gefangen Blasen erweitern und kann den Wafer kippen, wenn die Schale in den Ofen gelegt wird. Jetzt gießen entgastem PDMS auf die Schablone auf der Oberseite des Wafers, bis sie von etwa 5-8 mm von PMDS bedeckt ist. Wenn eine Luftblase mit einer Pipettenspitze sanft entfernen auftritt.
- Bake die PMDS in einem Ofen bei 60 ° C für 3 Stunden. Stellen Sie sicher, dass das Gericht Ebene ist.
- Verwenden Sie einen neuen Skalpellklinge ein PDMS-Block auszuschneiden die geformten Kanalstrukturen enthalten. Das ausgeschnittene Block muss auf einem Deckglas zu passen (siehe Schritt 4.1.9).
- Ziehen Sie aus dem Block herausgeschnitten aus der Schale und place es auf ein Stück sauberer Aluminiumfolie.
Anmerkung: Die vernetzten PMDS Blöcke können für ein paar Monate aufbewahrt werden. Auf einem einzelnen Wafer gibt es 6 Sätze von Strömungskanälen (1A), so 6 PDMS - Blöcke können in einem einzigen PDMS Verfahren hergestellt werden. Nach dem Schneiden alle 6 der PDMS-Blöcke heraus, sauber lose Stücke von PDMS, aber ansonsten überspringen Sie die restlichen PDMS vor dem Gießen und Vernetzen der nächsten Charge nicht entfernen, die für die erste Charge verwendet Hälfte nur über die PDMS benötigen. Eine gute Vorlage kann ein paar Jahre dauern. - Verwenden Sie ein Loch Puncher in einer geraden Bewegung (Abbildung 1B) durch die PDMS - Block zu bohren. Starten Sie auf der Seite der Kanalrinnen. Achten Sie darauf, das gestanzt Stück PDMS zu entfernen. Wiederholen Sie diesen Vorgang für alle acht Löcher des Vier-Kanal-Design.
- Platzieren Sie den PDMS-Block Kanalseite nach unten auf eine neue und knitterfrei Stück Aluminiumfolie. Lagern Sie den Block für bis zu ein paar Monate in einem trockenen Feld, idealerweise in einem Exsikkator.
- Schieben Sie den Schlauch (0,25 mm ID, 0,76 mm OD) etwa ein Drittel in das gestanzte Loch mit einer Pinzette (Abbildung 1C) verwendet wird . Schneiden Sie den Schlauch zum leichteren Einführen schräg. Lassen Sie Rohre lang genug, um die SUV-Reservoir und die Spritzenpumpe zu erreichen sind. Nach dem montierten Chip auf das Mikroskop legen, schneiden Sie die Rohre wieder, wenn sie zu lang sind.
- zu den Spritzen der Pumpe, schneiden Sie ein kurzes Stück größer Silikonschlauch (0,51 mm ID, 2,1 mm Außendurchmesser) und legen Sie die dünneren Rohr in eine Seite zu verbinden. Diese ready-to-use-Block von PDMS kann für ein paar Monate lang aufbewahrt werden.
2. Coverslip Reinigung
- Saubere Deckgläser nach dem Protokoll in Karatekin und Rothman 25 beschrieben , eine starke oxidierende Gemisch aus Schwefelsäure (H 2 SO 4) und unter Verwendung von Wasserstoffperoxid (H 2 O 2). Führen Sie dieses Verfahren in einem Reinraum, und beachten Sie den entsprechenden Sicherheitsvorkehrungen. Alternativ können Sie einen Reinraum gereinigt coverslip, dass im Handel erhältlich ist (siehe Materialliste).
3. Herstellung von v-SUVS, die sowohl Lipid und Inhalt Labels

Figur 2 Schematische Darstellung der SUV Zubereitung. Lipids in einem Glasrohr (1) gemischt und das Lösungsmittel verdampft wird durch Drehen des Rohrs in einem Wasserbad (2) einen Lipidfilm zu bilden. Verbleibende Spuren von Lösungsmittel unter Hochvakuum (3) entfernt. Der Lipidfilm wird in Rekonstitution Puffer, enthaltend Detergens und Protein während gevortext (4) hydratisiert. Wenn Inhalte Farbstoff werden verkapselt ist, wird in diesem Schritt als auch in dem Verdünnungsschritt (5) enthalten. Verdünnung der Detergenskonzentration unter seiner kritischen Mizellenkonzentration führt Bildung Liposom. Waschmittel wird über Nacht dialysiert (6) weg. Für t-SUVs für SBL Bildung einschließlich NBD-PE (grün), Vesikel werden in einem Dichtegradienten schwebten und gesammelt andie Grenzfläche zwischen zwei Schichten (7a). Zur Trennung v-SUVs mit gekapselter Inhalt Marker aus freiem Farbstoff die Probe über einen Größenausschluss - Säule laufen gelassen wird und in 0,5 - ml - Fraktionen (7b) gesammelt. Bitte klicken Sie hier , um eine größere Version dieser Figur sehen.
- Herstellung von 4 l Rekonstitution Puffer, enthaltend 25 mM HEPES-KOH, 140 mM KCl, 100 uM EGTA und 1 mM DTT, pH 7,4. Verwenden die meisten der Puffer für die Dialyse und 100 ml für andere Schritte.
Hinweis: Andere Puffer können verwendet werden, aber es ist wichtig , den Puffer Osmolarität konsistent während des Experiments zu halten und zu wählen , Bedingungen , bei denen die Proteine funktionsfähig sind. Zur Herstellung von v-SUVs Lipid - Etiketten , die nur und T-SUVs, folgen Karatekin und Rothman 25. - Als Lipid Bestände bei -20 ° C in Chloroform oder Chloroform gespeichert sind: Methanol (2: 1, v / v), lassen Fläschchen Raumtemperatur vor dem Öffnen erreichen zuVermeidung von Kondensation.
- Spülen mit einem Glasrohr mit Chloroform Spuren von Detergenz zu entfernen, und die Lipide mischen im gewünschten Verhältnis mit einer Endmenge von 1 & mgr; mol in einem Gemisch aus Chloroform und Methanol (2: 1, v / v). verwenden Glasspritzen / Röhrchen Nur organische Lösungsmittel / gelösten Lipide zu behandeln.
Hinweis: Die verwendeten Lipide sind hier POPC: SAPE: DOPS: Cholesterin: PEG2000-DOPE: DiD bei einem Molverhältnis von 57,4: 15: 12: 10: 4,6: 1 für v-SNARE enthaltende Vesikel und POPC: SAPE: DOPS: Cholesterin : Gehirn PI (4,5) P 2: PEG2000-DOPE: NBD-PE (54,9: 15: 12: 10: 3: 4,6: 0,5) für t-SNARE SUVs. Siehe Werkstoff für weitere Einzelheiten. Andere Lipidzusammensetzungen verwendet werden. - Dampfe das Lösungsmittel unter einem leichten Stickstoffstrom oder in einem Rotationsverdampfer. Tauchen Sie die Spitze des Röhrchens in einem Wasserbad (37 ° C) oberhalb der höchsten Schmelztemperatur der Lipide erhitzt, um eine homogene Lipidfilm zu erhalten und große Temperaturänderungen zu vermeiden, während das Lösungsmittel verdampft. Starten Sie Vakuum bei rund300 mbar, bis der Lipidfilm gebildet wird, dann weiter für ~ 2 min bei möglichst hohen Vakuum im Rotationsverdampfer.
- Entfernen Sie das Glasrohr und wickeln Sie es in Alufolie Lichtexposition vermeiden und entfernen verbleibenden Spuren des Lösungsmittels durch das Rohr in einem Exsikkator unter Hochvakuum Platzierung für mindestens 2 Stunden. Dieser Schritt kann auch über Nacht laufen.
- Zu dem getrockneten Lipidfilm rehydrieren, Herstellung einer Mischung aus Detergens (n-Octyl-β-D-glucopyranosid, OG), Protein (v-SNARE) und SRB in Rekonstitution Puffer (500 ul Endvolumen). Halten die endgültige Detergenskonzentration ~ 2-fach über der kritischen Mizellenkonzentration (CMC, 20-25 mM OG) und stellen das Endvolumen, so dass das Detergens zu Lipid-Verhältnis> 10. Auflösen Sulforhodamin B (SRB) -Pulver in dem Protein-Detergens-Lösung in einer Endkonzentration von 10-50 mM SRB zu erhalten durch vorsichtiges Schütteln der Lösung. Stellen Sie die Osmolarität der Rekonstitution Puffer bei hohen Konzentrationen von SRB Zugabe durch die Co zu reduzierenncentration Kaliumchlorid entsprechend.
Hinweis: Lipid-zu-Protein - Verhältnis (LP) für t-SNAREs hoch ist (LP ~ 20.000 ~ 70 t-SNAREs pro Quadratmikrometer . 25). Deutlich höhere t-SNARE Dichten in der SBL kann inaktive Proteinaggregate führen mit deutlich 17,24 die Fusionsraten reduziert. LP für v-SNARE ist viel niedriger (LP ~ 200 ~ 7000 v-SNAREs pro & mgr; m 2), in der Nähe der v-SNARE Dichten auf synaptischen Vesikeln 46,47 gefunden. Proteinfunktion nach rekombinanter Expression und Reinigung von v- und T-SNAREs Um zu gewährleisten , ist es sinnvoll , eine einfache Massenfusionstest 48 , bevor er durch alle Schritte des einzelnen Vesikelfusion Assay durchzuführen. - Schütteln Sie die den getrockneten Lipidfilm vorgewärmten Glasröhre enthält, während das Protein-Reinigungsmittel-SRB-Lösung aus Schritt 3.6 hinzugefügt wird. Versuchen Sie zu vermeiden Blasen zu schaffen. Weiterhin bei 37 ° C für 15 min zu schütteln.
- Verdünnen Sie das Reinigungsmittel das Fünffache durch Rekonstitution ZugabePuffer, der SRB (2 ml, 2,5 ml Endvolumen), während schnell Konzentrationsgradienten zu vermeiden verwirbelt. Weiterhin bei 37 ° C für 15 min bis 1 h schütteln.
Hinweis: Längere Inkubation Erhöhung Protein Rekonstitution Effizienz. Die schnelle Verdünnung verringert die Waschmittelkonzentration unterhalb der CMC und führt zur Bildung von kleinen Liposomen. - Dialysiere die Vesikelsuspension zuerst gegen ~ 1 l Rekonstitution Puffer für 1-2 Stunden bei Raumtemperatur und dann gegen 3 l Rekonstitution Puffer über Nacht bei 4 ° C mit 4 g Polystyrol-Adsorbens, eine 20.000 MWCO Dialyseschlauch oder Kassette verwendet wird. Verwenden verschiedene Bechergläser für die Dialyse von Vesikeln mit und ohne SRB Kreuzkontamination zu verhindern.
- Äquilibrieren eine Gelfiltrationssäule mit Rekonstitutionspuffer. Führen Sie die Vesikelsuspension durch die Säule frei SRB aus den V-SUVs mit gekapselten SRB zu trennen. Verwenden Sie die Rekonstitution Puffer ohne SRB als Eluent einmal die gesamte Probe hatdie Spalte eingetragen. Sammeln v-SUVs in 0,5-ml-Fraktionen.
- Überprüfen der erfolgreichen SRB Verkapselung bei selbst gequencht Konzentrationen durch Messung SRB Fluoreszenz vor und nach der Zugabe von Detergens zu einem Aliquot von Vesikeln 16. Mit Hilfe eines Fluoreszenz-Spektrometer, regen sie die Probe bei 550 nm und scannen die SRB-Emission zwischen 570 nm und 630 nm. Erwarten Sie eine 4-8 fache Erhöhung der SRB Fluoreszenz auf dem eingekapselten Menge auf Detergenzzugabe abhängig als die Membran solubilisierten ist und das freigesetzte SRB verdünnt.
- Charakterisieren Sie die Lipid- und Proteingewinnung unter Verwendung von Fluoreszenzspektroskopie 24 und SDS-PAGE - Gelelektrophorese sind. Verwenden Sie eine empfindliche Färbeverfahren, welches speziell für die LP t-SUV Proben (siehe Materialliste). Typischerweise etwa 50% sowohl der Lipid- und Protein-Eingang wird während der Herstellung verloren in einem LP nahe dem Nominalwert führt.
- Charakterisieren SUV Größen unter Verwendung der dynamischen Lichtstreuung 24 oder Elektronen microscopy 48. Shop SRB enthält v-SUVs bei 4 ° C bis ~ 3-4 Tage. Nicht einfrieren, wie Einfrieren und Auftauen der Membran und gibt verkapselt SRB bricht.
4. SUV-SBL Fusion Assay zur Überwachung Nur Lipid Mitteilung
- Die Bildung der tethered supported bilayer innerhalb der mikrofluidischen Strömungskanäle
- Platzieren Sie die PDMS-Block (Schritt 1.11) unter hohem Vakuum für mindestens 20 min vor dem Experiment, um gelöste Gase zu entfernen. Dies reduziert das Risiko von Luftblasen in den Mikrofluidik-Kanälen während des Fusionsexperiment.
- Schalten Sie den Mikroskopaufbau und erhitze die Bühne und Probenhalter (3 und 4) auf die gewünschte Temperatur.
- Filtern Sie die Rekonstitution verwendete Puffer zu verdünnen, die Vesikel durch einen Filter mit 0,45 um oder kleinere Porengröße.
- Verdünnen ~ 30 ul der NBD-PE-markierten T-SUVs oder proteinfrei (pf-SUVs) Kontrolle Liposomen (LagerLösung 0,5-1 mM Lipid) mit ~ 60 & mgr; l Puffer. Die endgültige Konzentration ist hier nicht entscheidend.
- Entgasen diese Mischung eine 3 ml Spritze. Drücken Sie den größten Teil der Luft über der Probe, während die Spritze senkrecht halten. Verschließen Sie die (Nadelfrei) Spitze Paraffinfilm mit und ein Vakuum erzeugen, indem der Kolben nach unten. Tippen den Spritzenzylinder Entgasung der Lösung zu beschleunigen. Wiederholen Sie diesen Vorgang einige Male, bis keine Blasen mehr auftreten, wenn Vakuum angelegt wird.
- Verwenden einer Injektionsnadel mit einem Durchmesser, der etwas größer ist als der Schlauch, die an dem PDMS-Block angebracht ist, ein Loch in der Kappe eines Mikrozentrifugenröhrchens zu stanzen. Stellen Sie sicher, dass das ausgestanzte Stück Plastik nicht innerhalb des Rohres ist, da sie den Schlauch später an die mikrofluidische Kanaleinlaß verbunden könnten verstopfen.
- Füllen Sie das entgast SUV-Lösung in das Reaktionsgefäß ein Loch in der Kappe mit und legen Sie sie in die Halterung auf dem Mikroskoptisch auf die eingestellte Temperatur zu äquilibrieren.
- Place eine zuvor gereinigte Deckglas (Abschnitt 2) in einem Plasma-Reiniger und laufen Luft-Plasma für ca. 5 min. Legen Sie das Plasma behandelt Deck (behandelte Seite nach oben) auf der Oberseite von wenigen fusselfreien Gewebe als Polster dienen.
- Entfernen Sie die Aluminiumfolie aus dem entgasten PDMS-Block und legen Sie den Block auf der Oberseite des Deckglases. Bei Wiederverwendung der PDMS-Block ein Stück Klebeband auf der Kanalseite aufsetzen und abnehmen zu reinigen. Drücken Sie die PDMS-Block auf das Deckglas mit einer Pinzette verwenden, um es halten, aber nicht drücken zu hart, da das Glas brechen könnte.
- Den montierten Flusszelle auf dem Mikroskoptisch und schließen Sie den Schlauch mit dem SUV Reservoir und der Spritzenpumpe verbunden. Band das Deckglas in die Stufe (3B).
- Beginnen Aspirieren SUVs bei 3 & mgr; l / min, bis die Lösung die Kanäle vollständig ausfüllt (~ 2,25 mm / sec für den 75 um x 300 um Kanalquerschnitt). Wenn Lösungen für alle Kanäle in Bewegung setzen auf the Schlauch auf der Austrittsseite, reduzieren Strömungs bis 0,5 & mgr; l / min und Inkubation für 30-45 min.
Hinweis: Die Zeit zwischen dem Plasma die Glasobjektträger zu behandeln und den SUVs in den Kanal fließt , nicht 10-20 min als die Wirkung der Plasmabehandlung übersteigen sollte , ist vorübergehend. - Überprüfen Sie die Kanäle für jede undichte Stelle. Verwenden Sie ein 10-20x Luft Ziel NBD-PE-Fluoreszenz von der NBD-PE enthalten, t- oder pf-SUVs zu beobachten. Anregungs-NBD Fluorophore die 488 nm-Laser verwendet wird. Leaks sind schwierig mit Hellfeldbeleuchtung zu erkennen.
- Setzen Sie ein Rohr mit entgastem Rekonstitution Puffer in die Halterung ein und lassen Sie die Temperatur so schnell Veränderungen in der Temperatur ins Gleichgewicht könnten Defekte in der SBL verursachen. Stoppen Sie den Fluss und warten ~ 1 min um sicherzustellen, dass der Fluss gestoppt vollständig und keine Luftblase wird in das Rohr abgesaugt werden, bevor die Einlassrohre an den entgastem Puffer umgeschaltet wird.
- Spülen Sie alle Kanäle mit entgastem Puffer ungebundenen SUVs abzuwaschen.
- Wechseln Sie zu einem higher Vergrößerung TIRF Ziel (60X, Öl, NA 1,45-1,49) und stellen Sie sicher, dass die Doppelschicht homogen aussieht und ist frei von irgendwelchen offensichtlichen großen Defekte wie dunkle Flecken oder Lipid-Tubuli aus der SBL heraus erstreckt.
- Überprüfen Sie die Fluidität der Doppelschicht
- Wenn eine eigene FRAP Gerät nicht verfügbar ist, oder wenn eine FRAP-Sequenz nicht programmiert werden kann, dann Fluidität qualitativ Testmembran wie folgt.
- Schließen der Feldblende auf eine kleine Größe (~ 40 & mgr; m Durchmesser) und Einstellen der 488 nm Anregungslichtintensität , die durch die Software (20-80 & mgr; W oder 15-60 nW / & mgr; m 2) der NBD-PE - Fluoreszenz in den freiliegenden zu bleichen Bereich deutlich, jedoch nicht vollständig. Für eine Fluid bilayer, im stationären Zustand, die Fluoreszenzintensität in der Mitte des freiliegenden Bereich sollte niedriger sein als an den Rändern, als intakte NBD-PE Moleküle die belichtete Fläche und diffundieren in einem bestimmten Abstand vor dem Bleichen ein. Im Gegensatz dazu, wenn die Oberfläche anhaftenden SUVs gescheitert zu platzen, oder aus einem anderen Grund die supported bilayer nicht flüssig ist, werden alle Fluorophore im belichteten Bereich bleichen sollte.
Hinweis: Laser - Intensitätswerte in dieser und den nachfolgenden Schritten werden als grobe Ausgangspunkt gegeben und sollte für einen gegebenen Satz von Bedingungen optimiert werden. - Stoppen Sie die Beleuchtung und starten Sie es wieder ein paar Minuten später die Ergebnisse der Steady-State-Messungen zu überprüfen
- Schließen der Feldblende auf eine kleine Größe (~ 40 & mgr; m Durchmesser) und Einstellen der 488 nm Anregungslichtintensität , die durch die Software (20-80 & mgr; W oder 15-60 nW / & mgr; m 2) der NBD-PE - Fluoreszenz in den freiliegenden zu bleichen Bereich deutlich, jedoch nicht vollständig. Für eine Fluid bilayer, im stationären Zustand, die Fluoreszenzintensität in der Mitte des freiliegenden Bereich sollte niedriger sein als an den Rändern, als intakte NBD-PE Moleküle die belichtete Fläche und diffundieren in einem bestimmten Abstand vor dem Bleichen ein. Im Gegensatz dazu, wenn die Oberfläche anhaftenden SUVs gescheitert zu platzen, oder aus einem anderen Grund die supported bilayer nicht flüssig ist, werden alle Fluorophore im belichteten Bereich bleichen sollte.
- Programmieren einer FRAP-Sequenz für eine quantitative Messung, falls möglich. Siehe Zusatzdateien und die entsprechende Legende für weitere Einzelheiten.
Hinweis: Manchmal SUVs haften auf das Deckglas, stellen aber keine Flüssigkeit Bilayer zu platzen und bilden. Wenn dies der Fall ist , spülen Sie die Kanäle mit entgastem Rekonstitution Puffer, der 10 mM Mg 2+ supported bilayer Bildung zu helfen. Verwenden Fluoreszenzbleich Bilayer Fluidität wie in 4.2 zu beurteilen. Sobald eine Flüssigkeit Bilayer gebildet wird, spülen Sie mit Mg 2+ -freien Reconstitution Puffer.
- Wenn eine eigene FRAP Gerät nicht verfügbar ist, oder wenn eine FRAP-Sequenz nicht programmiert werden kann, dann Fluidität qualitativ Testmembran wie folgt.
- Einführung in v-SUVs in den mikrofluidischen Strömungskanäle
- Degas Rekonstitutionspuffer und es verwenden , um einen Faktor von ungefähr 3. Oktober - 5. Oktober in Abhängigkeit von der v-SUV Lager Konzentration der V-SUV - Stammlösung zu verdünnen. Ziel für eine Verdünnung, die innerhalb von 60 Sekunden in das Sichtfeld in etwa 10-100 Fusionsereignissen zur Folge hat.
Hinweis: Zu viele Fusionen erhöhen , um die Hintergrundfluoreszenz (da jedes Ereignis Einlagen LR-PE oder DiD Lipid - Etiketten in den SBL) und machen Erkennung und Analyse von Fusionsereignissen schwierig. Im Gegensatz dazu ist eine Fusionsrate, die zu niedrig führt zu einer schlechten Statistik ist oder erfordert Erwerb von vielen weiteren Filmen. Für einen v-SUV-Konzentration von 0,1 mM Lipid, starten durch Verdünnen von 5 & mgr; l SUV Lager in 995 ul Rekonstitution Puffer und dann 5-50 ul dieser in 950-995 ul Rekonstitution Puffer verdünnen. - Lassen Sie Temperatur äquilibrieren, bevor die Strömung zu stoppen und inserting das Einlassrohr in die verdünnte v-SUV-Lösung.
- Stellen Sie den TIRF Winkel und Polarisation wie folgt.
- Nachdem die gewünschte Polarisations Einstellen des Anregungsstrahls durch Drehen, passen die Neigung des Spiegels um die Position des Strahls durch einen Schrittmotor über die Software befehlen. Langsam bewegen die Position des Laserstrahls, der von der Mitte des Ziels in der hinteren Brennebene auf der einen Seite nicht in der Mitte. Beachten Sie, das Licht von dem Ziel an der Vorderseite entstehen mit zunehmendem Winkel in Bezug auf die Zielachse als die Position weiter außerhalb der Mitte bewegt wird.
- Beachten Sie die Motorposition , wenn der austretende Strahl zuerst das Ziel verschwindet, das heißt, wenn TIR zuerst erreicht.
- Langsam halten die Strahlposition weiter außermittig zu bewegen, während die Fluoreszenz von der Oberfläche zu überwachen. Beachten Sie die Motorposition, wenn die Oberfläche Fluoreszenz verschwindet, wenn der Strahl zu weit außerhalb der Mitte bewegt hat.
- Wählen Sie einen Strahl positio n zwischen den beiden Grenzen oben bestimmt. Für beste Signal-Rausch-Verhältnis und mehr Signalverstärkung bei der Fusion, wählen Sie eine flache Eindringtiefe (Strahlposition näher an den Rand des Objektivs), die durch die immer noch eine gleichmäßige Ausleuchtung des Sichtfeld bietet.
Hinweis: Am besten ist es die gleiche TIR Strahlposition (gleiche Eindringtiefe) für alle Experimente zu halten , nachdem die Einstellungen optimiert sind. Achten Sie auf die Polarisationsrotations führt nicht zu Veränderungen in der Strahlposition.
- Strom V-SUVs in den Kanal bei einer Flussrate von 2 & mgr; l / min, entsprechend einer durchschnittlichen linearen Fließgeschwindigkeit von ~ 1,5 mm / sec für einen Querschnitt von 75 um x 300 um. Wechseln Sie auf die Anregungs- / Emissions-Einstellungen zu überwachen Lipid Mischen (LR oder DiD nur).
- Degas Rekonstitutionspuffer und es verwenden , um einen Faktor von ungefähr 3. Oktober - 5. Oktober in Abhängigkeit von der v-SUV Lager Konzentration der V-SUV - Stammlösung zu verdünnen. Ziel für eine Verdünnung, die innerhalb von 60 Sekunden in das Sichtfeld in etwa 10-100 Fusionsereignissen zur Folge hat.
- Beobachten Fusion zwischen einzelnen V-SUVs und der SBL
.jpg "/>
Abbildung 3. Die experimentelle pTIRF Setup. (A) Schematische Darstellung eines V-SUV und ein T-SBL auf einem Glassubstrat. Falschfarben TIRFM Bilder einzelner SUV-SBL-Fusionsereignis zeigt Lipid docking (1) und die Freisetzung von Lipid Farbstoff in die SBL (2), gefolgt von Bleichen und Abnahme der Fluoreszenzintensität (3). Die Gesamtfluoreszenzintensität (Summe der Pixelwerte in dem 5,3 um x 5,3 um Box) -Signals gezeigt. (B) Das Deckglas mit dem PDMS - Block verbunden ist , auf die erhitzte Bühne geklebt. Das Einlaßrohr für mikrofluidische Kanäle ziehen Proben aus Rohr in Metallprobenhalter (rechts) von der Spritzenpumpe abgesaugt (links). Unter der Pumpe die Doppelemissionseinheit ist. Bitte hier klicken , um eine größere Version dieser Figur zu sehen.
OAD / 54349 / 54349fig4.jpg "/>
Abbildung 4. Schematische Darstellung des experimentellen Aufbaus. Die abklingende Welle wird an der Glas Puffer - Schnittstelle in der mikrofluidischen Kanal erstellt. Die SBL wird auf dem Glas gebildet und v-SUV aus dem Metallprobenhalter (oben rechts) durch den Kanal in die Spritze (oben links) abgesaugt. M, Spiegel; DM, dichroitische Spiegel; L, Linse; F, Filter; P, Polarisator. Bitte klicken Sie hier , um eine größere Version dieser Figur zu sehen.
- Kontinuierlich erregen und v-SUV Fluoreszenz unter Verwendung eines 561 nm (für LR-PE) oder 638 nm (für DiD) Laser in Abhängigkeit von der in v-SUVs eingebaut Fluorophor überwachen. Als V-SUVs den Strömungskanal und Dock auf und verschmelzen mit der SBL erreichen, beginnt die Hintergrundfluoreszenzsignal zu akkumulieren.
- Stellen Sie die Anregungslaserintensität durch die Software kontinuierlich den Hintergrund zu bleichenFluoreszenz, so dass im stationären Zustand neue Andocken und Fusionsereignisse leicht beobachtet werden kann.
Hinweis: Wenn Bleichen zu langsam ist, Hintergrundfluoreszenz zu hoch sein wird. Wenn Bleich zu schnell ist, Signale von gedockt SUVs und von Fluorophoren in den SBL freigesetzt wird schnell verblassen, Verkürzen des Fensters, während der Fusion von gedockt SUVs detektiert werden kann oder ein Fluorophor verfolgt werden kann. Für LR-PE bei 561 nm angeregt, 2,5-7,5 mW Leistung eines 190 & mgr; m Durchmesser - Kreis Beleuchtung (100-250 nW / & mgr; m 2) ist ein vernünftiger Wert zu starten. Für 638 nm Anregung von DiD, 0,8-1,6 mW Leistung über einen 190 & mgr; m Durchmesser - Kreis (30-60 nW / & mgr; m 2) für erste Tests verwendet werden. - Erwerben Sie mehrere Filme an verschiedenen Positionen in einem bestimmten Kanal. Prüfen Sie NBD-PE-Fluoreszenz zu überprüfen, ob die SBL nicht für Mängel an diesen Positionen hat. Erwerben Sie Full-Frame-Filme mit der maximalen Geschwindigkeit (~ 50 Bilder / s) oder einem beschnittenen Bereich von Interesse an höheren Bildrate (bis zu~ 100 Hz) für 60 sec.
- Wechseln Sie zu einem anderen mikrofluidischen Kanal und wiederholen Sie die Aufnahmen für andere Bedingungen. Sind negative Kontrollen, wie beispielsweise einem proteinfreien SBL oder SUVs, die lösliche cytoplasmatische Domäne des v-SNARE VAMP2 (CDV) als Inhibitor hinzuzufügen oder v-SUV mit Tetanus-Neurotoxin (30 min bei 37 ° C) in einem oder mehreren behandeln der Kanäle auf dem gleichen Deckglas.
- Aufräumen und PDMS Recycling
- Um die PDMS-Block, spülen Sie die mikrofluidischen Kanäle mit ~ 200 & mgr; l 70% Ethanol bei einer Durchflussrate von 5 ul / min wiederzuverwenden. Schließlich Luft durch die Rohre und Kanäle anzusaugen.
- Nehmen Sie die PDMS vorsichtig aus dem Deck blockieren, indem sie leicht zusammendrücken. Legen Sie es auf ein sauberes Stück Aluminiumfolie. Lagern Sie in einem Vakuumexsikkator. Es kann mehrmals wiederverwendet werden.
- Für eine gründliche Reinigung spülen, die Kanäle mit einem Detergens oder Natronlauge vor der 70% Ethanol. Alternativ entfernen Sie alle Rohre her m die PDMS und beschallen den Block für 30 Minuten in Isopropanol, vor dem Trocknen und neuen Schlauch einführen.
5. SUV-SBL Fusion Assay Veröffentlichung Lipid und Inhalt zu überwachen gleichzeitig
- Bei Dual-Farbüberwachung von simultanen Lipid und löslichen Inhalt freigeben, folgen die gleichen Schritte wie in Abschnitt 4, außer Verwendung von Liposomen, markiert mit sowohl ein Lipid (DID) und einem löslichen Inhalt (SRB) Etikett, wie in Abschnitt 3 SRB erklärt gekapselt und 10 mM ist anfänglich hoch selbst gequencht.
- Mit der gezeigten Konfiguration schematisch in Abbildung 4, erregt SRB und DiD Fluoreszenz 561 nm und 638 nm Laser gleichzeitig jeweils unter Verwendung. Ein dichroitischer Spiegel (640 nm) spaltet die Emission in zwei Strahlen, die durch eine kurze (595/50 nm) laufen und eine lange (700/75 nm) Wellenlängenfilter die SRB und DiD-Emissionen bzw. zu erfassen. Die beiden Emissionsstrahlen werden projiziert, die Seite an Seite auf den EM-CCD-Chip.
- Analyse von FRAP Daten
- Verwenden Sie das MATLAB - Programm in der Zusatzinformationen bereitgestellt , um die Lipid - Diffusionskoeffizienten abzuschätzen, D. Das Programm liest eine Liste von OME-TIFF - Dateien 49, detektiert die gebleichte Fläche, zeichnet die mittleren Pixelwerte in dem gebleichten Bereich als eine Funktion der Zeit, und passt die resultierende Erholungskurve zu einem Modell von Soumpasis 50 die Wiederherstellungszeit zu extrahieren,
 , Wobei w der Radius des gebleichten Kreises.
, Wobei w der Radius des gebleichten Kreises.
Hinweis: Ein MATLAB - Programm wurde früher 25 für die Analyse von FRAP Filmen mit Nikon ND2 Dateien zur Verfügung gestellt. Das aktuelle Programm liest OME-TIFF - Dateien 49, weil die meisten Dateiformate leicht zu OME-TIFF umgewandelt werden können. Eine quantitative Analyse von FRAP Daten ist am einfachsten und genauesten, wenn ist momentane Bleichen, die bleacHED Bereich ein Kreis ist, und das Bleichen beim Auslesen ist vernachlässigbar. Obwohl diese Kriterien nicht streng in den einfachen FRAP Messungen hier beschrieben erfüllt sind, kann eine vernünftige Schätzung des Diffusionskoeffizienten erhalten werden. Für eine genauere Schätzung verwenden Tracking einzelner Lipid Farbstoffe (Abschnitt 6.3).
- Verwenden Sie das MATLAB - Programm in der Zusatzinformationen bereitgestellt , um die Lipid - Diffusionskoeffizienten abzuschätzen, D. Das Programm liest eine Liste von OME-TIFF - Dateien 49, detektiert die gebleichte Fläche, zeichnet die mittleren Pixelwerte in dem gebleichten Bereich als eine Funktion der Zeit, und passt die resultierende Erholungskurve zu einem Modell von Soumpasis 50 die Wiederherstellungszeit zu extrahieren,
- Die Analyse der Docking - Rate, Fusionsrate und Docking-to-Fusion Verzögerungszeiten
- Verwenden Sie ImageJ, um einen Film zu öffnen, zu analysieren und die Helligkeit und den Kontrast zu deutlich Vesikel Docking- und Verschmelzen Ereignisse zu identifizieren. Starten Sie das SpeckleTrackerJ 26 - Plugin. Siehe Smith et al. 26 und Online - Dokumentation zu SpeckleTrackerJ Anweisungen zur Verwendung.
- Identifizieren Sie alle neu dockt SUVs in SpeckleTrackerJ. Zur Unterscheidung SUVs, die fest von denen andocken, die von der SBL prallen, verhängen eine minimale Docking Dauer von ein paar Frames. Speichern Sie die Spuren, und wiederholen Sie für alle Filme.
Hinweis: Für die docKönig Rate, alles, was zählt, ist, zu identifizieren, wenn ein SUV dockt, so dass nur das erste Bild, in dem die SUVs Bedürfnisse koppelte in diesen Spuren aufgezeichnet werden. Der Rest der Tracks werden nicht berücksichtigt bei der Analyse entnommen werden. Allerdings Auto Tracking jedes SUV, bis es bleicht oder Sicherungen hilft Marke SUVs, die bereits verfolgt wurden. - Identifizieren Sie alle fusionierenden Vesikel. Für diese sollten die Spuren umfassen alle Rahmen von dem ersten Rahmen ein SUV, bis der erste Rahmen verankert, in der Fusion durch einen plötzlichen Anstieg in der Fluoreszenz des verfolgten Stelle ersichtlich ist. Die Dauer dieser Spuren werden verwendet, um die Docking-to-Fusion Verzögerungen zu berechnen. Speichern Sie alle Spuren. Wiederholen Sie für alle Filme.
- Für Batch - Analyse von Daten Docking oder Fusion, stellt eine Liste der Trajektorie Dateien aus den jeweiligen Filmen und die MATLAB - Programme mit Karatekin und Rothman 25 vorgesehen laufen. Die Anweisungen, die zusammen mit den Programmen bereitgestellt werden.
Hinweis: Die Programme zeichnen die cumkulative Andocken und Fusionsereignisse als Funktion der Zeit, basierend auf Informationen aus den Bahn Dateien extrahiert. Docking und Fusionsraten werden aus den Steigungen dieser Parzellen geschätzt. Für Fusionsdaten werden die Docking-to-Fusion Verzögerungen ebenfalls berechnet und deren Verteilung als Überleben Grundstück aufgetragen, das heißt, die Wahrscheinlichkeit , dass die Fusion noch nicht von einer bestimmten Verzögerung nach dem Andocken aufgetreten ist.
- Lipid Diffusität
- Für jede Fusion Ereignis verfolgen einzelnen fluoreszierenden Lipide , wie sie erkennbar werden , wenn sie aus der Fusionsstelle (Abbildung 6) ausreichend entfernt diffundiert sind. Verwenden Sie SpeckleTrackerJ für die Verfolgung, und speichern Sie die Tracks für die weitere Analyse mit MATLAB. Je nach Größe des Schmelz Vesikel kann typischerweise 3-30 einzelne Fluorophore nachgeführt werden. Weil mehr Spuren sind wünschenswert für die Berechnung von
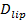 , Versuchen einzelne Moleküle so lange zu verfolgenwie möglich mit der manuellen Korrektur von Trajektorien, wenn nötig.
, Versuchen einzelne Moleküle so lange zu verfolgenwie möglich mit der manuellen Korrektur von Trajektorien, wenn nötig. - Berechnen Sie die mittlere Verschiebung im Quadrat (MSD) für einzelne Lipidmarker Trajektorien> 40-50 Frames dauern (ca. ~ 1,5 sec). Verwenden Sie die MSD des Lipiddiffusionskoeffizienten zu berechnen, . Siehe Smith et al. 26 und Stratton et al. 27 für weitere Einzelheiten.
- Für jede Fusion Ereignis verfolgen einzelnen fluoreszierenden Lipide , wie sie erkennbar werden , wenn sie aus der Fusionsstelle (Abbildung 6) ausreichend entfernt diffundiert sind. Verwenden Sie SpeckleTrackerJ für die Verfolgung, und speichern Sie die Tracks für die weitere Analyse mit MATLAB. Je nach Größe des Schmelz Vesikel kann typischerweise 3-30 einzelne Fluorophore nachgeführt werden. Weil mehr Spuren sind wünschenswert für die Berechnung von
- Einzellipid Farbstoff Intensität, SUV-SBL Intensität Reduktionsfaktor, und Vesikelgröße
- Messen der Summe der Pixelwerte eines Lipidmarker in einem 3 x 3 Pixel (0,8 um x 0,8 um) Bereich um den Marker für ~ 15 Bilder vor und nach den Markierungsbleichmittel in einem einzigen Schritt. Subtrahieren Sie die Hintergrundintensität über die post-Bleichrahmen aus dem Vorbleiche Intensität gemittelt gemittelt werden, bevor das Bleichen der Intensität des verfolgten Lipidmarker zu erhalten. Wiederholen Sie die Messung für so viele Markierungen wie praktisch für einen bestimmten Film.
- Zeichnen Sie die Verteilung der einzelnen Lipid Label Intensitäten,
 passen, und eine Gaußsche den Mittelwert abzuschätzen.
passen, und eine Gaußsche den Mittelwert abzuschätzen. - Berechnen der Verzögerung zwischen dem Fusions aufgetreten ist, und wenn die Flugbahn eines einzelnen in diesem Fall freigesetzt Lipidmarker endete in einem einstufigen Bleichen. Beziehen Sie die Bleichzeit für einen Fluorophors in der SBL,
 , Durch die Überlebensfunktion der Verzögerungen und passend zu einem exponentiellen Plotten.
, Durch die Überlebensfunktion der Verzögerungen und passend zu einem exponentiellen Plotten.
Hinweis: Da die polarisierten Anregungsfeld Paare schwächer an den Fluorophoren auf einem SUV, ist die Bleichzeit eines SUV in der Regel viel langsamer 27. - Schätzen
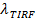 Die Intensität Reduktionsfaktor für einen Lipid Farbstoff , wenn sie in dem SUV relativ zu , wenn es in der SBL ist, folgende Stratton 27 (tp_upload / 54349 / 54349eq8.jpg "/> ist die Intensität eines einzelnen Farbstoffes in dem SUV).
Die Intensität Reduktionsfaktor für einen Lipid Farbstoff , wenn sie in dem SUV relativ zu , wenn es in der SBL ist, folgende Stratton 27 (tp_upload / 54349 / 54349eq8.jpg "/> ist die Intensität eines einzelnen Farbstoffes in dem SUV).
Hinweis: Wenn Bleichen waren vernachlässigbar, würde angedockten Intensität gleich  (Punkt (1) in 3A, rechts), durch die Gesamtintensität geteilt erreicht , nachdem alle Fluorophore in die SBL beim Schmelzen abgeschieden werden (gestrichelte Linie markiert
(Punkt (1) in 3A, rechts), durch die Gesamtintensität geteilt erreicht , nachdem alle Fluorophore in die SBL beim Schmelzen abgeschieden werden (gestrichelte Linie markiert  in Abbildung 3). Jedoch aufgrund der schnellen Bleichen in der SBL, typischerweise nicht erreicht wird und eine genaue Schätzung der erfordert auf einen Ausdruck 27 , dass incl die Freisetzungskinetik Montage udes beide (Siehe 6.4.3) und . Separate Ausdrücke werden in Stratton et al 27 für die Fälle von poren begrenzt und Diffusion begrenzte Freisetzungskinetik. die beste Lösung Fall variiert von Fall zu Fall (siehe 6.5.1 für den Fall poren begrenzt Kinetik).
in Abbildung 3). Jedoch aufgrund der schnellen Bleichen in der SBL, typischerweise nicht erreicht wird und eine genaue Schätzung der erfordert auf einen Ausdruck 27 , dass incl die Freisetzungskinetik Montage udes beide (Siehe 6.4.3) und . Separate Ausdrücke werden in Stratton et al 27 für die Fälle von poren begrenzt und Diffusion begrenzte Freisetzungskinetik. die beste Lösung Fall variiert von Fall zu Fall (siehe 6.5.1 für den Fall poren begrenzt Kinetik). - Berechnen Sie die Vesikel Bereich für individuelle Veranstaltungen von 27
 , woher ist die dockt SUV Intensität, wird die Intensität eines einzelnen Lipid Farbstoff in der SBL bedeuten, ist die Intensität Reduktionsfaktor für einen Lipid Farbstoff, wenn er in dem SUV relativ zu ist, wenn es in der SBL ist, undation 1 "src =" / files / ftp_upload / 54349 / 54349eq12.jpg "/> ist die bekannte Flächendichte von Lipid-Farbstoffe.
, woher ist die dockt SUV Intensität, wird die Intensität eines einzelnen Lipid Farbstoff in der SBL bedeuten, ist die Intensität Reduktionsfaktor für einen Lipid Farbstoff, wenn er in dem SUV relativ zu ist, wenn es in der SBL ist, undation 1 "src =" / files / ftp_upload / 54349 / 54349eq12.jpg "/> ist die bekannte Flächendichte von Lipid-Farbstoffe.
- Fusion Poreneigenschaften
- Unter der Annahme , Mitteilung ist poren beschränkt, passen die Lipid - Label Freisetzungskinetik bis 27
 , woher
, woher  ist die SUV gedockt Intensität gerade vor der Fusion und die anderen Parameter, wie zuvor definiert sind. Verwenden Sie den Wert von in 6.4.3 als fester Parameter erhalten, und extrahieren Sie die beste Lösung Schätzungen für und
ist die SUV gedockt Intensität gerade vor der Fusion und die anderen Parameter, wie zuvor definiert sind. Verwenden Sie den Wert von in 6.4.3 als fester Parameter erhalten, und extrahieren Sie die beste Lösung Schätzungen für und  .
. - Schätzen Sie den Bruchteil der Zeit die Poren offen ist , P 0, unter der Annahme , Verzögerung der Lipid Freisetzung zurückzuführen ist 27 Poren Flimmern:60;
 =
=  , Wobei A ves das Vesikel - Bereich (Abschnitt 6.4.5) ist, b ist die Poren Höhe ( in der Regel genommen ~ 15 nm zu sein), r p ≈ 3 nm die effektive Porenradius ist , wie durch die streuende Lipid Etiketten gesehen und beinhaltet Halb die Dicke der Doppelschicht (~ 2 nm), Diffusität ist das Lipid (berechnet in 6.3), und die Zeit ist, für die Lipide aus dem SUV in die SBL (von 6.5.1) freigegeben werden.
, Wobei A ves das Vesikel - Bereich (Abschnitt 6.4.5) ist, b ist die Poren Höhe ( in der Regel genommen ~ 15 nm zu sein), r p ≈ 3 nm die effektive Porenradius ist , wie durch die streuende Lipid Etiketten gesehen und beinhaltet Halb die Dicke der Doppelschicht (~ 2 nm), Diffusität ist das Lipid (berechnet in 6.3), und die Zeit ist, für die Lipide aus dem SUV in die SBL (von 6.5.1) freigegeben werden. - Um zu bestätigen , dass eine nominale P 0> 1 zeigt eine vollständig offene Pore, P 0 = 1, passen die Intensität zeitlichen Verlauf der Gleichung 4 von Stratton et al. 27, die vorhergesagten Kinetik für eine permanent offene Pore. Diese passen sollte besser sein als fitting der Ausdruck in 6.5.1 für eine permanent offene Pore.
- Unter der Annahme , Mitteilung ist poren beschränkt, passen die Lipid - Label Freisetzungskinetik bis 27
 , Wobei w der Radius des gebleichten Kreises.
, Wobei w der Radius des gebleichten Kreises.  , Durch die Überlebensfunktion der Verzögerungen und passend zu einem exponentiellen Plotten.
, Durch die Überlebensfunktion der Verzögerungen und passend zu einem exponentiellen Plotten.  (Punkt (1) in 3A, rechts), durch die Gesamtintensität geteilt erreicht , nachdem alle Fluorophore in die SBL beim Schmelzen abgeschieden werden (gestrichelte Linie markiert
(Punkt (1) in 3A, rechts), durch die Gesamtintensität geteilt erreicht , nachdem alle Fluorophore in die SBL beim Schmelzen abgeschieden werden (gestrichelte Linie markiert  in Abbildung 3). Jedoch aufgrund der schnellen Bleichen in der SBL,
in Abbildung 3). Jedoch aufgrund der schnellen Bleichen in der SBL,  , woher
, woher  , woher
, woher  ist die SUV gedockt Intensität gerade vor der Fusion und die anderen Parameter, wie zuvor definiert sind. Verwenden Sie den Wert von
ist die SUV gedockt Intensität gerade vor der Fusion und die anderen Parameter, wie zuvor definiert sind. Verwenden Sie den Wert von  =
=  , Wobei A ves das Vesikel - Bereich (Abschnitt 6.4.5) ist, b ist die Poren Höhe ( in der Regel genommen ~ 15 nm zu sein), r p ≈ 3 nm die effektive Porenradius ist , wie durch die streuende Lipid Etiketten gesehen und beinhaltet Halb die Dicke der Doppelschicht (~ 2 nm),
, Wobei A ves das Vesikel - Bereich (Abschnitt 6.4.5) ist, b ist die Poren Höhe ( in der Regel genommen ~ 15 nm zu sein), r p ≈ 3 nm die effektive Porenradius ist , wie durch die streuende Lipid Etiketten gesehen und beinhaltet Halb die Dicke der Doppelschicht (~ 2 nm), Subscription Required. Please recommend JoVE to your librarian.
Representative Results
SBL - Qualität
Es ist wichtig, die Qualität und die Fluidität des SBL vor dem Fusionsexperiment zu verifizieren. Die Fluoreszenz an der Unterseite, Glasseite eines mikrofluidischen Kanal sollte ohne offensichtliche Mängel einheitlich sein. Wenn eine Luftblase, obwohl der Kanal passiert, lässt es in der Regel sichtbare Narben auf der SBL. Wenn es eine solche groß angelegte Narben / Defekte sind, nicht, dass die Kanal verwenden. Manchmal kann SUVs auf dem Substrat haften, sondern eine kontinuierliche SBL zu platzen und bilden scheitern. Wenn das der Fall ist, kann die Fluoreszenz noch sehr einheitlich erscheinen, wird aber nicht mehr erholen, nachdem eine kleine Region Bleichen. Einführung in 10 mM Mg 2+ oft hilft , dieses Problem zu lösen. Andere Mittel können eingeführt werden , um die SUVs bersten, wie andere divalente Kationen 51, einer Polymerlösung 52 oder osmotischen Schock 25.
Temperaturänderungen führen zu Änderungen der Flächen SBL weil die mittlere Fläche von einer Lipid besetzt von der Temperatur abhängt. Erhöhen der Temperatur oder um 5-10 ° C oder mehr abnimmt führen über Bereich Röhren gehen in von der Oberfläche extrudiert wird , oder dunkel, SBL freie Flecken auf der Oberfläche erscheinen , bzw. 53. Es ist daher wichtig, Temperaturschwankungen auf ein Minimum zu halten.
Abbildung 5 für ein SBL gezeigt , die relativ fehlerfrei und flüssig war.

Abbildung 5. FRAP SBL Fließfähigkeit zu untersuchen. Eine Sequenz von Bildern eine beleuchtete Fläche , begrenzt durch die geschlossene Feldblende (Durchmesser 49 & mgr; m) vor der (ersten Rahmen) und nach Bleichen mit einer starken Lichtimpuls zeigt. Das Gebiet ist bei niedriger Belichtung abgebildet wird die Fluoreszenz Erholung durch Diffusion von NBD markierte Lipide zu folgen. Die Grafik zeigt die normalisierten Fluoreszenz als Funktion der Zeit für mehrere Kanäle von verschiedenen Deck die hohe Reproduzierbarkeit des Verfahrens angibt. Die Lipidzusammensetzung der t-SBL wurde POPC: SAPE: DOPS: Cholesterol: Gehirn PI (4,5) P 2: PEG2000-DOPE: NBD-PE (54,9: 15: 12: 10: 3: 4,6: 0,5 Mol-% ) und LP = 20.000. Die Bleichbereich war 984,203 μm 3. Die Erholungszeit und der Diffusionskoeffizient von einem Anfall berechnet (siehe 6.1) waren τ = 67,3 sec und D = 1,99 & mgr; m 2 / s auf. Bitte hier klicken , um eine größere Version dieser Figur zu sehen.
Einfarbige Erkennung von Docking und Fusion Events Mit einem Lipid - Aufkleber
Manuelles Zählen von Docking- und Fusionsereignisse ist eine mühsame Aufgabe als 100-400 Ereignisse pro Bedingung für gute Statistiken benötigt werden. Da SUVs auf der SBL vor dem Verschmelzen bewegen kann, wird eine Tracking-Software benötigt. SpeckleTrackerJ 26 - Plugin für ImageJ verfügt über Funktionen , die für die Verfolgung von SUVs für die Analyse von Docking- und Fusionsereignisse speziell entwickelt wurden. Oft nur ein Bruchteil der angedockten Vesikel Ende Verschmelzen;das Andocken Rate viel mehr SUVs als die zu verfolgenden müssen zu berechnen, oder nur ein Teil des Films verschmelzen muss analysiert werden, bis eine ausreichende Anzahl von Ereignissen für eine zuverlässige Schätzung detektiert werden. Wenn der Schwerpunkt ist die Fusionsrate und / oder die Docking-to-Fusion Verzögerungen nur die Fusion SUVs müssen identifiziert und verfolgt werden.
Nachdem die Qualität der SBL zu gewährleisten, SUVs sind in die Kanäle eingeführt und ihre Fluoreszenz unter TIR mit s-polarisiertem Licht kontinuierlich erregt. Für die ersten Versuche, muss der Einfallswinkel des TIR Anregungsstrahl kann die Eindringtiefe der abklingenden Welle resultierenden abzustimmen angepasst werden. Ein Vertreter einzigen SUV Andocken und Fusion - Ereignis wird in 6A gezeigt. Das Vesikel während des Rahmens dockt markiert D und Fusion begann im Rahmen F. Über 1,3 Sekunden später (Frame S) einzelne Lipide wurde erkennbar, wie sie aus der Fusion diffuses entfernt sitzene. Die Gesamtfluoreszenzintensität (Summe der Pixelwerte) in jedem Feld wird als eine Funktion der Zeit in 6B aufgetragen, mit den Rahmen , an dem Andocken und Fusions angegeben aufgetreten ist . Die kumulative Zahl von Fusionsereignissen als eine Funktion der Zeit in 6C aufgetragen ist, für fünf verschiedene Filme t-SNARE Verwendung rekonstituiert SBLS (t-SBL, LP = 20.000, etwa 70 t-SNAREs pro sq. Micron). Die Fusionsrate von jedem Film wurde aus einer linearen Anpassung der Steigung berechnet. Diese durchschnittliche Fusionsrate ± SEM wird als Balkendiagramm in Platte D. Die Docking-to-Fusionsverzögerung für das Beispiel in Figur 6A, B, gezeigt betrug 0,07 sec. Die Verteilung der Verzögerungen in ähnlicher Weise für insgesamt 158 Ereignisse berechnet wird in 6D als Überlebender Stück gezeigt, das heißt, die Wahrscheinlichkeit , dass ein koppelte Vesikel durch eine gegebene Verzögerung noch nicht geschmolzen ist. Über 1/2 dieser Vesikel innerhalb von 100 msec fusioniert. Mit zunehmender Cholesterinspiegel, die distribution von Docking-to-Fusion Verzögerungen verkürzt, obwohl Lipid Diffusion 27 verlangsamt.
Es ist wichtig, Kontrollexperimente parallel laufen zu lassen. Auf dem gleichen Deckglas , aus dem die Daten in 6B, D erworben wurden Kontrollbedingungen in benachbarten Kanälen getestet. In einem Kanal, umfassen die SBL brachte keine t-SNAREs. Von 5 Filme dauerhafte 60 sec jeweils nur 7 Fusionsereignisse wurden im Vergleich zu 158 erfasst , wenn die t-SNAREs enthalten waren (6C). In einem anderen Kanal wurde die lösliche cytoplasmatische Domäne des v-SNARE VAMP2 (CDV) enthalten. CDV bindet die t-SNAREs auf der SBL, die Bindung des in voller Länge VAMP2 zu verhindern, die auf den SUVs ist. In fünf 60 sec Filme wurden keine Fusionsereignisse in Gegenwart von CDV (6C) nachgewiesen.

Figur 6. SUV-SBL Andocken und Fusion Ereignisse. (A) Sequenz von Bildern eines V-SUV - Fusionsereignis mit dem T-SBL durch pTIRF Mikroskopie abgebildet die Verbreitung von LR-PE folgen. Die Bilder werden alle 18 ms, nur jedes zweite Bild genommen wird gezeigt. D und F markieren den Beginn der Andocken und Fusion sind. Einzelne Lipid Etiketten werden erkennbar, wie sie von der Fusionsstelle und miteinander (S) diffundieren. Rahmenmaße: 16,7 um · 16,7 um. (B) Fluoreszenzintensität als Funktion der Zeit für den Fall fusion in (A) gezeigt ist . (C) Kumulierte Anzahl der Fusionsereignisse als Funktion der Zeit für die v-SUVs , markiert mit einem anderen Satz von Experimenten DiD. (D) die normierte Fusionsrate (Mittelwert ± SEM, sx & mgr; m 2 x pM SUV) -1, unter der Annahme , 2 x 10 4 Lipide pro SUV und (E) die Überlebenswahrscheinlichkeit für eine bestimmte Zeit nach dem Andocken der Vesikel. Die Zusammensetzung für alle SBLs waren POPC: SAPE: DOPS: Cholesterin: Gehirn PI (4,5) P 2: PEG2000-DOPE: NBD-PE (54,9: 15: 12: 10: 3: 4,6: 0,5 Mol%) und LP = 20.000. v-SUVs von POPC zusammengesetzt waren: SAPE: DOPS: Cholesterol: PEG2000-DOPE: LR-PE (57,4: 15: 12: 10: 4,6: 1 Mol%) und LP = 200 für A und B. Für CE, die v -SUV Zusammensetzung war ähnlich, außer DiD verwendet wurde anstelle von LR-PE. Bitte hier klicken , um eine größere Version dieser Figur zu sehen.
Lipid Diffusionsvermögen, Vesicle Größe und Fusion Poreneigenschaften
Die Einzelmolekülempfindlichkeit des Assays ermöglicht die Extraktion von mehreren Parametern zusätzlich zu den Docking-und Fusionsraten und die Docking-to-Fusion Verzögerungsverteilungen. Single, werden fluoreszenzmarkierte Lipide erkennen , wie sie von der Fusionsstelle (6A) diffundieren. Tracking-tiese liefert direkt Lipid Diffusität 26, 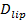 Und eine digitale Messung der Bleichrate für Fluorophore in der SBL 27. Wie in der Einleitung erwähnt, unterscheiden sich die Fluoreszenzemission der Etiketten in dem SUV Vergleich in der SBL durch drei Faktoren , die zu entwirren im allgemeinen schwierig sind: (i) Dequenching von Etiketten beim Schmelzen aufgrund ihrer Verdünnung wenn sie in die SBL übertragen , (ii) eine höhere Anregungsintensität in der SBL aufgrund des Zerfalls des evaneszenten Feldes durch TIR erzeugt, und (iii) die Orientierung der Fluorophor Übergangsdipole in Bezug auf die Polarisation des Anregungsstrahls. Die Größe des Faktors (ii) hängt von den relativen Größen des abklingenden Feldes Zerfallslänge und Vesikel Größe und unterscheidet sich für jede Fusion Veranstaltung aufgrund der Dispersität in SUV Größen. Faktor (iii) abhängig von dem Fluorophor und dem Polarisations verwendet. Anstatt zu versuchen, di sentangle diese Effekte eine die Größe der Fluoreszenzänderung durch Vergleich der Gesamtpixelwerte in einem Bereich rund um einen SUV vor der Fusion und nach alle Fluorophore wurden in die folgenden SBL Fusions (3A abgeschieden wird , direkt zu messen (1) vs. ( 2)). Anders als in früheren Arbeiten 29,44, sollte die Region ausreichend groß gewählt werden , so dass keine Fluorophore wird es für die Zeiten durch Diffusion verlassen haben , nach dem Verschmelzen betrachtet. Ein 30 x 30 Pixel - Bereich (8 & mgr; m × 8 & mgr; m) für die Analyse von bis zu 1,6 Sekunden nach der Fusion Die Wahl ist in der Regel ausreichend 27. Ignoriert Bleich für den Moment, das Verhältnis von
Und eine digitale Messung der Bleichrate für Fluorophore in der SBL 27. Wie in der Einleitung erwähnt, unterscheiden sich die Fluoreszenzemission der Etiketten in dem SUV Vergleich in der SBL durch drei Faktoren , die zu entwirren im allgemeinen schwierig sind: (i) Dequenching von Etiketten beim Schmelzen aufgrund ihrer Verdünnung wenn sie in die SBL übertragen , (ii) eine höhere Anregungsintensität in der SBL aufgrund des Zerfalls des evaneszenten Feldes durch TIR erzeugt, und (iii) die Orientierung der Fluorophor Übergangsdipole in Bezug auf die Polarisation des Anregungsstrahls. Die Größe des Faktors (ii) hängt von den relativen Größen des abklingenden Feldes Zerfallslänge und Vesikel Größe und unterscheidet sich für jede Fusion Veranstaltung aufgrund der Dispersität in SUV Größen. Faktor (iii) abhängig von dem Fluorophor und dem Polarisations verwendet. Anstatt zu versuchen, di sentangle diese Effekte eine die Größe der Fluoreszenzänderung durch Vergleich der Gesamtpixelwerte in einem Bereich rund um einen SUV vor der Fusion und nach alle Fluorophore wurden in die folgenden SBL Fusions (3A abgeschieden wird , direkt zu messen (1) vs. ( 2)). Anders als in früheren Arbeiten 29,44, sollte die Region ausreichend groß gewählt werden , so dass keine Fluorophore wird es für die Zeiten durch Diffusion verlassen haben , nach dem Verschmelzen betrachtet. Ein 30 x 30 Pixel - Bereich (8 & mgr; m × 8 & mgr; m) für die Analyse von bis zu 1,6 Sekunden nach der Fusion Die Wahl ist in der Regel ausreichend 27. Ignoriert Bleich für den Moment, das Verhältnis von  vor und nach der Fusion bietet die Fluoreszenzintensität Reduktionsfaktor,
vor und nach der Fusion bietet die Fluoreszenzintensität Reduktionsfaktor, 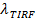 . Die Vesikel Größe kann die Fluoreszenzintensität eines einzelnen Etiketts in der SBL gegeben geschätzt werden (iles / ftp_upload / 54349 / 54349eq6.jpg "/>), würde der Faktor, mit dem die Intensität reduziert, wenn es in einem SUV waren ( ), Die bekannte Dichte von Etiketten in der SUV, und der Bereich von einer Lipid besetzt. In der Praxis ist Bleichen in der Regel schnell genug, dass durch die Zeit, alle Marker in die SBL hinterlegt sind, haben einige bereits gebleicht. Aus diesem Grund ist für eine genaue Schätzung, die Fluoreszenzintensität Reduktionsfaktor, Und die Lipidfreigabezeit,
. Die Vesikel Größe kann die Fluoreszenzintensität eines einzelnen Etiketts in der SBL gegeben geschätzt werden (iles / ftp_upload / 54349 / 54349eq6.jpg "/>), würde der Faktor, mit dem die Intensität reduziert, wenn es in einem SUV waren ( ), Die bekannte Dichte von Etiketten in der SUV, und der Bereich von einer Lipid besetzt. In der Praxis ist Bleichen in der Regel schnell genug, dass durch die Zeit, alle Marker in die SBL hinterlegt sind, haben einige bereits gebleicht. Aus diesem Grund ist für eine genaue Schätzung, die Fluoreszenzintensität Reduktionsfaktor, Und die Lipidfreigabezeit,  , Werden am besten aus einer Anpassung an den zeitlichen Verlauf der Gesamtpixelwerte extrahiert , die 27 berücksichtigt Bleichen erfolgt. Die Bleichrate von Etiketten in der SBL ist unabhängig von einzelnen Lipid Farbstoff-Tracking geschätzt oder durch eine exponentielle auf das Gesamtintensitätsprofil passend,
, Werden am besten aus einer Anpassung an den zeitlichen Verlauf der Gesamtpixelwerte extrahiert , die 27 berücksichtigt Bleichen erfolgt. Die Bleichrate von Etiketten in der SBL ist unabhängig von einzelnen Lipid Farbstoff-Tracking geschätzt oder durch eine exponentielle auf das Gesamtintensitätsprofil passend,  Bei langen Zeiten (zB Regime (3) in Figur 3). Die Kenntnis der Größe eines Verschmelzen Vesikel und Lipiddiffusionsvermögen ermöglicht einen Vergleich der Ist-Lipid Freigabezeit, An die Diffusionszeit eines Lipids um die Vesikel
Bei langen Zeiten (zB Regime (3) in Figur 3). Die Kenntnis der Größe eines Verschmelzen Vesikel und Lipiddiffusionsvermögen ermöglicht einen Vergleich der Ist-Lipid Freigabezeit, An die Diffusionszeit eines Lipids um die Vesikel  , Wobei A ves ist das Vesikel - Bereich und Diffusionsvermögen ist das Lipid. Wenn die beiden Zeitskalen vergleichbar sind, stellt die Poren wenig Widerstand gegen Lipid-Freisetzung und die Freisetzung diffusionsbegrenzt. Wenn im Gegensatz
, Wobei A ves ist das Vesikel - Bereich und Diffusionsvermögen ist das Lipid. Wenn die beiden Zeitskalen vergleichbar sind, stellt die Poren wenig Widerstand gegen Lipid-Freisetzung und die Freisetzung diffusionsbegrenzt. Wenn im Gegensatz  , Dann verzögert die Poren deutlich Lipidfluss. Ein quantitatives Maß der Verzögerung ist die "Poren Offenheit", P 0 ist , gleich dem Verhältnis
, Dann verzögert die Poren deutlich Lipidfluss. Ein quantitatives Maß der Verzögerung ist die "Poren Offenheit", P 0 ist , gleich dem Verhältnis  mal ein Faktor of , um die Einheit im Zusammenhang mit 27 Geometrie Pore. Für eine Zwei-Staaten, offene / geschlossene Pore, P 0 ist der Bruchteil der Zeit im offenen Zustand. Für eine flackernde Pore , deren Größe kontinuierlich variiert, P 0 den zeitlich gemittelten Radius relativ zu der vollständig geöffneten Radius.
mal ein Faktor of , um die Einheit im Zusammenhang mit 27 Geometrie Pore. Für eine Zwei-Staaten, offene / geschlossene Pore, P 0 ist der Bruchteil der Zeit im offenen Zustand. Für eine flackernde Pore , deren Größe kontinuierlich variiert, P 0 den zeitlich gemittelten Radius relativ zu der vollständig geöffneten Radius.
Das Signal-Rausch-Verhältnis muss für die Verfolgung von einzelnen Lipid-Fluorophore ausreichend gut zu sein. Dies ist leichter in einfarbige Experimente erreicht, eine helle Markierung wie LR-PE verwendet.
Gleichzeitige Detektion von Lipid und Lösliche Frachtmitteilung
Eine repräsentative Ereignis wird in 7 gezeigt. Bei den hohen Konzentrationen für die Verkapselung verwendet wird , die SRB Fluoreszenz ist zunächst selbst gequencht. Daher sind die meisten dockt v-SUVs sind nur nachweisbar durch ihre DiD Signal 27. Ein starker Anstiegin DiD Fluoreszenz Lipidmischung markiert, wie in einfarbige Lipid-Label-Experimente. Der Beginn der Lipid Freisetzung fiel mit dem Auftreten einer Erhöhung der SRB Fluoreszenz. Dieser Anstieg war höchstwahrscheinlich auf SRB Dequenching als die SRB-Moleküle die SUV und der verbleibende SRB entkommen wurde unter selbstlöschenden Konzentrationen verdünnt. Interessanterweise erreichte die SRB Fluoreszenz ein stabiles Plateau. Wenn die Fusionspore offen geblieben, würde man das SRB-Signal erwarten aufgrund Dequenching auf ein Maximum zu erhöhen, durch eine Abnahme in den Hintergrund gefolgt wie die restlichen SRB den SUV links, oder wenn der SUV in die SBL brach zusammen. Die stabiles Plateau meisten zeigt wahrscheinlich die Pore wieder verschlossen, nachdem alle Lipid-Etiketten und eine Fraktion des löslichen Ladung freizusetzen.
In einigen Fällen wurde die Lipid-Freigabesignal durch irgendwelche nachweisbaren Inhalt nicht begleitet die Freigabe-Signale. Dies könnte entweder, weil die Poren zu klein waren Passage von SRB zu ermöglichen, oder einigewie der SUV hatte bereits seine lösliche Ladung verloren. Zur Unterscheidung zwischen diesen beiden Möglichkeiten, Bleichen von gedockt SUVs analysiert werden. Das Bleichen der Lipid Etikett DiD reduziert seine Fluoreszenzsignal innerhalb von Sekunden, während Bleichen der anfänglich selbst abgeschreckt SRB führt zu einer Erhöhung des SRB-Signal als einen Bruchteil der SRB sind Foto umgewandelt zu anderen Produkten und Selbstlöschung wird entlastet. Während die meisten dockt SUVs (48 von 58 Ereignisse analysiert) angezeigt, um den erwarteten Anstieg der SRB Fluoreszenz über längere Zeiträume Beleuchtung, ein kleiner Bruchteil (10/58) zeigten keine SRB Signale überhaupt. So verlieren ein kleiner Bruchteil der SUVs können ihre löslichen Gehalt in der Zeit zwischen ihrer Herstellung und Verwendung in den Experimenten.
In mehr als 80% der Ereignisse (74 von 91) , für die beide SRB und DiD Signale verfolgt werden konnte, erhöhte sich die SRB - Fluoreszenz und blieb auf einem stabilen Plateau 27. In ~ 20% tiese Fälle die restlichen SRB-Fluoreszenz wurde plötzlich später auf ein paar Sekunden freigegeben werden. Dies könnte entweder durch eine Wiederaufnahme der Pore die eine schnelle, vollständige Freigabe innerhalb von 1-2 Frames (18-36 ms) oder einem Bersten des SUV durch Ansammlung von Foto-Schaden 16. Unabhängig von ihrer Herkunft, diese zweite, schnelle Freisetzung von SRB die Möglichkeit aus, dass die Rest SRB Signal nach dem ersten Release sein könnte aufgrund der SRB in dem Raum eingeschlossen bleibt zwischen der SBL und dem Glassubstrat nach der vollständigen Freigabe. Ein solcher Mechanismus wurde in einer früheren Studie vorgeschlagen , in dem die SBL direkt auf Glas unterstützt wurde, mit wenig Raum zwischen den beiden 16. Im Gegensatz dazu sollte 4-5 nm Abstand zwischen dem SBL und dem Glas 25, so dass der SRB aus der Fusionsstelle bieten das pegylierte Lipide in diesem Test verwendeten weg zu diffundieren.

Abbildung 7. Simultan eous Lipid und Content - Release. Fluoreszenzsignale aus Lipid (blau) und Inhalt (rot) Marker für ein SUV-SBL - Fusionsereignis (Einzelheiten siehe Text). Schnappschüsse von jedem Kanal sind an der Unterseite (4,0 & mgr; m breit) gezeigt. Cartoons (Mitte) zeigen die verschiedenen Stufen. (1) SUV-Docks, DiD Fluoreszenz ist gering. (2) Fusion ermöglicht die Übertragung von in die SBL DiD und DiD Fluoreszenz erhöht. SRB-Signal erhöht sich auch als Selbstlöschung durch Entweichen von SRB durch die Pore entlastet wird. (3) SRB-Signal bleibt stabil, während DiD Moleküle innerhalb der SBL diffundieren eine mögliche Wiederverschließbarkeit der Pore anzeigt. (4) SRB Signale verringern sehr schnell, aufgrund der schnellen vollständigen Fusion oder Liposom Platzen. Die Zusammensetzung von v-SUVs war POPC: SAPE: DOPS: PEG2000-DOPE: DiD (67: 15: 12: 5: 1 Mol%), LP 200, während T-SBLS wurden die aus POPC: SAPE: DOPS: Gehirn PI (4,5) P 2: PEG2000-DOPE: NBD-PE (64,5: 15: 12: 3: 5: 0,5 Mol%) und LP = 20.000. = "_ Blank"> Bitte hier klicken, um eine größere Version dieser Figur zu sehen.
FRAP_instructions.pdf -. Hinweise zum Einrichten eines FRAP - Sequenz nach oben Bitte hier klicken , um diese Datei herunterzuladen.
ReadMe_FRAP.txt -. Textdatei mit der Prozedur beschreibt FRAP Recovery - Daten zu analysieren , um die Matlab - Code verwenden , die zur Verfügung gestellt wird , bitte hier klicken um diese Datei herunterzuladen.
EK_FRAPbatch.m - MATLAB - Datei Batch-Analyse Frap. Diese ruft den MATLAB-Subroutinen EK_getFRAPvals.m und EK_SoumpassisFit.m."_blank"> Bitte hier klicken, um diese Datei herunterzuladen.
EK_getFRAPvals.m -. MATLAB - Datei, Unterprogramm von EK_FRAPbatch.m genannt Bitte hier klicken , um diese Datei herunterzuladen.
EK_SoumpassisFit.m -. MATLAB - Datei, Unterprogramm von EK_FRAPbatch.m genannt Bitte hier klicken , um diese Datei herunterzuladen.
JN150923c1_FRAP_list.txt -. Ein Beispiel für eine Liste von Dateinamen durch den Haupt MATLAB Skript EK_FRAPbatch.m chargen analysiert werden Bitte hier klicken , um diese Datei herunterzuladen.
DasFolgende Dateien sind TIF-Stacks und die entsprechenden Textdateien für fünf Beispiele von FRAP-Sequenzen, die durch Ausführen EK_FRAPbatch.m und die Wahl der JN150923c1_FRAP_list.txt Datei analysiert werden kann, wenn aufgefordert, eine Dateiliste zu wählen.
JN150923c1_F1_MMStack_Pos0.ome.tif -. FRAP Bildstapel Bitte hier klicken , um diese Datei herunterzuladen.
JN150923c1_F1_MMStack_Pos0_metadata.txt -. Entsprechenden Textdatei mit Metadaten Bitte hier klicken , um diese Datei herunterzuladen.
JN150923c1_F2_MMStack_Pos0.ome.tif -. FRAP Bildstapel Plerleichtern klicken hier, um diese Datei herunterzuladen.
JN150923c1_F2_MMStack_Pos0_metadata.txt -. Entsprechenden Textdatei mit Metadaten Bitte hier klicken , um diese Datei herunterzuladen.
JN150923c1_F3_MMStack_Pos0.ome.tif -. FRAP Bildstapel Bitte hier klicken , um diese Datei herunterzuladen.
JN150923c1_F3_MMStack_Pos0_metadata.txt -. Entsprechenden Textdatei mit Metadaten Bitte hier klicken , um diese Datei herunterzuladen.
JN150923c1_F4_MMStack_Pos0.ome.tif - FRAP Bildstapel. Bitte hier klicken , um diese Datei herunterzuladen.
JN150923c1_F4_MMStack_Pos0_metadata.txt -. Entsprechenden Textdatei mit Metadaten Bitte hier klicken , um diese Datei herunterzuladen.
JN150923c1_F5_MMStack_Pos0.ome.tif -. FRAP Bildstapel Bitte hier klicken , um diese Datei herunterzuladen.
JN150923c1_F5_MMStack_Pos0_metadata.txt -. Entsprechenden Textdatei mit Metadaten Bitte hier klicken , um diese Datei herunterzuladen.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Die erfolgreiche Umsetzung des SUV-SBL Fusionstest hier beschrieben hängt entscheidend von mehreren wichtigen Schritten, wie funktionelle Rekonstitution von Proteinen in Liposomen, gute Qualität SBLS zu erhalten, und die Wahl der richtigen Abbildungsparameter einzelner Moleküle zu detektieren. Obwohl es einige Zeit und Mühe in Anspruch nehmen , um zu folgen, sobald der Test erfolgreich durchgeführt wird, liefert es eine Fülle von Informationen über den Schweißvorgang nicht bei jedem anderen in vitro Fusionstest in Einleitung diskutiert. Die Geschwindigkeiten, mit denen SUVs andocken und verschmelzen mit der SBL, die Verteilung der Docking-to-Fusionszeiten, Lipid Diffusität, die Größe jedes Verschmelzen Liposom, und ob Lipid-Release für ein Ereignis gegeben Fusion ist Diffusions- oder poren gesteuert Dose bestimmt werden. Wenn Release ist viel langsamer als von Diffusion zu erwarten, dass ein Modell Poren Flimmern annimmt liegt unter der Verzögerung der Lipid Freisetzung Ausbeuten, P 0, den Anteil der Zeit , die Poren is offen bei der Fusion 27.
Frühere Arbeiten einzigen SUV-SUV Fusion 18-20,28 oder Fusion von Liposomen und Viruspartikel zu SBLS Sondieren 15-17,21,22,33,44 auf FRET und Selbstlöschung zwischen Farbstoffmolekülen verlassen. Im Gegensatz vermeiden wir weitgehend farbstoff Dequenching Kinetik der Lipidetikettentransfer-Sonde in die SBL, aus zwei Gründen. Erstens ist FRET-Effizienz eine hoch nichtlineare reporter of Lipid-Release. Kleine Änderungen in der Zwischenfarbstoff Abstand d zu großen Veränderungen in der FRET - Effizienz, jedoch nur für einen begrenzten Bereich von d / R & sub0 ; -Werte, wobei R 0 ist der Förster - Abstand. Das heißt, eine Verzögerungszeit zwischen der tatsächlichen Farbstoffverdünnungs und einer beobachteten Anstieg des Dequenching Signal auftreten können, aufgrund der Zeit , für die erforderlich ist, um einen Wert nahe R 0 zu fallen. In ähnlicher Weise gibt es wenig Änderung in der Dequenching Signal, sobald die Farbstoffdichte ausreichend auf Lipid Freisetzung verringert, so dass  1.4, selbst wenn eine beträchtliche Menge an Farbstoff bleibt in dem Vesikel. Diese Eigenschaften machen Dequenching ein gutes Werkzeug ein Dorn im Signal zu erhalten Fusionsereignisse zu erfassen, sondern erschweren quantitative kinetische Analyse von Lipid Freisetzung hoher Zeitauflösung verwendet wird. Im Gegensatz dazu durch Intensitätsänderungen zu Dipol Umorientierung und evaneszente Feldeffekte wie ein Fluorophor Transfers von einem SUV in die SBL werden der Anteil des Farbstoffes in der SUV verbleibenden noch linear bezogen. Zweitens hohe Konzentrationen von Lipid-Etiketten für Selbstlöschung erforderlich kann das Fusionsprozess selbst beeinflussen.
1.4, selbst wenn eine beträchtliche Menge an Farbstoff bleibt in dem Vesikel. Diese Eigenschaften machen Dequenching ein gutes Werkzeug ein Dorn im Signal zu erhalten Fusionsereignisse zu erfassen, sondern erschweren quantitative kinetische Analyse von Lipid Freisetzung hoher Zeitauflösung verwendet wird. Im Gegensatz dazu durch Intensitätsänderungen zu Dipol Umorientierung und evaneszente Feldeffekte wie ein Fluorophor Transfers von einem SUV in die SBL werden der Anteil des Farbstoffes in der SUV verbleibenden noch linear bezogen. Zweitens hohe Konzentrationen von Lipid-Etiketten für Selbstlöschung erforderlich kann das Fusionsprozess selbst beeinflussen.
Für polarisierte TIRF - Anregung ein Eigenbau-Setup wie in Abbildung 4 ist kostengünstig und bietet eine hervorragende Kontrolle über Polarisationseigenschaften. Allerdings ist ein Eigenbau-Setup ist nicht erforderlich, da zumindest einige kommerzielle TIRF-Mikroskope polarisierten Anregung verwenden und off-the-shelfZweifarben-Emissions Splitting-Module zur Verfügung. Ein handelsübliches Mikroskop , das eine polarisationserhaltende (PM) Faser zu koppeln , von dem Laser in das Mikroskop das polarisierte Anregungslicht verwendet erfolgreich zuvor 25-27 wurde Schwellen verwendet. Auf ein solches Instrument kann die Polarisation durch Drehen des PM-Fasereingang zu der TIRF der Mikroskopeinheit gedreht einfach werden. Im Gegensatz dazu ist ein polarisierender Strahlteiler in der TIRF Einheit Polarisations Anpassungen erschweren oder unmöglich machen können verwenden einige andere kommerzielle Setups. Leider Polarisationseigenschaften sind oft nicht für kommerzielle Systeme angegeben. Daher ist es von entscheidender Bedeutung für Benutzer, die nicht wollen, ein Haus gebaut System zu konstruieren Polarisationseigenschaften zu diskutieren und eine Demo-Einheit, um zu versuchen, bevor sie in einer bestimmten kommerziellen Option zu investieren.
Gleichzeitige Überwachung von Lipid und löslichen Fracht-Version bietet weitere Informationen über den Fusionsprozess. Viele Poren scheinen nach rele wieder zu verschließenAsing nur ein Bruchteil der löslichen Markers in eine 27 SUV eingekapselt. Normalerweise werden alle Lipid-Etiketten werden durch die Zeit veröffentlicht die Pore wieder abdichtet, so Pore Wiederverschließbarkeit war bisher nicht im Verdacht von der Überwachung Lipid Freisetzung allein. Tatsächlich Lipid Release Überwachung allein, Kiessling et al. 29 hatte der SBL vorgeschlagen Übertragung markierter Lipide in aufgetreten über extrem schnelle Zusammenbruch des gesamten SUV - Membran in die SBL.
Wiederverschließen der Fusions Poren nach der teilweisen Freigabe von Inhalten zuvor in einem Liposom-tethered Bilayer Patch Fusionstest beobachtet wurde. Rawle et al. 54 vorbereitet , freistehende Doppelschicht - Patches, die von ~ 8 nm langen DNA - Abstandshalter auf einem Deckglas gebunden wurden. Sie führten dann Liposomen, deren Fusion mit dem Bilayer-Patch durch Hybridisierung komplementärer Einzelstrang-DNA-Lipid-Konjugate zu verankernden das Liposom und Patch-Membranen angetrieben wurde. 12% aller Ereignisse waren vorübergehend Fusionen, was teilial Freisetzung der Liposomeninhalte. Liposome Platzen Ereignisse, 44 in einigen früheren SUV-SBL - Fusion - Assays gesehen, waren sehr selten. Die Autoren schlugen vor, die ~ 8 nm Raum unterhalb der Doppelschicht-Patch, Bilayer-Substrat-Wechselwirkungen zu verhindern, haben unter der Doppelschicht in den Raum nicht undicht Übertragung der Liposomen Inhalte erlaubt. Zusätzlich Diffusion der freigesetzten löslichen Etiketten wurde in den Raum zwischen der planaren Bilayer und dem Glassubstrat nicht wesentlich behindert. In dem Test hier beschrieben, wird die planar bilayer ähnlich gehalten ~ 5 nm entfernt von der Deckglases durch die Anwesenheit von Poly (ethylenglykol) -Lipid-Konjugate.
Die zusätzlichen Informationen durch Zweifarben-Überwachung von löslichen und Lipid-Ladung vorgesehenen kommt mit einigen Kompromissen. Zunächst Herstellung von SUVs mit den beiden Labels ist etwas komplizierter als Lipid-Etiketten allein einschließlich. Zweitens gibt es eine unvermeidliche Verlust an Signal-zu-Rauschen, wenn bei der T-Signale von den Lipid Etiketten Überwachungser gleiche Erfassungsrate wie in einfarbige Lipidfreisetzungsversuche (15-20 ms pro Bild). Ein Teil dieser Verluste ist aufgrund der Tatsache, dass als Lipid Etikett zusammen mit dem löslichen SRB Marker genutzt, als LR-PE in einfarbige Experimenten verwendet nicht so hell ist. Zusätzliche Verluste ergeben sich aus höheren Hintergrund aufgrund simultanen Zweilaseranregung und zusätzliche Komponenten in dem optischen Pfad (Abbildung 4). Bisher ist das untere Signal-zu-Rausch-hinderten uns zuverlässige Verfolgung einzelner DiD Etiketten in Zweifarben-Experimenten. Dies wiederum verhindert eine gründliche Analyse alle Informationen zu extrahieren, die mit Single-Farb-LR-PE-Messungen sein kann. Diese Herausforderung kann durch die Optimierung der Aufnahmebedingungen und Test andere Paare von kompatiblen Etiketten mit verbesserten Eigenschaften in der Zukunft überwunden werden.
Gleichzeitige Überwachung von löslichen und Lipid-Ladung Freisetzung kann auch hemifusion erkennen, ein Zustand, in dem die proximalen Blättchen verschmolzen, aber das distaleFaltblätter nicht. Tatsächlich können solche zweifarbige Experimente kann die einzige abschließende Beurteilung der Anwesenheit von hemifusion in anderen In - vitro - Assays Fusion 18,20 bereitzustellen. Allerdings kann hemifusion zuverlässig in einfarbige SUV-SBL Experimente Überwachung Lipid Freisetzung allein 22,44 nachgewiesen werden. Bei solchen Versuchen, etwa 1/2 der anfänglichen Lipid Label Intensität eines SUV bleibt an der Fusion vor Ort. Die verbleibende Intensität kann verschwinden, wenn die distalen Blättchen einige Zeit später verschmelzen, wenn der Fleck noch nicht gebleicht.
Die Verwendung von pegyliertem Lipide ein weiches Kissen zwischen dem SBL einzuführen und dem Glasträger wurde bei der Reproduktion des SNAP25 Anforderung der Exozytose Schlüssel gewesen. Das Protokoll führt zu einer PEG bürsten Sie die Seite des SBL abdeckt, die auch den Strömungskanal zugewandt ist. Dies hat einige Vorteile, wie nicht-spezifische Wechselwirkungen zwischen SUVs zu reduzieren und die SBL, wie in Introduction erläutert. Wenn Proteine, die mit den Phospholipiden der interagierenSBL werden in den Assay eingebracht, jedoch wird die PEG Bürste eine sterische Barriere gegen solche Wechselwirkungen darzustellen. Somit für einige Anwendungen wäre es wünschenswert sein, die PEG-Schicht asymmetrisch zu umfassen, nur zwischen dem SBL und dem Deckglas. Die Seite des SBL den Strömungskanal zugewandten würde dann mit Phospholipid-Bindungsproteine, wie synaptotagmin können zu interagieren. Dies kann durch Verwendung bifunktionellen PEG-Ketten erreicht werden, die kovalent an das Deckglas an einem Ende verbunden werden kann und einen Lipidanker am anderen trägt. Langmuir-Blodgett - Verfahren können dann verwendet werden , 55 eine Lipid - Monoschicht auf die PEG - Schicht abzuscheiden. Liposome über diesem hydrophoben Oberflächen Sicherung eingeführt mit ihm unter Bildung einer Doppelschicht 56. Es ist zu hoffen, dass dieser Ansatz mit der Mikrofluidik in Zukunft kombiniert wird.
Es wird erwartet, dass die SUV-SBL-Test hier vorgestellt wird hilfreich sein, die Rolle von zusätzlichen Proteinen zu erforschen, die mit SNAREs in Wechselwirkung treten,wie die Munc18 / Sec1 Familie, dem Calciumsensor synaptotagmin-1 und Complexin.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Die Autoren erklären, dass sie keine finanziellen Interessen haben.
Materials
| Name | Company | Catalog Number | Comments |
| Reagents | |||
| Milli-Q (MQ) water | Millipore | ||
| KOH | J.T. Baker | 3040-05 | |
| Ethanol 190 Proof | Decon | ||
| Isopropanol | Fisher Chemical | A416P4 | |
| HEPES | AmericanBio | AB00892 | |
| Sodium Cholride (KCl) | AmericanBio | AB01915 | |
| Dithiothreitol | AmericanBio | AB00490 | |
| N-[2-hydroxyethyl] piperazine-N'-[2-ethanesulfonic acid] (HEPES) | AmericanBio | AB00892 | |
| EGTA | Acros Organics | 409911000 | |
| Buffers | |||
| HEPES-KOH buffer (pH 7.4) | 25 mM HEPES-KOH, 140 mM KCl, 100 μM EGTA, 1 mM DTT | ||
| Solvents | |||
| Chloroform | J.T. Baker | 9180-01 | in glass bottle, CAUTION, wear PPE |
| Methanol | J.T. Baker | 9070-03 | in glass bottle, CAUTION, wear PPE |
| Liposome preparation | |||
| Gastight Hamilton syringe | Hamilton | var. sizes | only use glass sringe with solents (Chlorophorm/ Methanon, 2:1, v/v) |
| Glass tubes Pyrex Vista 11 ml, 16x100 mm screw cap culture tube | Pyrex | 70825-16 | clean thoroughly, rinse with chloroform |
| 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine, 16:0-18:1 PC (POPC) | Avanti Polar Lipids | 850457 | Lipids come dissolved in CHCl3 or as lyphilized powder in sealed vials. Aliquot upon opening. Store extra as dried lipid films under inert atmosphere at -20 °C. Keep stocks in CHCl3/MeOH (2:1, v/v) at -20 °C. let come to RT before opening |
| 1,2-dioleoyl-sn-glycero-3-phospho-L-serine (sodium salt), 18:1 PS (DOPS) | Avanti Polar Lipids | 840035 | Same as above. |
| 1-stearoyl-2-arachidonoyl-sn-glycero-3-phosphoethanolamine, 18:0-20:4 PE (SAPE) | Avanti Polar Lipids | 850804 | Same as above. |
| L-α-phosphatidylinositol-4,5-bisphosphate (Brain, Porcine) (ammonium salt), Brain PI(4,5)P2 | Avanti Polar Lipids | 840046 | Same as above. |
| 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(7-nitro-2-1,3-benzoxadiazol-4-yl) (ammonium salt), 18:1 NBD PE | Avanti Polar Lipids | 810145 | Same as above. |
| 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (ammonium salt), 18:1 PEG2000 PE | Avanti Polar Lipids | 880130 | Same as above. |
| cholesterol (ovine wool, >98%) | Avanti Polar Lipids | 700000 | Same as above. |
| DiD' oil; DiIC18(5) oil (1,1'-Dioctadecyl-3,3,3',3'-Tetramethylindodicarbocyanine Perchlorate) | Molecular Probes | D-307 | |
| Rotavapor R-210 | Buchi | R-210 | heat bath above Tm of lipids used |
| OG n-Octyl-β-D-Glucopyranoside | Affymetrix | 0311 | store at -20 °C, let come to RT before opening |
| Shaker - Eppendorf Thermomixer R | Eppendorf | ||
| Slide-A-Lyze Dialysis Cassettes, 20k MWCO, 3 ml | life technologies | 66003 | |
| Bio-Beads SM-2 Adsorbents | Bio-Rad | 1523920 | |
| OptiPrep Density Gradient Medium | Sigma-Aldrich | D1556 | |
| Ultracentrifugation tube, Thinwall, Ultra-Clear, 13.2 ml, 14 x 89 mm | Beckman Coulter | 41121703 | |
| Beckman SW41 Ti rotor | |||
| SuflorhodamineB | Molecular Probes | S-1307 | |
| Econo-Column Chromatography Columns, 2.5 × 10 cm | Bio-Rad | 7372512 | |
| Sepharose CL-4B | GE Healthcare | 17-0150-01 | |
| SYPRO Orange Protein Gel Stain | Molecular Probes | S-6650 | 5,000x Concentrate in DMSO |
| PDMS block | |||
| Sylgard 184 Silicone elastomer kit, PDMS | Dow Corning | 3097358-1004 | |
| Pyrex glass petri dish, 150 x 20 mm, complete with cover | Corning | 3160-152 | |
| Hole puncher - Reusable Biopsy Punch, 0.75 mm | World Precision Instruments | 504529 | |
| Manual Hole Punching Machine | SYNEO | MHPM-UNV | |
| Drill .035 x .026 x 1.5 304 SS TiN coated round punch | SYNEO | CR0350265N20R4 | drill diameter: 0.9 mm, punch bore size: 0.74 mm |
| Tygon Microbore tubing, 0.25 mm ID, 0.76 mm OD | Cole-Parmer | 06419-00 | 0.010" ID, 0.030" OD |
| Silicone Tubing (0.51 mm ID, 2.1 mm OD | Cole-Parmer | 95802-00 | 0.020" ID, 0.083" OD |
| Cover glass - cleanroom cleaned | |||
| Schott Nexterion cover slip glass D | Schott | 1472305 | |
| plasma cleaner | Harrick | PDC-32G | |
| pTIRF setup and accessories | |||
| IX81 microscope body | Olympus | IX81 | |
| EM CCD camera | Andor | ixon-ultra-897 | |
| Thermo Plate, heated microscope stage | Tokai Hit | MATS-U52RA26 | |
| 1 ml hamilton glass syringes (4x) | Hamilton | 81365 | |
| syringe pump | kd Scientific | KDS-230 | |
References
- Sudhof, T. C., Rothman, J. E. Membrane fusion: grappling with SNARE and SM proteins. Science. 323, 474-477 (2009).
- Wickner, W., Schekman, R. Membrane fusion. Nat Struct Mol Biol. 15, 658-664 (2008).
- Harrison, S. C. Viral membrane fusion. Nat Struct Mol Biol. 15, 690-698 (2008).
- Jahn, R., Scheller, R. H. SNAREs--engines for membrane fusion. Nat Rev Mol Cell Biol. 7, 631-643 (2006).
- Lindau, M., Alvarez de Toledo, G. The fusion pore. Biochim Biophys Acta. 1641, 167-173 (2003).
- Staal, R. G., Mosharov, E. V., Sulzer, D. Dopamine neurons release transmitter via a flickering fusion pore. Nat Neurosci. 7, 341-346 (2004).
- Wu, Z., et al. Nanodisc-cell fusion: Control of fusion pore nucleation and lifetimes by SNARE protein transmembrane domains. Sci. Rep. 6, 27287 (2016).
- Alabi, A. A., Tsien, R. W. Perspectives on kiss-and-run: role in exocytosis, endocytosis, and neurotransmission. Ann Rev Physiol. 75, 393-422 (2013).
- Rossetto, O., Pirazzini, M., Montecucco, C. Botulinum neurotoxins: genetic, structural and mechanistic insights. Nature Rev Microbiol. 12, 535-549 (2014).
- Weber, T., et al. SNAREpins: minimal machinery for membrane fusion. Cell. 92, 759-772 (1998).
- Nickel, W., et al. Content mixing and membrane integrity during membrane fusion driven by pairing of isolated v-SNAREs and t-SNAREs. Proc Natl Acad Sci U S A. 96, 12571-12576 (1999).
- McNew, J. A., et al. Compartmental specificity of cellular membrane fusion encoded in SNARE proteins. Nature. 407, 153-159 (2000).
- Melia, T. J., You, D. Q., Tareste, D. C., Rothman, J. E. Lipidic antagonists to SNARE-mediated fusion. J Biol Chem. 281, 29597-29605 (2006).
- Hernandez, J. M., et al. Membrane fusion intermediates via directional and full assembly of the SNARE complex. Science. 336, 1581-1584 (2012).
- Fix, M., et al. Imaging single membrane fusion events mediated by SNARE proteins. Proc Natl Acad Sci U S A. 101, 7311-7316 (2004).
- Bowen, M. E., Weninger, K., Brunger, A. T., Chu, S. Single molecule observation of liposome-bilayer fusion thermally induced by soluble N-ethyl maleimide sensitive-factor attachment protein receptors (SNAREs). Biophys J. 87, 3569-3584 (2004).
- Liu, T., Tucker, W. C., Bhalla, A., Chapman, E. R., Weisshaar, J. C. SNARE-driven, 25-millisecond vesicle fusion in vitro. Biophys J. 89, 2458-2472 (2005).
- Yoon, T. Y., Okumus, B., Zhang, F., Shin, Y. K., Ha, T. Multiple intermediates in SNARE-induced membrane fusion. Proc Natl Acad Sci U S A. 103, 19731-19736 (2006).
- Diao, J., et al. A single-vesicle content mixing assay for SNARE-mediated membrane fusion. Nat Commun. 1, 1-6 (2010).
- Kyoung, M., et al. In vitro system capable of differentiating fast Ca2+-triggered content mixing from lipid exchange for mechanistic studies of neurotransmitter release. Proc Natl Acad Sci U S A. 108, E304-E313 (2011).
- Domanska, M. K., Kiessling, V., Stein, A., Fasshauer, D., Tamm, L. K. Single vesicle millisecond fusion kinetics reveals number of SNARE complexes optimal for fast SNARE-mediated membrane fusion. J Biol Chem. 284, 32158-32166 (2009).
- Kreutzberger, A. J., Kiessling, V., Tamm, L. K. High Cholesterol Obviates a Prolonged Hemifusion Intermediate in Fast SNARE-Mediated Membrane Fusion. Biophys J. 109, 319-329 (2015).
- Schwenen, L. L., et al. Resolving single membrane fusion events on planar pore-spanning membranes. Sci Rep. 5, 12006 (2015).
- Karatekin, E., et al. A fast, single-vesicle fusion assay mimics physiological SNARE requirements. Proc Natl Acad Sci U S A. 107, 3517-3521 (2010).
- Karatekin, E., Rothman, J. E. Fusion of single proteoliposomes with planar, cushioned bilayers in microfluidic flow cells. Nat Protoc. 7, 903-920 (2012).
- Smith, M. B., et al. Interactive, computer-assisted tracking of speckle trajectories in fluorescence microscopy: application to actin polymerization and membrane fusion. Biophys J. 101, 1794-1804 (2011).
- Stratton, B. S., et al. Cholesterol Increases the Openness of SNARE-mediated Flickering Fusion Pores. Biophysical journal. 110, (2016).
- Diao, J., et al. Synaptic proteins promote calcium-triggered fast transition from point contact to full fusion. Elife. 1, e00109 (2012).
- Kiessling, V., Domanska, M. K., Tamm, L. K. Single SNARE-mediated vesicle fusion observed in vitro by polarized TIRFM. Biophys J. 99, 4047-4055 (2010).
- Blasi, J., et al. Botulinum neurotoxin A selectively cleaves the synaptic protein SNAP-25. Nature. 365, 160-163 (1993).
- Washbourne, P., et al. Genetic ablation of the t-SNARE SNAP-25 distinguishes mechanisms of neuroexocytosis. Nat Neurosci. 5, 19-26 (2002).
- Diaz, A. J., Albertorio, F., Daniel, S., Cremer, P. S. Double cushions preserve transmembrane protein mobility in supported bilayer systems. Langmuir. 24, 6820-6826 (2008).
- Floyd, D. L., Ragains, J. R., Skehel, J. J., Harrison, S. C., van Oijen, A. M. Single-particle kinetics of influenza virus membrane fusion. Proc Natl Acad Sci U S A. 105, 15382-15387 (2008).
- Albertorio, F., et al. Fluid and air-stable lipopolymer membranes for biosensor applications. Langmuir. 21, 7476-7482 (2005).
- Daniel, S., Albertorio, F., Cremer, P. S. Making lipid membranes rough, tough, and ready to hit the road. Mrs Bulletin. 31, 536-540 (2006).
- Gao, Y., et al. Single reconstituted neuronal SNARE complexes zipper in three distinct stages. Science. 337, 1340-1343 (2012).
- Kenworthy, A. K., Hristova, K., Needham, D., Mcintosh, T. J. Range and Magnitude of the Steric Pressure between Bilayers Containing Phospholipids with Covalently Attached Poly(Ethylene Glycol). Biophys J. 68, 1921-1936 (1995).
- Knoll, W., et al. Solid supported lipid membranes: New concepts for the biomimetic functionalization of solid surfaces. Biointerphases. 3, Fa125-Fa135 (2008).
- Quinn, P., Griffiths, G., Warren, G. Density of newly synthesized plasma membrane proteins in intracellular membranes II. Biochemical studies. J Cell Biol. 98, 2142-2147 (1984).
- Sund, S. E., Swanson, J. A., Axelrod, D. Cell membrane orientation visualized by polarized total internal reflection fluorescence. Biophys J. 77, 2266-2283 (1999).
- Johnson, D. S., Toledo-Crow, R., Mattheyses, A. L., Simon, S. M. Polarization-controlled TIRFM with focal drift and spatial field intensity correction. Biophys J. 106, 1008-1019 (2014).
- Anantharam, A., Onoa, B., Edwards, R. H., Holz, R. W., Axelrod, D. Localized topological changes of the plasma membrane upon exocytosis visualized by polarized TIRFM. J Cell Biol. 188, 415-428 (2010).
- Axelrod, D. Carbocyanine dye orientation in red cell membrane studied by microscopic fluorescence polarization. Biophys J. 26, 557-573 (1979).
- Wang, T., Smith, E. A., Chapman, E. R., Weisshaar, J. C. Lipid mixing and content release in single-vesicle, SNARE-driven fusion assay with 1-5 msec resolution. Biophys J. 96, 4122-4131 (2009).
- Chernomordik, L. V., Frolov, V. A., Leikina, E., Bronk, P., Zimmerberg, J. The pathway of membrane fusion catalyzed by influenza hemagglutinin: restriction of lipids, hemifusion, and lipidic fusion pore formation. J Cell Biol. 140, 1369-1382 (1998).
- Takamori, S., et al. Molecular anatomy of a trafficking organelle. Cell. 127, 831-846 (2006).
- Wilhelm, B. G., et al. Composition of isolated synaptic boutons reveals the amounts of vesicle trafficking proteins. Science. 344, 1023-1028 (2014).
- Scott, B. L., et al. Liposome fusion assay to monitor intracellular membrane fusion machines. Methods Enzymol. 372, 274-300 (2003).
- Linkert, M., et al. Metadata matters: access to image data in the real world. J Cell Biol. 189, 777-782 (2010).
- Soumpasis, D. M. Theoretical analysis of fluorescence photobleaching recovery experiments. Biophys J. 41, 95-97 (1983).
- Ohki, S. A mechanism of divalent ion-induced phosphatidylserine membrane fusion. Biochim Biophys Acta. 689, 1-11 (1982).
- Berquand, A., et al. Two-step formation of streptavidin-supported lipid bilayers by PEG-triggered vesicle fusion. Fluorescence and atomic force microscopy characterization. Langmuir. 19, 1700-1707 (2003).
- Tamm, L. K., McConnell, H. M. Supported phospholipid bilayers. Biophys J. 47, 105-113 (1985).
- Rawle, R. J., van Lengerich, B., Chung, M., Bendix, P. M., Boxer, S. G. Vesicle fusion observed by content transfer across a tethered lipid bilayer. Biophys J. 101, L37-L39 (2011).
- Wagner, M. L., Tamm, L. K. Reconstituted syntaxin1a/SNAP25 interacts with negatively charged lipids as measured by lateral diffusion in planar supported bilayers. Biophys J. 81, 266-275 (2001).
- Kalb, E., Frey, S., Tamm, L. K. Formation of Supported Planar Bilayers by Fusion of Vesicles to Supported Phospholipid Monolayers. Biochimica Et Biophysica Acta. 1103, 307-316 (1992).
