

Summary
Qui, vi presentiamo un protocollo per rilevare, eventi di fusione singoli SNARE-mediata tra liposomi e bistrati supportati in canali microfluidica utilizzando TIRFM polarizzata, con sensibilità singola molecola e ~ 15 risoluzione temporale msec. Lipidi e il rilascio del carico solubili possono essere rilevati simultaneamente. dimensioni liposomi, diffusività dei lipidi, e poro di fusione proprietà sono misurate.
Abstract
Nel processo onnipresente di fusione della membrana l'apertura di un poro di fusione stabilisce il primo collegamento tra due compartimenti precedentemente separate. Durante neurotrasmettitore o il rilascio di ormoni tramite esocitosi, il poro di fusione può transitoriamente aprire e chiudere più volte, che regolano carico cinetica di rilascio. la dinamica dei pori determinano anche la modalità di riciclo delle vescicole; risultati richiusura irreversibili nella fusione transitoria, "kiss-and-run", mentre la dilatazione porta alla completa fusione. Per capire meglio quali fattori governano la dinamica dei pori, abbiamo sviluppato un test per monitorare la fusione delle membrane mediante microscopia polarizzata riflessione interna totale in fluorescenza (TIRF) con sensibilità singola molecola e ~ 15 msec risoluzione temporale in una biochimica ben definito sistema in vitro. Fusione di fluorescente piccole vescicole contenenti proteine unilamellari v-SNARE (v-SUV) con un cuscinetto doppio strato planare t-SNARE, appoggiato su un cuscino di polimero morbido (t-SBL, doppio strato t-supportato), È monitorata. Il test utilizza canali di flusso microfluidica che assicurano il consumo di campione minimo mentre fornisce una densità costante di SUV. Sfruttando il potenziamento del segnale rapida al momento del trasferimento di etichette lipidi dal SUV per la SBL durante la fusione, la cinetica di trasferimento della tintura dei lipidi è monitorato. La sensibilità di microscopia TIRF permette il monitoraggio etichette lipidici fluorescenti singoli, da cui lipidi diffusività e la dimensione SUV possono essere dedotte per ogni evento di fusione. Lipidi tempi di rilascio di colorante può essere molto più lungo del previsto per il passaggio senza ostacoli attraverso i pori permanentemente aperti. Utilizzando un modello che assume il ritardo del rilascio di lipidi è dovuta a pori sfarfallio, un poro "apertura", la frazione di tempo poro rimane aperto durante la fusione, può essere stimato. Un marcatore solubile può essere incapsulato in SUV per il monitoraggio simultaneo di lipidi e il rilascio del carico solubile. Tali misurazioni indicano alcuni pori possono richiudere dopo aver perso una frazione del carico solubile.
Introduction
Fusione della membrana è un processo biologico universale necessario per il traffico intracellulare di lipidi e proteine, la secrezione, la fecondazione, lo sviluppo, e avvolto entrata del virus in organismi ospiti 1-3. Per la maggior parte delle reazioni di fusione intracellulare, tra cui il rilascio di ormoni e neurotrasmettitori tramite esocitosi, l'energia per fondere due doppi strati lipidici è fornito dalla formazione di un fascio di quattro elica tra cognate solubile N-ethylmaleimide sensibile proteina recettore attaccamento fattore (militare) proteine, ancorato a la vescicola (v-militare) e la membrana di destinazione (t-SNARE) 4, rispettivamente. Sinaptica vescicole esocitosi è la reazione di fusione più strettamente regolata e si verifica all'interno di un millisecondo dopo l'arrivo di un potenziale 1,4,5 azione. Il poro di fusione, la connessione iniziale tra i due comparti di fusione, può sfarfallio aperto e chiuso più volte prima di richiudere o espandere in maniera irreversibile 5-7. Gli ex risultatinel transitorio, "kiss & run" di fusione, mentre il secondo porta alla piena fusione. I fattori che regolano l'equilibrio tra queste due modalità di fusione e dei meccanismi che regolano pori sfarfallamento non sono ben compresi 5,8.
proteine SNARE sono necessari per esocitosi; sinaptica fusione delle vescicole è abolita sulla scissione di insidie da neurotossine 9. Esperimenti di fusione di massa utilizzando piccole vescicole unilamellari (SUV) ha mostrato che SNAREs non solo sono necessari, ma anche sufficiente per guidare membrana fusione 10. In questo saggio rinfusa, SUV ricostituiti con v-SNARE (v-SUV) sono stati drogato con fosfolipidi fluorescenti (N - (7-nitro-2-1,3-benzoxadiazol-4-il) -phosphoethanolamine (NBD-PE) e ( N -. (lissamina rodamina B sulfonil) -phosphoethanolamine (LR-PE) e mescolato con vescicole non etichettati contenenti t-SNARE (t-SUV) Inizialmente la fluorescenza di NBD-PE in V-SUV si spegne da Förster trasferimento di energia di risonanza (FRET ) a LR-PE. Come laboratorioELED v-SUV fondono con etichettati t-SUV, la densità della superficie fluoroforo nella membrana ora combinato si riduce e il conseguente aumento della fluorescenza NBD-PE riporta la portata di lipidi miscelazione 10. Come il saggio di massa è facile da configurare e analizzare, è stato ampiamente utilizzato per studiare i meccanismi di SNARE mediata fusione 10-14. Tuttavia, ha diverse limitazioni, come la bassa sensibilità e risoluzione temporale poveri. Ancora più importante, come una misura insieme, è in media risultati per tutti gli eventi che fanno discriminazione tra attracco e la fusione, così come il rilevamento di intermedi hemifusion difficili.
Negli ultimi dieci anni diversi gruppi, compreso il nostro, hanno sviluppato nuovi metodi per monitorare gli eventi di fusione a singolo livello di vescicole 15-27. Ha e colleghi hanno utilizzato v-SUV impastoiati su una superficie e monitorato la loro fusione con i libero t-SUV 18,19. Lipid miscelazione è stata monitorata utilizzando FRET tra una coppia di fluorofori lipidi-bound emletti nel V e t-SUV, rispettivamente, utilizzando la microscopia totale 18 di riflessione a fluorescenza interna (TIRF). Più tardi, il laboratorio di Brunger utilizzato una singola specie di lipidi-label insieme con un pennarello contenuti per la rilevazione simultanea di lipidi e contenuti miscelazione 20,28. Sia i lipidi e gli indicatori contenuti sono stati inclusi a concentrazioni elevate, auto-spegnimento; fusione con SUV senza etichetta portato a fluorescenza dequenching 20,28.
Altri hanno fuso v-SUV di doppi strati planari ricostituiti con t-SNARE 15-17,21-27,29. La geometria planare del target (t-SNARE contenente) imita doppio strato meglio il processo di fusione fisiologica di piccole vescicole molto curve con una membrana piatto al plasma. Il gruppo Steinem impiegato membrane poro-spanning ricostituiti con t-SNARE, sospesi su un substrato di nitruro di silicio poroso e rilevata la fusione con i singoli v-SUV con laser confocale microscopia a scansione di 23. altri Fusato v-SUV a doppi strati planari ricostituiti con t-SNARE, supportati su un substrato di vetro 15-17,21,22,24-27,29. Il grande vantaggio di utilizzare bistrati supportati (SBLs) è che microscopia TIRF può essere utilizzato per rilevare attracco e fusion eventi con un eccellente rapporto segnale-rumore e senza interferenze da trasporto V-SUV, anche se con microfluidica offre anche la risoluzione single-evento utilizzando standard di campo lontano epifluorescenza 24.
Una delle principali preoccupazioni è se e come le interazioni substrato-doppio strato incidere supportati qualità doppio strato e il processo di fusione. I primi lavori ha fatto uso di SBLs planari che sono stati direttamente supportati su un substrato di vetro o quarzo 15-17. Queste SBLs sono state fatte da adsorbimento, di rottura, la diffusione e la fusione delle membrane t-SUV sul substrato. E 'stato ben presto si rese conto, tuttavia, che omettendo una componente chiave t-SNARE, SNAP25, da SBLs preparati in questo modo portato a v-SUV di docking e fusion cinetica indistinguisHable da quelli ottenuti con il completo t-SNARE 17. Perché SNAP25 è assolutamente necessaria per la fusione in vivo 30,31, la rilevanza fisiologica di questi primi tentativi è stata messa in discussione. Il gruppo di Tamm ha superato questa sfida utilizzando la formazione di doppio strato controllato meglio supportato 21. Ha usato la deposizione Langmuir-Blodgett per il libero di proteine prima illustrativo del SBL, seguita dalla fusione di che monostrato con t-SUV 21. Ciò ha provocato la fusione SNAP25-dipendente.
Per evitare potenziali artefatti associati con un doppio strato supportato direttamente su un substrato di vetro, senza necessità di utilizzare metodi di Langmuir-Blodgett, Karatekin et al. Introdotto un poli idrato (glicole etilenico) morbido cuscino, (PEG) tra il doppio strato e il substrato 24. Questa modifica ha portato anche in SNAP25-dipendente fusione 24. Doppi strati imbottiti su uno strato di polimero morbido erano stati conosciuti per meglio conservare transmembranala mobilità e la funzione delle proteine 32, ed erano stati utilizzati negli studi di fusione con i virus 33. Inoltre, doppi strati PEG sembrano mantenere una certa capacità di auto-guarigione e sono molto robusti 34,35. Innanzitutto, una frazione di catene lipidiche PEG-linked disponibili in commercio sono inclusi nella membrana t-SUV. Quando queste t-SUV scoppiano e formano un doppio strato planare su un substrato di vetro, una spazzola PEG riguarda sia volantini del doppio strato planare. Perché la formazione planare doppio strato è guidato dalla adesione delle catene PEG circostanti t-SUV sulla superficie di vetro idrofila, liposomi scoppio e la formazione di doppio strato planare sono relativamente insensibili alla composizione lipidica utilizzata. Tuttavia, quando sono compresi grandi quantità di colesterolo, aumentando le proprietà coesive dei SUV, SUV non possono scoppiare spontaneamente. Se questo è il caso, osmotici ioni d'urto o bivalenti possono essere impiegati per aiutare a planare formazione doppio strato 25.
Come accennato in precedenza, in questa approccio un pennello PEG copre entrambi i lati della planare, bistrato supportato. La spazzola di fronte al canale di flusso microfluidica aiuta a prevenire l'adesione non specifica del v-SUV in arrivo che sono di solito ricoperti da uno strato PEG. La formazione di complessi V- e t-SNARE inizia dalla membrana-distale N-Termini e procede a tappe verso i domini di membrana-prossimale 36. Per il V-SUV di interagire con il t-SBL, il V- e necessità t-SNARE N-termini a sporgere al di sopra delle spazzole PEG, che sembra essere il caso nelle condizioni del saggio. Altezza spazzola può essere adattato per studiare proteine diverse dalle SNAREs variando la densità di lipidi peghilato e la lunghezza della catena PEG 37,38. Un altro vantaggio delle spazzole PEG che coprono le superfici prossimali dei doppi strati di fusione è che imitano l'ambiente affollato delle membrane biologiche, che sono ricchi di 30.000-40.000 proteine integrali di membrana per quadrato micron 39. Proprio come le catene di PEG in questo saggio, la Repustrato proteico lsive copre membrane biologiche deve essere messo da parte per consentire il contatto tra le due doppi strati fosfolipidi per la fusione a verificarsi.
canali di flusso microfluidica sono utilizzati in questo test, in quanto offrono vantaggi unici. In primo luogo, il flusso microfluidica consente più la deposizione uniforme delle t-SUV a diffondersi e si fondono per formare il t-SBL. In secondo luogo, il volume del canale piccolo (<1 ml) minimizza il consumo campione. In terzo luogo, i piccoli volumi necessari consentono l'intero esperimento possa essere condotto sotto flusso costante. Flusso rimuove debolmente, presumibilmente non specifico, aderito v-SUV dal SBL 16. Mantiene inoltre una densità costante di v-SUV sopra la t-SBL, semplificando l'analisi cinetica 17. Infine, vescicole attraccate sono facilmente distinguibili da quelli gratuiti effettuate dal flusso 25. Quarto, diversi canali microfluidici possono essere utilizzati sullo stesso vetrino, ognuna sondando una condizione diversa. Ciò permette il confronto delle condizioni during stesso periodo sperimentale. Un approccio simile è stato utilizzato dal gruppo van Oijen per studiare la fusione tra il virus influenzale e SBLs imbottiti 33.
In microscopia TIRF, il decadimento esponenziale del campo evanescente (con una costante di decadimento ~ 100 nm) limita eccitazione di fluorescenza a quelle molecole che sono in stretta vicinanza dell'interfaccia di vetro-buffer. Questo minimizza il contributo di molecole fluorescenti che sono più lontani, aumenta il rapporto segnale-rumore, e permette sensibilità singola molecola con tempi di esposizione telaio di 10-40 msec. Il campo evanescente comporta anche un aumento del segnale dopo la fusione: il trasferimento lipidi etichettata SUV nel SBL, si trovano, in media, in un campo di eccitazione più forte. Questo aumento della fluorescenza è più forte per liposomi grandi.
Se la luce polarizzata viene utilizzato per generare il campo evanescente, effetti aggiuntivi contribuiscono a variazioni nella fluorescenza upon transfer di etichette dal SUV nella SBL. Alcuni coloranti lipidi hanno un dipolo di transizione orientato con un angolo medio preferito rispetto al doppio strato in cui sono inserite. Questo crea una differenza nella quantità di fluorescenza emessa da fluorofori quando sono in SUV rispetto al SBL, poiché il fascio polarizzato ecciterà coloranti nei due membrane diverso. Per il primo, il fascio di eccitazione interagirà con dipoli di transizione orientate in tutto il SUV sferica, mentre per il secondo, orientamenti dipolo saranno limitati dalla geometria SBL piatta. Ad esempio, quando si utilizza s-polarizzata luce incidente (polarizzata normale al piano di incidenza), eccitazione è più efficace quando il colorante è nella SBL rispetto al SUV per un colorante lipidi transizione dipolo orientato parallelamente alla membrana 29,40 (come quella di DII o DID 41-43). Un SUV drogato con una tale fluoroforo appare scura quando esso si fissa su l'SBL (Figura 7, Representat ive risultati). Come un poro di fusione apre e collega le membrane SUV e SBL, sonde fluorescenti diffondono nel SBL e diventano più probabilità di essere eccitato dal campo evanescente s-polarizzati 25,27,29. Di conseguenza, il segnale di fluorescenza integrato intorno al sito di fusione aumenta bruscamente durante il trasferimento di colorante dal SUV nella SBL 27 (Figura 3 e Figura 7). Un ulteriore fattore che contribuisce alle variazioni del segnale che accompagnano la fusione è dequenching di etichette fluorescenti come vengono diluiti quando trasferito nella SBL. Il contributo di dequenching è generalmente minore rispetto al campo di decadimento e di polarizzazione effetti evanescenti nel test qui descritto, perché solo una piccola frazione (  ) Dei lipidi sono etichettati.
) Dei lipidi sono etichettati.
L'aumento del segnale dopo fusione può essere sfruttata per dedurre proprietà poro di fusione confrontando il tempo,1 "src =" / files / ftp_upload / 54349 / 54349eq2.jpg "/>, richiesto per un lipide di fuggire attraverso un poro che è liberamente permeabile ai lipidi per il tempo di rilascio effettivo,  . Se i due scale temporali sono comparabili, sarebbe concludere che il poro presenta poca resistenza al flusso dei lipidi. Tuttavia, se il tempo di rilascio effettivo è notevolmente più lungo del tempo di immissione a diffusione limitata, ciò indica un processo, come pori sfarfallio, ritardando il rilascio dei lipidi. Il tempo di rilascio di diffusione limitata,
. Se i due scale temporali sono comparabili, sarebbe concludere che il poro presenta poca resistenza al flusso dei lipidi. Tuttavia, se il tempo di rilascio effettivo è notevolmente più lungo del tempo di immissione a diffusione limitata, ciò indica un processo, come pori sfarfallio, ritardando il rilascio dei lipidi. Il tempo di rilascio di diffusione limitata,  , Dipende dalle dimensioni del liposoma fusione e lipidi diffusività; la stima richiede questi due parametri da quantificare. Il singolo sensibilità molecola del test permette di lipidi diffusività da misurare, monitorando diversi fluorofori singolo lipidi dopo la loro immissione in SBL per ogni evento di fusione 26. Le dimensioni di ogni vescicole fusionepuò essere stimato 27 combinando (i) l'intensità di un singolo colorante lipidi, (ii) la variazione di fluorescenza totale circa una docking sito dopo tutti fluorofori sono trasferiti nel SBL sulla fusione, (iii) la nota densità etichettatura dei SUV lipidi, e (iv) la superficie per lipidica. Per molti eventi di fusione, l'attuale tempi di rilascio dei lipidi sono risultati essere molto più lento di quanto previsto dal rilascio diffusione controllato 27, come è stato notato in precedenza assumendo dimensioni SUV uniforme 44. Assumendo il ritardo del rilascio di lipidi è dovuta a pori sfarfallio, un modello quantitativo permette di stimare "apertura dei pori", la frazione di tempo poro rimane aperta durante la fusione 27.
, Dipende dalle dimensioni del liposoma fusione e lipidi diffusività; la stima richiede questi due parametri da quantificare. Il singolo sensibilità molecola del test permette di lipidi diffusività da misurare, monitorando diversi fluorofori singolo lipidi dopo la loro immissione in SBL per ogni evento di fusione 26. Le dimensioni di ogni vescicole fusionepuò essere stimato 27 combinando (i) l'intensità di un singolo colorante lipidi, (ii) la variazione di fluorescenza totale circa una docking sito dopo tutti fluorofori sono trasferiti nel SBL sulla fusione, (iii) la nota densità etichettatura dei SUV lipidi, e (iv) la superficie per lipidica. Per molti eventi di fusione, l'attuale tempi di rilascio dei lipidi sono risultati essere molto più lento di quanto previsto dal rilascio diffusione controllato 27, come è stato notato in precedenza assumendo dimensioni SUV uniforme 44. Assumendo il ritardo del rilascio di lipidi è dovuta a pori sfarfallio, un modello quantitativo permette di stimare "apertura dei pori", la frazione di tempo poro rimane aperta durante la fusione 27.
Ogniqualvolta pratica, è importante testare meccanismi di fusione utilizzando sia lipidi e contenuto solubili etichette. Ad esempio, il rilascio dei lipidi potrebbe essere ritardato con metodi diversi da pori tremolante, come la restrizione di lipidi diffusione dalle proteine SNARE che circondano tegli poro. Se così fosse, quindi rilasciare dei contenuti sarebbe precedere supporto di etichette lipidi, purché il poro è sufficientemente grande da consentire il passaggio di sonde solubili. Un difetto più fondamentale in approccio potrebbe essere nella ipotesi che il trasferimento dei lipidi etichettati al SBL avviene attraverso uno stretto poro di fusione collega la SBL ad una vescicola che ha sostanzialmente mantenuto la sua forma pre-fusione. Trasferimento dei lipidi nel SBL potrebbe anche derivare da una rapida dilatazione del poro di fusione con un concomitante, estremamente rapido collasso del SUV nella membrana SBL, come suggerito in precedenza sulla base dei dati di rilascio dei lipidi solo 29. Monitoraggio sia lipidi e contenuto rilasciare contemporaneamente, si è constatato che molti pori richiuse dopo aver rilasciato tutte le loro etichette di lipidi, ma mantenuto alcune delle loro carico solubile 27. Questo indica che almeno alcuni liposomi non collassano nel SBL dopo la fusione, e che il trasferimento di colorante dei lipidi nel SBL avviene attraverso un poro di fusione. Inoltre, lIPID e contenuto liberazione è avvenuta contemporaneamente 27, il che rende improbabile che il ritardo del rilascio di lipidi è dovuto alla impedimento di lipidi diffusione dalle proteine SNARE che circondano il poro 45.
Un protocollo di fusione SUV-SBL che non monitorare il rilascio contenuti solubili è stato precedentemente pubblicato da Karatekin e Rothman 25. Qui, gli sviluppi più recenti sono incluse, il monitoraggio e cioè simultaneo di lipidi e contenuti rilascio e la stima delle proprietà SUV, lipidi, e poro di fusione 27. Il protocollo inizia con le istruzioni per la preparazione delle cellule microfluidica, realizzati da incollaggio un poli (dimetil silossano) (PDMS) elastomero blocco contenente scanalature con un vetrino di vetro 25. Successivamente, la preparazione di v-SUV con sia lipidi e contenuto marcatori è spiegato. Sezioni 4 e 5 forniscono istruzioni per l'assemblaggio delle celle microfluidica, formando le SBLs in situ e il controllo di difetti e la fluidità, l'introduzione di vSUVS nelle celle di flusso e il rilevamento degli eventi di fusione. Sezione 6 fornisce le istruzioni per l'analisi dei dati.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. Preparazione di un PDMS blocco per formare il canale Microfluidic

Figura 1. Microfabbricazione del modello di cella di flusso e la preparazione del blocco PDMS. (A) Progetto di un cella di flusso a quattro canali che si inserisce su un vetrino di vetro 24 x 60 mm (in basso). Sei disegni identici sono disposti ad adattarsi su un wafer di silicio 10 cm (in alto). (B) Tagliare blocco di PDMS spessore di circa 5-8 mm su una perforatrice. (C) Inserimento del tubo nel foro perforato con un paio di pinzette. Clicca qui per vedere una versione più grande di questa figura.
- Ottenere un modello di cella di flusso come quella nella Figura 1A. canali tipici sono larghe 0,3-1 mm, alta 70-100 micron e lunghezza 1-2 cm. Realizzare un modello di noiing tecniche di fotolitografia standard, in una struttura di camera bianca 25 o ordine se l'accesso camera sterile non è disponibile. In alternativa, la macchina un modello con grandi dimensioni da un materiale adatto.
Nota: il personale del locale senza polvere può formare e guidare gli utenti inesperti (l'accesso a un impianto di camera bianca è sempre limitato agli utenti con una formazione adeguata) nella progettazione e ordinamento di una maschera, pulizia di wafer, e fotolitografia. Una volta che un modello viene ottenuta, può essere utilizzato al di fuori della camera bianca volte, se conservato in un piatto chiuso e si ha cura di eliminare la polvere. - Preparare una miscela di ~ 100 ml PDMS (silicone elastomero di base) e ~ 10 ml di cross-linker (induritore) in un bicchiere di plastica usa e getta. Spezzare la punta di una pipetta monouso per gestire i PDMS viscosi più facilmente. Pesare il PDMS nella tazza di plastica come pipettaggio non è preciso.
- Mescolare bene il composto. Rimuovere le numerose bolle d'aria di degasaggio in un essiccatore sotto vuoto (circa 20min). Poiché la coppa viene posto sotto vuoto, le bolle inizialmente crescere in dimensioni, aumentando il volume della miscela notevolmente. Controllare il vuoto che viene applicato e assicurarsi che la coppa è profondo abbastanza per evitare una fuoriuscita.
- Versare una grande goccia della miscela degasato PDMS in una piastra di Petri in vetro (150 mm x 20 mm) e premere il wafer sui PDMS con il modello rivolto verso l'alto. Questo consente di evitare bolle d'essere intrappolato sotto il wafer. la formazione di bolle si espande e possono inclinare il wafer quando il piatto viene posto in forno. Ora versare PDMS degassati sulla dima sulla parte superiore della fetta fino è coperto da circa 5-8 mm di PMDS. Se una bolla d'aria si verifica rimuovere delicatamente con un puntale.
- Cuocere i PMDS in forno a 60 ° C per 3 ore. Assicurarsi che il piatto è di livello.
- Utilizzare una nuova lama di bisturi per tagliare un blocco PDMS che contiene le strutture di canale stampati. Il blocco di tagliare fuori ha bisogno di adattarsi su un vetrino (vedi punto 4.1.9).
- Sbucciare fuori il blocco tagliato fuori dal piatto e place è su un pezzo di foglio di alluminio pulito.
Nota: I blocchi reticolati PMDS possono essere conservati per un paio di mesi. Su un singolo wafer, ci sono 6 gruppi di canali di flusso (Figura 1A), in modo da 6 blocchi PDMS può essere prodotto in un unico stampaggio PDMS. Dopo il taglio di tutti e 6 i blocchi PDMS, pulito pezzetti di PDMS, ma per il resto non rimuovere i rimanenti PDMS prima di versarlo e cross-linking la prossima partita, che richiederà solo circa la metà dei PDMS utilizzati per il primo lotto. Un buon modello può durare alcuni anni. - Utilizzare una perforatrice per perforare attraverso il blocco PDMS in un unico movimento rettilineo (Figura 1B). Avviare sul lato delle scanalature canale. Assicurarsi di rimuovere il pezzo perforato di PDMS. Ripetere questa operazione per tutti gli otto fori del disegno a quattro canali.
- Posizionare il lato canale di blocco PDMS giù su un nuovo e senza rughe foglio di alluminio. Conservare il blocco fino a pochi mesi in una scatola asciutta, idealmente in un essiccatore.
- Inserire il tubo (0,25 mm di diametro 0,76 mm di diametro) circa un terzo nel foro punzonato usando un paio di pinzette (Figura 1C). Tagliare il tubo inclinato per facilitare l'inserimento. Lasciare tubi abbastanza lungo per raggiungere il serbatoio SUV e la pompa a siringa, rispettivamente. Dopo aver posizionato il chip montato sul microscopio, tagliare i tubi di nuovo se sono troppo lunghi.
- Per connettersi a le siringhe della pompa, tagliare un breve pezzo di grandi tubi in silicone (ID 0,51 millimetri, 2,1 millimetri OD) e inserire il tubo più sottile in un lato. Questo blocco pronto per l'uso di PDMS può essere conservato per alcuni mesi.
2. Coverslip Pulizia
- Coprioggetti puliti secondo il protocollo descritto in Karatekin e Rothman 25 usando un forte ossidante miscela di acido solforico (H 2 SO 4) e perossido di idrogeno (H 2 O 2). Eseguire questa procedura in una camera sterile, ed osservare le misure di sicurezza adeguate. In alternativa, utilizzare una camera bianca pulita coverslip che è disponibile in commercio (vedi Materiali List).
3. Preparazione del v-SUV che contengono sia lipidi e le etichette di contenuti

Figura 2. schematiche di preparazione SUV. I lipidi vengono miscelati in un tubo di vetro (1) e solvente viene evaporato per formare un film lipidico ruotando la provetta in un bagno d'acqua (2). Rimanenti tracce di solvente vengono rimossi sotto alto vuoto (3). Il film lipidico è idratata in tampone di ricostituzione contenente detergente e proteine, mentre in agitazione (4). Se il contenuto colorante è da incapsulare, è incluso in questo passaggio e nel passaggio di diluizione (5). Diluizione della concentrazione del detersivo di sotto della sua concentrazione micellare critica porta alla formazione di liposomi. Detergente è dializzata via durante la notte (6). Per T-SUV per la formazione SBL compresi NBD-PE (verde), vescicole sono galleggiare in un gradiente di densità e raccolti al'interfaccia tra due strati (7a). Per separare v-SUV con pennarello contenuti incapsulato dal colorante libero il campione viene eseguito su una colonna dimensione-esclusione e raccolto in 0,5 ml frazioni (7b). Clicca qui per vedere una versione più grande di questa figura.
- Preparare 4 L di tampone di ricostituzione, contenente 25 mM HEPES-KOH, 140 mM KCl, 100 mM EGTA e 1 mM DTT, pH 7,4. Utilizzare la maggior parte del buffer per dialisi e 100 ml per le altre fasi.
Nota: Altri tamponi possono essere utilizzati, ma è importante mantenere l'osmolarità del buffer costante durante l'esperimento e selezionate condizioni in cui le proteine sono funzionali. Per la preparazione di v-SUV che contengono solo le etichette di lipidi e t-SUV, seguire Karatekin e Rothman 25. - Dato che gli stock di lipidi vengono conservati a -20 ° C in cloroformio o cloroformio: metanolo (2: 1, v / v), lascia flaconi a temperatura ambiente prima aperturaevitare la condensa.
- Risciacquare un tubo di vetro con cloroformio per rimuovere tracce di detersivo e mescolare i lipidi al rapporto desiderato con una quantità finale di 1 mmol in una miscela di cloroformio e metanolo (2: 1, v / v). Utilizzare solo siringhe di vetro / tubi per gestire solventi organici / lipidi disciolti.
Nota: I lipidi utilizzati qui sono POPC: SAPE: DOPS: Colesterolo: PEG2000-DOPE: fatto a un rapporto molare di 57.4: 15: 12: 10: 4,6: 1 per v-SNARE contenenti vescicole e POPC: SAPE: DOPS: Colesterolo : PI cerebrale (4,5) P 2: PEG2000-DOPE: NBD-PE (54,9: 15: 12: 10: 3: 4,6: 0,5) per t-SNARE SUV. Vedere il materiale per i dettagli. Altre composizioni lipidiche possono essere usati. - Si evapora il solvente sotto leggera corrente di azoto, o in un evaporatore rotante. Immergere la punta del tubo in un bagno d'acqua (37 ° C) riscaldato oltre la più alta temperatura di fusione dei lipidi per produrre un film lipidico omogeneo ed evitare variazioni significative di temperatura evaporando il solvente. Inizia il vuoto intorno300 mbar fino a formazione del film lipidico, poi proseguire per ~ 2 min a più alto possibile vuoto nell'evaporatore rotante.
- Rimuovere il tubo di vetro e avvolgerla in un foglio di alluminio per evitare l'esposizione alla luce e rimuovere le tracce di solvente ponendo la provetta in un essiccatore sotto vuoto spinto per almeno 2 ore. Questo passaggio può anche eseguire durante la notte.
- Per reidratare il film lipidico essiccato, preparare una miscela di detergente (n-ottil-β-D-glucopiranoside, OG), proteine (v-SNARE), e SRB nel tampone di ricostituzione (500 microlitri volume finale). Mantenere la concentrazione di detergente finale ~ 2 volte al di sopra della concentrazione critica micellare (CMC, 20-25 mm per OG) e regolare il volume finale in modo tale che il detergente al rapporto di lipidi è> 10. Sciogliere solforodamina B (SRB) in polvere nella soluzione proteica detergente per ottenere una concentrazione finale di 10-50 mM SRB agitando delicatamente la soluzione. Regolare la osmolarità del tampone di ricostituzione quando si aggiunge alte concentrazioni di SRB riducendo il concentration di cloruro di potassio conseguenza.
Nota: il rapporto Lipid-to-proteine (LP) per t-SNARE è alta (LP ~ 20.000, ~ 70 t-SNARE al mq micron. 25). Significativamente più alta densità di t-SNARE nel SBL può portare ad aggregati proteici inattive con ridotto significativamente i tassi di fusione 17,24. LP per v-SNARE è molto più bassa (LP ~ 200 ~ 7000 v-SNARE per micron 2), vicino alle densità v-SNARE presenti sulla vescicole sinaptiche 46,47. Per garantire la funzione delle proteine dopo l'espressione ricombinante e purificazione di V- e t-SNARE è utile eseguire un semplice test di fusione di massa 48 prima di passare attraverso tutte le fasi del test singola fusione delle vescicole. - Agitare delicatamente il tubo di vetro pre-riscaldato contenente il film lipidico secca durante l'aggiunta della soluzione proteica detergente-SRB dal punto 3.6. Cercate di evitare la creazione di bolle. Continuare ad agitare per 15 minuti a 37 ° C.
- Diluire il detergente quintuplicata con l'aggiunta di ricostituzionetampone contenente SRB (aggiungere 2 ml, 2,5 ml di volume finale), mentre rapidamente vortex per evitare gradienti di concentrazione. Continuare ad agitare per 15 minuti a 1 ora a 37 ° C.
Nota: Longer aumento di incubazione di efficienza proteica ricostituzione. La rapida diluizione diminuisce la concentrazione di detersivo sotto CMC e porta alla formazione di piccoli liposomi. - Dializzare sospensione vescicola prima contro ~ 1 L di tampone di ricostituzione per 1-2 ore a temperatura ambiente e poi contro 3 L di tampone di ricostituzione notte a 4 ° C con 4 g di polistirene adsorbente, utilizzando un 20.000 MWCO tubo dialisi o cassette. Utilizzare diversi bicchieri per la dialisi di vescicole con e senza SRB per evitare la contaminazione incrociata.
- Equilibrare una colonna di gel filtrazione con tampone di ricostituzione. Eseguire la sospensione delle vescicole attraverso la colonna per separare SRB libero dal V-SUV con SRB incapsulato. Utilizzare tampone di ricostituzione senza SRB come eluente una volta l'intero campione haentrato colonna. Raccogliere v-SUV in 0,5 ml frazioni.
- Verificare successo incapsulamento SRB a concentrazioni autonomi quenched misurando SRB fluorescenza prima e dopo l'aggiunta di detersivo da una aliquota di vescicole 16. Usando uno spettrometro a fluorescenza, eccitare il campione a 550 nm e digitalizzare l'emissione SRB tra 570 nm e 630 nm. Aspettatevi una piega aumento 4-8 in SRB fluorescenza a seconda della quantità incapsulato in seguito all'aggiunta di detergente, la membrana è solubilizzato e la SRB rilasciato è diluito.
- Caratterizzare il recupero dei lipidi e delle proteine usando la spettroscopia di fluorescenza 24 e SDS-PAGE elettroforesi su gel, rispettivamente. Utilizzare un metodo di colorazione sensibile, soprattutto per i campioni t-SUV di alta LP (vedi Lista dei materiali). Tipicamente circa il 50% sia in ingresso lipidi e proteine viene perso durante la preparazione conseguente LP prossima al valore nominale.
- Caratterizzare dimensioni SUV che utilizzano la luce dinamica dispersione 24 o elettrone microscopy 48. SRB negozio contenente v-SUV a 4 ° C fino a ~ 3-4 giorni. Non congelare, come congelamento e scongelamento rompe la membrana e rilascia SRB incapsulato.
4. SUV-SBL Fusion test per il monitoraggio dei lipidi di uscita Solo
- La formazione del doppio strato supportato ancorato all'interno dei canali di flusso microfluidica
- Posizionare il blocco PDMS (passo 1.11) sotto alto vuoto per almeno 20 minuti prima dell'esperimento per rimuovere gas disciolti. Questo riduce notevolmente il rischio di bolle d'aria nei canali microfluidici durante l'esperimento di fusione.
- Accendere la configurazione microscopio e riscaldare la fase e portacampioni (figure 3 e 4) alla temperatura desiderata.
- Filtrare il tampone di ricostituzione utilizzato per diluire le vescicole attraverso un filtro con 0,45 micron o inferiore dimensione dei pori.
- Diluire ~ 30 ml di NBD-PE etichettati T-SUV o privi di proteine (PF-SUV) liposomi di controllo (magazzinosoluzione 0,5-1 mM lipidi) con ~ 60 ml di tampone. La concentrazione finale non è critico qui.
- Degas Questa miscela utilizzando una siringa da 3 ml. Premere fuori la maggior parte dell'aria sopra il campione tenendo la siringa verticale. Sigillare la punta (ago libero) con film di paraffina e creare un vuoto tirando verso il basso il pistone. Premere il corpo della siringa per accelerare degasaggio della soluzione. Ripetere questa operazione un paio di volte fino a quando non si verificano più bolle quando si applica il vuoto.
- Utilizzare un ago ipodermico con un diametro leggermente maggiore del tubo che è collegato al blocco PDMS per perforare un foro nel tappo di una provetta. Assicurarsi che il pezzo punzonato di plastica non è all'interno del tubo come potrebbe intasare il tubo collegato all'ingresso del canale microfluidico tardi.
- Riempire la soluzione degasata SUV nella provetta avente un foro nel tappo e posizionarlo nel supporto sul palco microscopio per equilibrare la temperatura impostata.
- Place un vetrino precedentemente puliti (sezione 2) in un pulitore plasma e correre plasma ad aria per circa 5 minuti. Mettere il vetrino trattati al plasma (lato trattato rivolto verso l'alto) sulla parte superiore di alcuni tessuti privo di lanugine che serve come un cuscino.
- Rimuovere il foglio di alluminio dal blocco PDMS degasata e posizionare il blocco sulla parte superiore del coprioggetto. Quando riutilizzando il blocco PDMS mettere e staccare un pezzo di nastro adesivo al lato del canale per pulirlo. Premere il PDMS bloccare sul vetrino utilizzando un paio di pinzette per farlo aderire, ma non premere troppo duro come il vetro potrebbe rompersi.
- Posizionare la cella di flusso montato sul palco microscopio e collegare il tubo al serbatoio SUV e la pompa a siringa, rispettivamente. Nastro il vetrino sul palco (Figura 3B).
- Inizia aspirazione SUV a 3 ml / min fino a che la soluzione riempie completamente i canali (~ 2,25 millimetri / sec per il x 300 micron canale sezione 75 micron). Quando le soluzioni per tutti i canali iniziano a muoversi fino esimoe tubo sul lato di uscita, ridurre la portata di 0,5 ml / min e incubare per 30-45 min.
Nota: Il tempo tra plasma trattare il vetrino e fluente SUV nel canale non deve superare i 10-20 min come l'effetto del trattamento al plasma è transitorio. - Controllare i canali per eventuali perdite. Utilizzare un obiettivo d'aria 10-20x di osservare la fluorescenza NBD-PE dal NBD-PE contenente T o PF-SUV. Excite NBD fluorofori usando il laser a 488 nm. Le perdite sono difficili da rilevare con l'illuminazione in campo chiaro.
- Mettere un tubo con tampone di ricostituzione degasato nel supporto e lasciare che la temperatura equilibrare i cambiamenti più velocemente di temperatura potrebbe causare difetti nel SBL. Arrestare il flusso ed attendere ~ 1 min per assicurarsi che il flusso si è fermato completamente e bolle d'aria verrà aspirata nel tubo prima di passare i tubi di ingresso al buffer degasato.
- Risciacquare tutti i canali con tampone degasato per lavare via i SUV non legati.
- Passare a un higher obiettivo di ingrandimento TIRF (60X, olio, NA 1,45-1,49) e verificare che il doppio strato appare omogeneo ed è privo di difetti evidenti su larga scala, come macchie scure o tubuli di lipidi che si estendono fuori dal SBL.
- Controllare la fluidità del bistrato
- Se un'unità FRAP dedicato non è disponibile, o se una sequenza FRAP non può essere programmato, quindi qualitativamente fluidità di membrana prova come segue.
- Chiudere il diaframma di campo per una piccola dimensione (~ 40 micron diametro) e regolare l'intensità della luce di eccitazione 488 nm attraverso il software (20-80? W, o 15-60 NW / micron 2) per sbiancare la fluorescenza NBD-PE in vista Area significativamente, ma non completamente. Per un doppio strato di fluido, a regime, l'intensità di fluorescenza nel mezzo dell'area esposta dovrebbe essere inferiore ai bordi, come molecole NBD-PE intatto entrano nell'area esposta e diffondono una certa distanza prima sbianca. Al contrario, se la superficie aderito SUV non è riuscito a scoppiare, o per qualche altra ragione il doppio strato supportato non è fluida, tutte fluorofori nella zona esposta deve candeggina.
Nota: valori di intensità laser in questo e successive operazioni sono come punto di partenza ruvida e dovrebbe essere ottimizzato per un dato insieme di condizioni. - Fermare l'illuminazione e iniziare di nuovo un paio di minuti più tardi per verificare i risultati delle misurazioni stato stazionario
- Chiudere il diaframma di campo per una piccola dimensione (~ 40 micron diametro) e regolare l'intensità della luce di eccitazione 488 nm attraverso il software (20-80? W, o 15-60 NW / micron 2) per sbiancare la fluorescenza NBD-PE in vista Area significativamente, ma non completamente. Per un doppio strato di fluido, a regime, l'intensità di fluorescenza nel mezzo dell'area esposta dovrebbe essere inferiore ai bordi, come molecole NBD-PE intatto entrano nell'area esposta e diffondono una certa distanza prima sbianca. Al contrario, se la superficie aderito SUV non è riuscito a scoppiare, o per qualche altra ragione il doppio strato supportato non è fluida, tutte fluorofori nella zona esposta deve candeggina.
- Programmare una sequenza FRAP per una misurazione più quantitativa, se possibile. Vedere file supplementari e la relativa leggenda per i dettagli.
Nota: A volte SUV aderiranno sul vetrino di vetro, ma non riescono a scoppiare e formare un doppio strato fluido. In questo caso, sciacquare i canali con tampone di ricostituzione degasato contenente 10 mM Mg 2 + per aiutare la formazione di doppio strato sostenuta. Utilizzare fluorescenza sbiancante per valutare doppio strato fluidità come in 4.2. Una volta che un doppio strato di fluido è formato, sciacquare con Mg 2+-free recoBuffer nstitution.
- Se un'unità FRAP dedicato non è disponibile, o se una sequenza FRAP non può essere programmato, quindi qualitativamente fluidità di membrana prova come segue.
- L'introduzione di v-SUV nei canali di flusso microfluidica
- Degas Tampone di ricostituzione e utilizzarlo per diluire la soluzione madre v-SUV di un fattore di circa 10 MARZO - 10 maggio a seconda della concentrazione stock v-SUV. Obiettivo per una diluizione che si traduce in circa 10-100 eventi di fusione entro 60 secondi nel campo visivo.
Nota: Troppi fusioni aumentare la fluorescenza di fondo (dal momento che ogni depositi eventi LR-PE o ha etichette di lipidi nel SBL) e rendere la rilevazione e l'analisi di eventi di fusione difficile. Al contrario, un tasso di fusione che è risultato troppo basso in cattive statistiche o richiede l'acquisizione di molti altri film. Per una concentrazione di v-SUV di 0,1 mm lipidica, avviare diluendo 5 ml SUV stock in tampone di ricostituzione 995 ml e quindi diluire 5-50 ml di questa in 950-995 ml di buffer di ricostituzione. - Lasciate che la temperatura equilibrare prima di fermare il flusso e inserting il tubo di ingresso nella soluzione v-SUV diluito.
- Regolare l'angolo TIRF e polarizzazione come segue.
- Dopo aver impostato la polarizzazione desiderata ruotando il fascio di eccitazione, regolare l'inclinazione dello specchio comandare la posizione del fascio tramite un motore passo-passo attraverso il software. Lentamente spostare la posizione del fascio laser dal centro dell'obiettivo al piano focale posteriore di lato fuori centro. Osservare la luce dall'obiettivo sul lato anteriore emerge con un angolo crescente rispetto all'asse oggettiva la posizione viene spostato ulteriormente fuori centro.
- Nota la posizione del motore quando il fascio uscente scompare prima nell'obiettivo, cioè quando TIR è raggiunto per primo.
- Lentamente continuare a muoversi la posizione del raggio ulteriormente fuori centro monitorando la fluorescenza dalla superficie. Nota la posizione del motore quando la fluorescenza della superficie scompare quando il fascio è spostato troppo fuori centro.
- Scegliere un fascio positio n tra i due limiti sopra determinato. Per una migliore rapporto segnale-rumore e più miglioramento del segnale sulla fusione, scegliere una profondità di penetrazione superficiale (posizione del fascio più vicino al bordo dell'obiettivo) che fornisce ancora un'illuminazione uniforme della Viewfield.
Nota: Si consiglia di mantenere la stessa posizione del fascio TIR (stessa profondità di penetrazione) per tutti gli esperimenti dopo le impostazioni sono ottimizzate. Assicurarsi che ruotando la polarizzazione non comporta cambiamenti nella posizione del fascio.
- Flusso v-SUV nel canale ad un flusso di 2 ml / min, corrispondente ad una velocità di flusso lineare media di circa 1,5 mm / sec per una sezione trasversale di 75 um x 300 micron. Passa alle impostazioni di eccitazione / emissione per monitorare lipidi miscelazione (LR o fatto solo).
- Degas Tampone di ricostituzione e utilizzarlo per diluire la soluzione madre v-SUV di un fattore di circa 10 MARZO - 10 maggio a seconda della concentrazione stock v-SUV. Obiettivo per una diluizione che si traduce in circa 10-100 eventi di fusione entro 60 secondi nel campo visivo.
- Osservando fusione tra singoli v-SUV e la SBL
.jpg "/>
Figura 3. L'apparato sperimentale pTIRF. (A) Rappresentazione schematica di un V-SUV e una t-SBL su un substrato di vetro. False-colore immagini TIRFM di singolo evento fusione SUV-SBL mostrano aggancio lipidi (1) e il rilascio di colorante lipidi nel SBL (2) seguito da sbianca e diminuzione dell'intensità di fluorescenza (3). Di seguito riportiamo l'intensità della fluorescenza totale (somma dei valori dei pixel in 5,3 micron x 5.3 scatola micron) del segnale. (B) il vetrino legato al blocco di PDMS è registrato sul palco riscaldato. tubo di ingresso per i canali microfluidici prelevino campioni di tubo portacampioni metallo (destra) aspirato dalla pompa siringa (a sinistra). Sotto la pompa è l'unità doppia emissione. Cliccate qui per vedere una versione più grande di questa figura.
OAD / 54349 / 54349fig4.jpg "/>
Figura 4. Schema del setup sperimentale. L'onda evanescente è creato all'interfaccia vetro-buffer nel canale microfluidica. La SBL è formato sul vetro e v-SUV vengono aspirati dal supporto campione metallico (in alto a destra) attraverso il canale nella siringa (in alto a sinistra). M, specchio; DM, specchio dicroico; L, obiettivo; F, filtro; P, polarizzatore. Clicca qui per vedere una versione più grande di questa figura.
- Continuamente eccitare e monitorare la fluorescenza v-SUV con un 561 nm (per LR-PE) o 638 nm (per DID) laser a seconda del fluoroforo incorporata in v-SUV. V-SUV raggiungono il canale di flusso e dock sopra e si fondono con il SBL, il segnale di fluorescenza di fondo inizia ad accumularsi.
- Regolare l'intensità del laser di eccitazione attraverso il software di candeggina continuamente lo sfondofluorescenza tale che a livello degli Stati nuovi eventi di docking e fusion costante possano essere prontamente rilevate.
Nota: Se lo sbiancamento è troppo lento, fluorescenza di fondo sarà troppo alto. Se lo sbiancamento è troppo veloce, i segnali provenienti da SUV attraccate e dal fluorofori rilasciati nel SBL si dissolvono rapidamente, abbreviando la finestra durante il quale la fusione di SUV attraccate può essere rilevata o un fluoroforo possono essere monitorati. Per LR-PE eccitato a 561 nm, 2,5-7,5 potere mW illuminante un cerchio 190 micron di diametro (100-250 NW / micron 2) è un valore ragionevole per iniziare. Per 638 nm di eccitazione di DID, 0,8-1,6 potere mW per un diametro del cerchio 190 micron (30-60 NW / micron 2) può essere utilizzato per i test iniziali. - Acquisire diversi film in posizioni diverse in un dato canale. Vedi fluorescenza NBD-PE per verificare che la SBL non ha difetti in queste posizioni. Acquisire filmati full-frame al tasso massimo (~ 50 fotogrammi / sec) o di una regione ritagliata di interesse a più alto frame rate (fino a~ 100 Hz) per 60 sec.
- Spostarsi in un altro canale microfluidica e ripetere le registrazioni per le altre condizioni. Include controlli negativi quali un SBL privo di proteine o SUV, aggiungere il dominio citoplasmatico solubile del v-SNARE VAMP2 (CDV) come inibitore o trattare v-SUV con il tetano neurotossina (30 min a 37 ° C) in uno o più dei canali sullo stesso vetrino.
- Pulire e riciclaggio PDMS
- Al fine di riutilizzare il blocco PDMS, lavare i canali microfluidica con ~ 200 ml di etanolo al 70% ad una velocità di flusso di 5 ml / min. Infine, aspirare aria attraverso il tubo e canali.
- Staccare il PDMS bloccano delicatamente dal vetrino premendo leggermente. Mettere su un pezzo di foglio di alluminio pulita. Conservare in un essiccatore a vuoto. Può essere riutilizzato più volte.
- Per una pulizia più accurata lavare i canali con un detergente o di sodio idrossido soluzione prima 70% di etanolo. In alternativa, rimuovere tutti i tubi fro m il PDMS e sonicare il blocco per 30 minuti in isopropanolo, prima dell'essiccazione e l'inserimento di nuovi tubi.
5. SUV-SBL Fusion test per il monitoraggio dei lipidi e contenuti di rilascio contemporaneamente
- Per il monitoraggio a due colori di lipidi simultanea e contenuti solubili rilasciare, seguire la stessa procedura come nella sezione 4, l'eccezione di usare liposomi etichettati sia con un lipide (DID) e un contenuto solubili (SRB) etichetta, come spiegato nella sezione 3. SRB è incapsulato a 10 mm ed è inizialmente molto auto-spento.
- Utilizzando la configurazione mostrata schematicamente in figura 4, eccitare SRB e ha fatto di fluorescenza contemporaneamente utilizzando 561 nm e 638 nm laser, rispettivamente. Uno specchio dicroico (640 nm) divide l'emissione in due fasci che attraversano un breve (595/50 nm) e una lunga filtro (700/75 nm) di lunghezza d'onda per rilevare la SRB e ha fatto di emissioni, rispettivamente. I due fasci di emissione vengono proiettati side-by-side nel chip EM-CCD.
- Analisi dei dati FRAP
- Utilizzare il programma MATLAB fornito nei prospetti supplementari per stimare il coefficiente di lipidi diffusione, D. Il programma legge un elenco di file OME-TIFF 49, rileva la zona sbiancato, traccia i valori medi dei pixel nell'area sbiancato in funzione del tempo, e si adatta alla curva di recupero risultante da un modello Soumpasis 50 per estrarre il tempo di recupero,
 , Dove w è il raggio del cerchio sbiancato.
, Dove w è il raggio del cerchio sbiancato.
Nota: Un programma MATLAB è stato fornito in precedenza 25 per l'analisi dei filmati FRAP che utilizzano i file Nikon ND2. L'attuale programma legge i file OME-TIFF 49, perché la maggior parte formati di file possono essere facilmente convertiti in OME-TIFF. Una analisi quantitativa dei dati FRAP è più facile e più preciso quando lo sbiancamento è istantanea, il bleacregione Hed è un cerchio, e candeggio durante la lettura-out è trascurabile. Anche se questi criteri non sono strettamente soddisfatti nelle semplici misurazioni FRAP qui descritte, una ragionevole stima del coefficiente di diffusione può essere ottenuto. Per una stima più accurata, utilizzare il monitoraggio di coloranti lipidi singola (punto 6.3).
- Utilizzare il programma MATLAB fornito nei prospetti supplementari per stimare il coefficiente di lipidi diffusione, D. Il programma legge un elenco di file OME-TIFF 49, rileva la zona sbiancato, traccia i valori medi dei pixel nell'area sbiancato in funzione del tempo, e si adatta alla curva di recupero risultante da un modello Soumpasis 50 per estrarre il tempo di recupero,
- Analisi di tasso di attracco, tasso di fusione e docking-to-fusion tempi di ritardo
- Utilizzare ImageJ per aprire un film di analizzare e regolare la luminosità e il contrasto di identificare chiaramente attracco delle vescicole e gli eventi di fusione. Avviare il plugin SpeckleTrackerJ 26. Vedere Smith et al. 26 e on-line la documentazione per SpeckleTrackerJ per le istruzioni per l'uso.
- Identificare tutti i SUV di nuova attraccate in SpeckleTrackerJ. Per distinguere i SUV che possono essere ancorati saldamente da quelle che rimbalzano l'SBL, imporre un attracco durata minima di pochi fotogrammi. Salvare le tracce, e ripetere per tutti i film.
Nota: Per il doctasso di re, tutto ciò che conta è quello di individuare quando un SUV agganciato, in modo che solo il primo fotogramma in cui i SUV ancorata deve essere registrato in queste tracce. Il resto dei brani non sarà preso in considerazione nell'analisi. Tuttavia, l'auto-monitoraggio di ogni SUV fino a quando non schiarisce o fusibili aiuta SUV marchio che sono già stati monitorati. - Identificare tutte le vescicole fusione. Per questi, le tracce dovrebbero includere tutti i fotogrammi dal primo fotogramma di un SUV agganciata fino a quando il primo fotogramma in cui la fusione è evidente da un improvviso aumento della fluorescenza della macchia cingolato. La durata di queste tracce sono utilizzati per calcolare i ritardi di docking-to-fusion. Salvare tutte le tracce. Ripetere l'operazione per tutti i film.
- Per l'analisi dei dati lotto di attracco o di fusione, compilare un elenco di file di traiettoria dai film importanti e di eseguire i programmi MATLAB forniti con Karatekin e Rothman 25. Seguire le istruzioni che vengono fornite insieme ai programmi.
Nota: I programmi di tracciare lo spermaulative attracco e fusion eventi come funzione del tempo, in base alle informazioni estratte dai file traiettoria. tassi di aggancio e di fusione sono stimati dalle piste di queste trame. Per i dati di fusione, i ritardi di aggancio-to-fusione sono calcolati e la loro distribuzione tracciati come una trama di sopravvivenza, cioè la probabilità che la fusione non è ancora avvenuta da un dato ritardo dopo aggancio.
- Lipid diffusività
- Per ogni evento di fusione, monitorare i lipidi fluorescenti singoli man mano che diventano discernibile quando hanno diffuso sufficientemente lontano dal sito di fusione (Figura 6). Utilizzare SpeckleTrackerJ per il monitoraggio, e salvare le tracce per ulteriori analisi con MATLAB. A seconda delle dimensioni della vescicola fusione, tipicamente 3-30 singoli fluorofori possono essere monitorati. Poiché brani più lunghi sono desiderabili per il calcolo
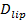 , Cercare di tenere traccia delle singole molecole come lungopossibile utilizzando correzione manuale delle traiettorie se necessario.
, Cercare di tenere traccia delle singole molecole come lungopossibile utilizzando correzione manuale delle traiettorie se necessario. - Calcolare la media al quadrato spostamento (MSD) per le singole traiettorie dei marker di lipidi che durano> 40-50 fotogrammi (circa ~ 1,5 sec). Utilizzare l'MSD per calcolare il coefficiente di lipidi diffusione, . Vedere Smith et al. 26 e Stratton et al. 27 per i dettagli.
- Per ogni evento di fusione, monitorare i lipidi fluorescenti singoli man mano che diventano discernibile quando hanno diffuso sufficientemente lontano dal sito di fusione (Figura 6). Utilizzare SpeckleTrackerJ per il monitoraggio, e salvare le tracce per ulteriori analisi con MATLAB. A seconda delle dimensioni della vescicola fusione, tipicamente 3-30 singoli fluorofori possono essere monitorati. Poiché brani più lunghi sono desiderabili per il calcolo
- Intensità colorante lipidi singolo, SUV-SBL fattore di riduzione di intensità, e le dimensioni delle vescicole
- Misurare la somma dei valori di pixel di un marcatore in un lipide 3 x 3 pixel (0,8 micron x 0,8 micron) area intorno al marcatore per ~ 15 fotogrammi prima e dopo i candeggianti marcatore in un unico passaggio. Sottrarre l'intensità bassa in media su telai post-candeggina dalla intensità pre-bleach media prima candeggio per ottenere l'intensità del marcatore lipidi cingolato. Ripetere la misurazione a molti marcatori come pratica per un determinato film.
- Tracciare la distribuzione di singolo lipidi intensità etichetta,
 , E montare una gaussiana per stimare la media.
, E montare una gaussiana per stimare la media. - Calcolare il ritardo tra quando si è verificata fusione e quando la traiettoria di un singolo marcatore lipidi rilasciato in tal caso finito in uno sbiancamento sola fase. Ottenere il tempo sbiancante per un fluoroforo nel SBL,
 , Tracciando la funzione sopravvissuto dei ritardi e il montaggio di un esponenziale.
, Tracciando la funzione sopravvissuto dei ritardi e il montaggio di un esponenziale.
Nota: Poiché i polarizzati coppie di campo di eccitazione più debolmente ai fluorofori su un SUV, il tempo di sbiancamento di un SUV è di solito molto più lento 27. - Stima
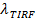 , Il fattore di riduzione dell'intensità di un colorante lipidico quando nella SUV rispetto a quando è nel SBL, seguendo Stratton 27 (tp_upload / 54349 / 54349eq8.jpg "/> è l'intensità di un singolo colorante nel SUV).
, Il fattore di riduzione dell'intensità di un colorante lipidico quando nella SUV rispetto a quando è nel SBL, seguendo Stratton 27 (tp_upload / 54349 / 54349eq8.jpg "/> è l'intensità di un singolo colorante nel SUV).
Nota: Se lo sbiancamento erano trascurabili, sarebbe uguale all'intensità ancorata  (punto (1) in Figura 3A, pannello di destra), divisa per l'intensità totale raggiunta dopo tutte fluorofori sono depositati nel SBL sulla fusione (linea tratteggiata etichettata
(punto (1) in Figura 3A, pannello di destra), divisa per l'intensità totale raggiunta dopo tutte fluorofori sono depositati nel SBL sulla fusione (linea tratteggiata etichettata  in figura 3). Tuttavia, a causa della rapida sbianca nel SBL, è in genere non raggiunto e una stima accurata dei richiede il montaggio dei cinetica di rilascio di una espressione che 27 incl udes sia (Vedi 6.4.3) e . Espressioni separate sono forniti in Stratton ed altri 27 per i casi di cinetica di poro-limitata e la diffusione versione limitata.; migliore dei casi in forma varia da un evento all'altro (vedi 6.5.1 per il caso della cinetica pori limitata).
in figura 3). Tuttavia, a causa della rapida sbianca nel SBL, è in genere non raggiunto e una stima accurata dei richiede il montaggio dei cinetica di rilascio di una espressione che 27 incl udes sia (Vedi 6.4.3) e . Espressioni separate sono forniti in Stratton ed altri 27 per i casi di cinetica di poro-limitata e la diffusione versione limitata.; migliore dei casi in forma varia da un evento all'altro (vedi 6.5.1 per il caso della cinetica pori limitata). - Calcolare l'area vescicola per i singoli eventi da 27
 , dove è l'intensità SUV ancorata, è l'intensità di un singolo colorante lipidi medio nel SBL, è il fattore di riduzione dell'intensità di un colorante lipidico quando è nella relativa SUV quando è nella SBL, ezione 1 "src =" / files / ftp_upload / 54349 / 54349eq12.jpg "/> è il noto densità areale di coloranti lipidi.
, dove è l'intensità SUV ancorata, è l'intensità di un singolo colorante lipidi medio nel SBL, è il fattore di riduzione dell'intensità di un colorante lipidico quando è nella relativa SUV quando è nella SBL, ezione 1 "src =" / files / ftp_upload / 54349 / 54349eq12.jpg "/> è il noto densità areale di coloranti lipidi.
- Proprietà poro di fusione
- Supponendo che il rilascio è pori limitata, montare la cinetica di rilascio dell'etichetta lipidi a 27
 , dove
, dove  è l'intensità ancorata SUV appena prima della fusione e gli altri parametri sono come definiti in precedenza. Utilizzare il valore di ottenuto in 6.4.3 come parametro fisso, ed estrarre le migliori stime atti e
è l'intensità ancorata SUV appena prima della fusione e gli altri parametri sono come definiti in precedenza. Utilizzare il valore di ottenuto in 6.4.3 come parametro fisso, ed estrarre le migliori stime atti e  .
. - Stima la frazione di tempo poro aperto, P 0, assumendo il ritardo del rilascio di lipidi è dovuta a pori tremolante 27:60;
 =
=  , Dove A è l'area Ves vescicole (sezione 6.4.5), b è l'altezza dei pori (tipicamente taken essere ~ 15 nm), r p ≈ 3 nm è il raggio dei pori effettiva vista dai diffusori etichette lipidi e comprende mezzo lo spessore doppio strato (~ 2 nm), è la diffusività lipidi (calcolato in 6.3), e è il momento per i lipidi per essere rilasciato dal SUV nella SBL (da 6.5.1).
, Dove A è l'area Ves vescicole (sezione 6.4.5), b è l'altezza dei pori (tipicamente taken essere ~ 15 nm), r p ≈ 3 nm è il raggio dei pori effettiva vista dai diffusori etichette lipidi e comprende mezzo lo spessore doppio strato (~ 2 nm), è la diffusività lipidi (calcolato in 6.3), e è il momento per i lipidi per essere rilasciato dal SUV nella SBL (da 6.5.1). - Per confermare che un P nominale 0> 1 indica un poro completamente aperto, P 0 = 1, montare il decorso temporale di intensità l'equazione 4 di Stratton et al. 27, la cinetica previsti per un poro aperto in modo permanente. Questa forma dovrebbe essere migliore di fitting l'espressione in 6.5.1 per un poro aperto in modo permanente.
- Supponendo che il rilascio è pori limitata, montare la cinetica di rilascio dell'etichetta lipidi a 27
 , Dove w è il raggio del cerchio sbiancato.
, Dove w è il raggio del cerchio sbiancato.  , Tracciando la funzione sopravvissuto dei ritardi e il montaggio di un esponenziale.
, Tracciando la funzione sopravvissuto dei ritardi e il montaggio di un esponenziale.  (punto (1) in Figura 3A, pannello di destra), divisa per l'intensità totale raggiunta dopo tutte fluorofori sono depositati nel SBL sulla fusione (linea tratteggiata etichettata
(punto (1) in Figura 3A, pannello di destra), divisa per l'intensità totale raggiunta dopo tutte fluorofori sono depositati nel SBL sulla fusione (linea tratteggiata etichettata  in figura 3). Tuttavia, a causa della rapida sbianca nel SBL,
in figura 3). Tuttavia, a causa della rapida sbianca nel SBL,  , dove
, dove  , dove
, dove  è l'intensità ancorata SUV appena prima della fusione e gli altri parametri sono come definiti in precedenza. Utilizzare il valore di
è l'intensità ancorata SUV appena prima della fusione e gli altri parametri sono come definiti in precedenza. Utilizzare il valore di  =
=  , Dove A è l'area Ves vescicole (sezione 6.4.5), b è l'altezza dei pori (tipicamente taken essere ~ 15 nm), r p ≈ 3 nm è il raggio dei pori effettiva vista dai diffusori etichette lipidi e comprende mezzo lo spessore doppio strato (~ 2 nm),
, Dove A è l'area Ves vescicole (sezione 6.4.5), b è l'altezza dei pori (tipicamente taken essere ~ 15 nm), r p ≈ 3 nm è il raggio dei pori effettiva vista dai diffusori etichette lipidi e comprende mezzo lo spessore doppio strato (~ 2 nm), Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Qualità SBL
È fondamentale verificare la qualità e la fluidità del SBL prima dell'esperimento di fusione. La fluorescenza sul lato inferiore, il vetro di un canale microfluidico deve essere uniforme, senza difetti evidenti. Se una bolla d'aria passa attraverso il canale, di solito lascia cicatrici visibili sulla SBL. Se ci sono così grandi cicatrici scala / difetti, non utilizzare quel canale. A volte SUV può aderire sul substrato ma non riescono a scoppiare e formare un continuo SBL. Se questo è il caso, la fluorescenza può ancora apparire molto uniforme, ma non si riprenderà dopo sbiancare una piccola regione. L'introduzione di 10 mM Mg 2+ spesso aiuta la soluzione di questo problema. Altri agenti possono essere introdotte per aiutare a scoppiare i SUV, come altri cationi bivalenti 51, una soluzione polimerica 52, o shock osmotico 25.
I cambiamenti di temperatura producono variazioni dell'area SBL perché l'area media occupata da un lipide dipende dalla temperatura. Aumentando la temperatura o diminuendolo di 5-10 ° C o più può provocare zona superiore andando in tubi estrusi dalla superficie, o scuro, macchie SBL-libere compaiono sulla superficie, rispettivamente, 53. E 'quindi importante mantenere le variazioni di temperatura al minimo.
Figura 5 per un SBL che era relativamente privo di difetti e fluida.

Figura 5. FRAP esaminare SBL fluidità. Una sequenza di immagini che mostrano un'area illuminata delimitata dalla membrana campo chiuso (diametro 49 micron) prima (primo fotogramma) e dopo candeggio con un forte impulso luminoso. L'area è ripreso a bassa esposizione alla luce di seguire il recupero di fluorescenza per diffusione di NBD lipidi etichettati. Il grafico mostra la fluorescenza normalizzata in funzione del tempo per diversi canali di diversi coprioggetti indicano il livello di riproducibilità del metodo. La composizione dei lipidi della t-SBL era POPC: SAPE: DOPS: PI cervello (4,5) P 2:: Colesterolo PEG2000-DOPE: NBD-PE (54,9: 15: 12: 10: 3: 4,6: 0,5 moli% ) e LP = 20.000. L'area di candeggina era 984.203 μm 3. Il tempo di recupero e il coefficiente di diffusione calcolata da una misura (vedi 6.1) sono stati τ = 67.3 sec e D = 1.99 micron 2 / sec, rispettivamente. Clicca qui per vedere una versione più grande di questa figura.
Rilevamento a un colore di attracco e Fusion eventi utilizzando un'etichetta lipidi
conteggio manuale di eventi di docking e fusion è un compito noioso come sono necessari 100-400 eventi per condizione di buone statistiche. Poiché SUV possono muoversi sul SBL prima di fondersi, è necessario un software di monitoraggio. SpeckleTrackerJ 26 plug-in per ImageJ ha caratteristiche che sono state appositamente sviluppate per il monitoraggio SUV per l'analisi degli eventi di docking e fusion. Spesso solo una frazione di vescicole attraccate finiscono per fusione;calcolare il tasso di espansione molti altri SUV devono essere monitorati quelli che fondono o solo una parte del film deve essere analizzato finché viene rilevato un numero sufficiente di eventi per una stima affidabile. Se l'obiettivo principale è il tasso di fusione e / o ritardi di docking-to-fusione, solo i SUV di fusione devono essere identificati e monitorati.
Dopo aver verificato la qualità del SBL, i SUV sono introdotti nei canali e la loro fluorescenza è di eccitazione continuo sotto TIR utilizzando la luce s-polarizzata. Per gli esperimenti iniziali, l'angolo di incidenza del fascio di eccitazione TIR può essere necessario regolare per regolare la profondità di penetrazione dell'onda evanescente risultante. Un rappresentante unico attracco SUV e la fusione evento è mostrato in Figura 6A. La vescicola ancorata durante l'intervallo segnato D e la fusione è iniziata nel telaio F. Circa 1,3 sec dopo (telaio S) singole lipidi è diventato riconoscibile in quanto diffusi via dalla fusione sederee. L'intensità della fluorescenza totale (somma dei valori di pixel) in ciascuna casella è tracciata in funzione del tempo nella Figura 6B, con le cornici in cui si è verificato indicati aggancio e fusione. Il numero cumulativo di eventi di fusione in funzione del tempo è tracciata nella Figura 6C, per cinque diversi film con t-SNARE ricostituita SBLs (t-SBL, LP = 20.000, circa 70 t-SNARE al mq. Micron). Il tasso di fusione da ogni film è stato calcolato da una misura lineare al pendio. Questo tasso medio di fusione ± SEM è mostrato come un grafico a barre nel pannello D. Il ritardo aggancio a fusione per l'esempio mostrato nella Figura 6A, B, era 0,07 sec. La distribuzione dei ritardi calcolati in modo simile per un totale di 158 eventi è illustrata nella Figura 6D come diagramma reversibilità, cioè la probabilità che una vescicola ancorata non ha ancora fusa da un certo ritardo. Circa 1/2 di queste vescicole fuso a 100 msec. Con l'aumento dei livelli di colesterolo, il distribution di ritardi di docking-to-fusione si accorcia, anche se la diffusione dei lipidi rallenta 27.
È importante eseguire esperimenti di controllo in parallelo. Sullo stesso coprioggetto da cui sono stati acquisiti i dati della figura 6B, D, condizioni di controllo sono stati testati in canali adiacenti. In un canale, la SBL non include alcun t-trappole. Fuori di 5 film della durata di 60 secondi ciascuno, solo 7 eventi di fusione sono stati registrati rispetto ai 158 quando le t-SNARE sono stati inclusi (Figura 6C). In un altro canale, il dominio citoplasmatica solubile del v-SNARE VAMP2 (CDV) è stato incluso. CDV lega le t-SNARE sulla SBL, impedendo vincolante del VAMP2 full-length che è sui SUV. In cinque film 60 sec, eventi di fusione sono stati rilevati in presenza di CDV (figura 6C).

Figura 6. SUV-SBL di docking e fusion eventi. (A) Sequenza di immagini di un evento di fusione v-SUV con la t-SBL imaged da pTIRF microscopio a seguito della diffusione della LR-PE. Le immagini sono prese ogni 18 msec, solo viene mostrato ogni secondo fotogramma. D e F segnano l'insorgenza di aggancio e fusione rispettivamente. Singole etichette lipidi diventano identificabili in quanto diffondono lontano dal sito di fusione e l'un l'altro (S). dimensioni del telaio: 16.7 x 16.7 micron micron. (B) l'intensità di fluorescenza in funzione del tempo per l'evento di fusione mostrato in (A). (C) il numero cumulativo di eventi di fusione in funzione del tempo per v-SUV etichettati con DID da un'altra serie di esperimenti. (D) il tasso di fusione normalizzata (media ± SEM, sx micron 2 x pM SUV) -1, assumendo 2 x 10 4 lipidi per SUV e (E) la probabilità di sopravvivenza per un tempo determinato dopo l'attracco della vescicola. La composizione per tutti SBLs erano POPC: SAPE: DOPS: PI cervello (4,5) P 2:: Colesterolo PEG2000-DOPE: NBD-PE (54,9: 15: 12: 10: 3: 4,6: 0,5 moli%) e LP = 20.000. v-SUV erano composti da POPC: SAPE: DOPS: Colesterolo: PEG2000-DOPE: LR-PE (57,4: 15: 12: 10: 4,6: 1 mole%) e LP = 200 per A e B. Per CE, la v composizione -SUV era simile, tranne che ha fatto è stato utilizzato al posto del LR-PE. clicca qui per vedere una versione più grande di questa figura.
Lipid Diffusività, vescicole dimensioni, e Fusion Pore Proprietà
La sensibilità singola molecola del test permette l'estrazione di vari parametri oltre ai tassi di aggancio e fusione e le distribuzioni di ritardo aggancio-to-fusione. Singolo, lipidi fluorescente diventano distinguibili in quanto diffondono lontano dal sito di fusione (figura 6A). monitoraggio tueste fornisce direttamente lipidi diffusività 26, 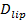 , Ed una misurazione digitale della velocità di sbiancamento per fluorofori nel SBL 27. Come menzionato nella introduzione, l'emissione di fluorescenza delle etichette differiscono in SUV rispetto al SBL, a causa di tre fattori che sono in generale difficile distinguere: (i) dequenching di etichette su fusione, a causa della loro diluizione quando trasferiti nel SBL , (ii) una intensità di eccitazione più elevato nel SBL causa del decadimento del campo evanescente generato da TIR, e (iii) l'orientazione dei dipoli fluoroforo transizione rispetto alla polarizzazione del fascio di eccitazione. L'entità del fattore (ii) dipende dalle dimensioni relative del evanescente lunghezza del campo di decadimento e la dimensione delle vescicole ed è diverso per ogni evento di fusione a causa del dispersità in dimensioni SUV. Factor (iii) dipende dal fluoroforo e la polarizzazione utilizzata. Piuttosto che cercare di di sentangle questi effetti, si può misurare la grandezza della variazione di fluorescenza direttamente confrontando i valori totali di pixel in una regione che circonda un SUV prima fusione e dopo tutte fluorofori sono stati depositati nel SBL seguito alla fusione (Figura 3A, (1) vs. ( 2)). Diversamente lavoro precedente 29,44, la regione dovrebbe essere scelto sufficientemente grande in modo che non fluorofori avranno lasciato tramite diffusione per tempi considerati dopo la fusione. La scelta di una regione di pixel 30 x 30 (8 micron x 8 micron) per l'analisi fino a 1,6 secondi dopo la fusione è di solito sufficiente 27. Ignorando sbiancante per il momento, il rapporto tra
, Ed una misurazione digitale della velocità di sbiancamento per fluorofori nel SBL 27. Come menzionato nella introduzione, l'emissione di fluorescenza delle etichette differiscono in SUV rispetto al SBL, a causa di tre fattori che sono in generale difficile distinguere: (i) dequenching di etichette su fusione, a causa della loro diluizione quando trasferiti nel SBL , (ii) una intensità di eccitazione più elevato nel SBL causa del decadimento del campo evanescente generato da TIR, e (iii) l'orientazione dei dipoli fluoroforo transizione rispetto alla polarizzazione del fascio di eccitazione. L'entità del fattore (ii) dipende dalle dimensioni relative del evanescente lunghezza del campo di decadimento e la dimensione delle vescicole ed è diverso per ogni evento di fusione a causa del dispersità in dimensioni SUV. Factor (iii) dipende dal fluoroforo e la polarizzazione utilizzata. Piuttosto che cercare di di sentangle questi effetti, si può misurare la grandezza della variazione di fluorescenza direttamente confrontando i valori totali di pixel in una regione che circonda un SUV prima fusione e dopo tutte fluorofori sono stati depositati nel SBL seguito alla fusione (Figura 3A, (1) vs. ( 2)). Diversamente lavoro precedente 29,44, la regione dovrebbe essere scelto sufficientemente grande in modo che non fluorofori avranno lasciato tramite diffusione per tempi considerati dopo la fusione. La scelta di una regione di pixel 30 x 30 (8 micron x 8 micron) per l'analisi fino a 1,6 secondi dopo la fusione è di solito sufficiente 27. Ignorando sbiancante per il momento, il rapporto tra  prima e dopo la fusione fornisce il fattore di riduzione dell'intensità di fluorescenza,
prima e dopo la fusione fornisce il fattore di riduzione dell'intensità di fluorescenza, 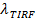 . La dimensione delle vescicole può essere stimata data l'intensità di fluorescenza di una singola etichetta nella SBL (iles / ftp_upload / 54349 / 54349eq6.jpg "/>), il fattore per il quale la sua intensità sarebbe ridotto se fosse in un SUV ( ), La densità noto di etichette in SUV, e l'area occupata da un lipide. In pratica, lo sbiancamento è di solito abbastanza veloce che per il momento tutti i marcatori sono depositati nel SBL, alcuni hanno già sbiancato. Per questo motivo, per una stima accurata, il fattore di riduzione dell'intensità di fluorescenza, , E il tempo di rilascio di lipidi,
. La dimensione delle vescicole può essere stimata data l'intensità di fluorescenza di una singola etichetta nella SBL (iles / ftp_upload / 54349 / 54349eq6.jpg "/>), il fattore per il quale la sua intensità sarebbe ridotto se fosse in un SUV ( ), La densità noto di etichette in SUV, e l'area occupata da un lipide. In pratica, lo sbiancamento è di solito abbastanza veloce che per il momento tutti i marcatori sono depositati nel SBL, alcuni hanno già sbiancato. Per questo motivo, per una stima accurata, il fattore di riduzione dell'intensità di fluorescenza, , E il tempo di rilascio di lipidi,  , Sono meglio estratto da una misura per l'andamento nel tempo dei valori dei pixel totali che tenga conto sbiancamento 27. Il tasso di sbiancamento delle etichette nella SBL si stima indipendentemente dal singolo monitoraggio della tintura dei lipidi, o inserendo un esponenziale al profilo totale intensità,
, Sono meglio estratto da una misura per l'andamento nel tempo dei valori dei pixel totali che tenga conto sbiancamento 27. Il tasso di sbiancamento delle etichette nella SBL si stima indipendentemente dal singolo monitoraggio della tintura dei lipidi, o inserendo un esponenziale al profilo totale intensità,  , A volte lunghi (ad esempio, regime (3) in Figura 3). Conoscendo le dimensioni di una vescicola di fusione e diffusività lipidi permette il confronto del tempo di rilascio lipidi effettiva, , Per il tempo di diffusione di un lipide intorno vescicola
, A volte lunghi (ad esempio, regime (3) in Figura 3). Conoscendo le dimensioni di una vescicola di fusione e diffusività lipidi permette il confronto del tempo di rilascio lipidi effettiva, , Per il tempo di diffusione di un lipide intorno vescicola  , Dove A è l'area ves vescicole e è la diffusività lipidi. Se le due scale temporali sono comparabili, il poro presenta poca resistenza al rilascio dei lipidi, e il rilascio è a diffusione limitata. Al contrario, se
, Dove A è l'area ves vescicole e è la diffusività lipidi. Se le due scale temporali sono comparabili, il poro presenta poca resistenza al rilascio dei lipidi, e il rilascio è a diffusione limitata. Al contrario, se  , Allora il poro ritarda significativamente flusso lipidico. Una misura quantitativa del ritardo è la "apertura dei pori", P 0, pari al rapporto
, Allora il poro ritarda significativamente flusso lipidico. Una misura quantitativa del ritardo è la "apertura dei pori", P 0, pari al rapporto  volte un fattore oPer f unità legato al poro geometria 27. Per un due stati, poro aperto / chiuso, P 0 è la frazione di tempo nello stato aperto. Per un poro tremolante cui dimensioni variano continuamente, P 0 rappresenta il raggio media nel tempo rispetto al raggio completamente aperta.
volte un fattore oPer f unità legato al poro geometria 27. Per un due stati, poro aperto / chiuso, P 0 è la frazione di tempo nello stato aperto. Per un poro tremolante cui dimensioni variano continuamente, P 0 rappresenta il raggio media nel tempo rispetto al raggio completamente aperta.
Il rapporto segnale-rumore deve essere sufficientemente buona per il monitoraggio singoli fluorofori lipidici. Ciò si ottiene facilmente in esperimenti solo colore, utilizzando un marcatore forte come LR-PE.
Rilevazione simultanea di lipidi e solubile Cargo di uscita
Un evento rappresentativo è mostrato in Figura 7. Alle alte concentrazioni usate per l'incapsulamento, la fluorescenza SRB è inizialmente auto-temprato. Pertanto, la maggior parte ancorate v-SUV sono rilevabili solo con il loro segnale DiD 27. Un forte aumentoin DiD fluorescenza segna lipidi miscelazione, come negli esperimenti di lipidi-label monocolore. L'insorgenza di rilascio lipidico coincide con l'inizio di un aumento SRB fluorescenza. Tale incremento è probabilmente dovuto al SRB dequenching come le molecole SRB sfuggito il SUV e il restante SRB è stato diluito al di sotto delle concentrazioni di auto-spegnimento. È interessante notare che la fluorescenza SRB ha raggiunto un plateau stabile. Se il poro di fusione rimasta aperta, ci si aspetterebbe segnale SRB per aumentare al massimo a causa dequenching, seguito da una diminuzione di sfondo come SRB residuo lasciato il SUV, o se il SUV crollata nel SBL. Il plateau stabile più probabile indica il poro richiusi dopo aver rilasciato tutte le etichette lipidici e una frazione del carico solubile.
In alcuni casi, il segnale di rilascio di lipidi non era accompagnato da alcun contenuto rilevabili rilasciare segnali. Questo potrebbe essere sia perché i pori sono troppo piccole per consentire il passaggio della SRB, o qualchecome il SUV aveva già perso il suo carico solubile. Per distinguere tra queste due possibilità, lo sbiancamento dei SUV attraccate può essere analizzato. Bleaching dell'etichetta lipidi DID riduce il segnale di fluorescenza in pochi secondi, mentre sbiancamento dei inizialmente auto-quenched risultati SRB in un aumento del segnale SRB come frazione della SRB sono foto-convertito in altri prodotti e autotempra è sollevato. Mentre la maggior parte SUV ancorate (48 su 58 analizzati eventi) visualizzati il previsto aumento della SRB fluorescenza per lunghi periodi di illuminazione, una piccola frazione (10/58) non ha mostrato alcun segnale SRB a tutti. Così, una piccola frazione dei SUV possono perdere il loro contenuto solubile nel periodo tra la loro preparazione e l'uso negli esperimenti.
In più del 80% degli eventi (74 su 91) per i quali sia SRB e segnali ha fatto potrebbe essere seguita, la fluorescenza SRB è aumentato ed è rimasto ad un plateau stabile 27. In ~ 20% di tueste casi il restante SRB fluorescenza è stato improvvisamente rilasciato fino a pochi secondi più tardi. Questo potrebbe essere dovuto ad una riapertura del poro consente una rapida, completa il rilascio entro 1-2 fotogrammi (18-36 msec) o una rottura del SUV a causa di accumulato foto-danni 16. Qualunque sia la sua origine, questa seconda, rapido rilascio di SRB esclude la possibilità che il segnale SRB residua dopo il rilascio iniziale potrebbe essere dovuto alla SRB rimanere intrappolato nello spazio tra la SBL ed il substrato di vetro dopo il rilascio completo. Tale meccanismo è stato suggerito in un precedente studio in cui la SBL è stato supportato direttamente sul vetro, con poco spazio tra i due 16. Al contrario, i lipidi PEG utilizzati in questo test devono fornire 4-5 nm di spazio tra il SBL e il vetro 25, permettendo la SRB per diffondere lontano dal sito di fusione.

Figura 7. Simultan dell'EOUS lipidi e il rilascio di contenuti. segnali di fluorescenza da lipidi (blu) e contenuti (rosso) marcatore per un evento di fusione SUV-SBL (per i dettagli vedi testo). Istantanee da ciascun canale vengono visualizzati nella parte inferiore (4,0 micron di larghezza). Cartoni animati (al centro) illustrano le diverse fasi. (1) banchine SUV, ha fatto la fluorescenza è basso. (2) Fusion consente il trasferimento del DiD nel SBL e ha fatto un aumento di fluorescenza. segnale SRB aumenta anche come auto-spegnimento è alleviato dalla fuga del SRB attraverso il poro. (3) segnale SRB rimane mentre molecole stabili ha fatto diffondere all'interno del SBL che indicano una possibile nuova chiusura del poro. (4) I segnali SRB diminuiscono molto rapidamente, a causa della rapida fusione completa o liposomi scoppio. La composizione del v-SUV era POPC: SAPE: DOPS: PEG2000-DOPE: DID (67: 15: 12: 5: 1 mole%), LP 200, mentre t-SBLs erano composti da POPC: SAPE: DOPS: cervello PI (4,5) P 2: PEG2000-DOPE: NBD-PE (64.5: 15: 12: 3: 5: 0,5 moli%) e LP = 20.000. = "_ Blank"> Clicca qui per vedere una versione più grande di questa figura.
FRAP_instructions.pdf -. Le istruzioni per la creazione di una sequenza di FRAP Cliccate qui per scaricare questo file.
ReadMe_FRAP.txt -. File di testo che descrive la procedura per analizzare i dati di recupero FRAP utilizzando il codice MATLAB che viene fornito Clicca qui per scaricare questo file.
EK_FRAPbatch.m - file di MATLAB a batch analizzare recupero di fluorescenza dopo photobleaching. Questo file chiama la subroutine EK_getFRAPvals.m MATLAB e EK_SoumpassisFit.m."_blank"> Clicca qui per scaricare il file.
EK_getFRAPvals.m -. File di MATLAB, subroutine chiamato da EK_FRAPbatch.m Cliccate qui per scaricare questo file.
EK_SoumpassisFit.m -. File di MATLAB, subroutine chiamato da EK_FRAPbatch.m Cliccate qui per scaricare questo file.
JN150923c1_FRAP_list.txt -. Un esempio di un elenco di nomi di file per essere batch analizzato dallo script MATLAB principale EK_FRAPbatch.m Clicca qui per scaricare questo file.
Ilseguenti file sono stack TIF e dei file di testo corrispondenti per cinque esempi di sequenze FRAP che possono essere analizzati eseguendo EK_FRAPbatch.m e scegliendo il file JN150923c1_FRAP_list.txt quando viene richiesto di scegliere un elenco di file.
JN150923c1_F1_MMStack_Pos0.ome.tif -. FRAP immagine Pila Cliccate qui per scaricare questo file.
JN150923c1_F1_MMStack_Pos0_metadata.txt -. File di testo che corrisponde con i metadati Cliccate qui per scaricare questo file.
JN150923c1_F2_MMStack_Pos0.ome.tif -. FRAP immagine Pila Plfacilità clicca qui per scaricare il file.
JN150923c1_F2_MMStack_Pos0_metadata.txt -. File di testo che corrisponde con i metadati Cliccate qui per scaricare questo file.
JN150923c1_F3_MMStack_Pos0.ome.tif -. FRAP immagine Pila Cliccate qui per scaricare questo file.
JN150923c1_F3_MMStack_Pos0_metadata.txt -. File di testo che corrisponde con i metadati Cliccate qui per scaricare questo file.
JN150923c1_F4_MMStack_Pos0.ome.tif - FRAP immagine Pila. Cliccate qui per scaricare questo file.
JN150923c1_F4_MMStack_Pos0_metadata.txt -. File di testo che corrisponde con i metadati Cliccate qui per scaricare questo file.
JN150923c1_F5_MMStack_Pos0.ome.tif -. FRAP immagine Pila Cliccate qui per scaricare questo file.
JN150923c1_F5_MMStack_Pos0_metadata.txt -. File di testo che corrisponde con i metadati Cliccate qui per scaricare questo file.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Il successo dell'attuazione del saggio di fusione SUV-SBL qui descritto dipende criticamente diversi passaggi chiave, come ad esempio la ricostituzione funzionale delle proteine nei liposomi, ottenendo SBLs di buona qualità, e scegliendo i parametri di imaging giusti per rilevare singole molecole. Anche se può richiedere un certo tempo e fatica per avere successo, una volta che il test viene implementato con successo, fornisce una serie di informazioni sul processo di fusione non disponibile da qualsiasi altro test in vitro in fusione discusso in Introduzione. Le aliquote dei SUV dock su e fusibile con il SBL, la distribuzione dei tempi di aggancio-to-fusione, diffusività lipidi, le dimensioni di ogni liposomi fusione, e se il rilascio di lipidi per un dato evento di fusione è can diffusion o pori controllati essere determinato. Se rilascio è molto più lento del previsto dalla diffusione, un modello che assume pori sfarfallio sottende il ritardo rendimenti rilascio lipidi, P 0, la frazione di tempo poro is aperto durante la fusione 27.
Impieghi precedenti sondare unico SUV-SUV fusione 18-20,28 o fusione dei liposomi e particelle virali per SBLs 15-17,21,22,33,44 si basava su FRET e di auto-spegnimento tra le molecole di colorante. Al contrario, abbiamo ampiamente evitare colorante dequenching per sondare cinetica di lipidi trasferimento dell'etichetta nel SBL, per due motivi. Innanzitutto, l'efficienza FRET è un reporter altamente non lineare del rilascio dei lipidi. Piccoli cambiamenti nella inter-dye risultato distanza d in grandi cambiamenti in termini di efficienza FRET, ma solo per un numero limitato di D / R 0 valori, dove R 0 è la distanza Forster. Cioè, un tempo di ritardo tra delle diluizione reale e un aumento nel segnale dequenching può verificarsi, a causa del tempo richiesto per far cadere ad un valore prossimo a R 0. Allo stesso modo, c'è poco cambiamento nel segnale dequenching volta la densità colorante diminuisce sufficientemente al rilascio lipidi tale che  1.4, anche se una notevole quantità di colorante rimane ancora in vescicole. Queste caratteristiche rendono dequenching un buon strumento per ottenere un picco nel segnale di rilevare eventi di fusione, ma complicare l'analisi quantitativa cinetica del rilascio dei lipidi con elevata risoluzione temporale. Al contrario, le variazioni di intensità a causa riorientamento dipolo e effetti di campo evanescente come trasferisce fluoroforo da un SUV nel SBL sono correlata linearmente alla frazione di colorante rimanendo in SUV. In secondo luogo, alte concentrazioni di etichette lipidi necessari per autotempra possono influenzare il processo di fusione.
1.4, anche se una notevole quantità di colorante rimane ancora in vescicole. Queste caratteristiche rendono dequenching un buon strumento per ottenere un picco nel segnale di rilevare eventi di fusione, ma complicare l'analisi quantitativa cinetica del rilascio dei lipidi con elevata risoluzione temporale. Al contrario, le variazioni di intensità a causa riorientamento dipolo e effetti di campo evanescente come trasferisce fluoroforo da un SUV nel SBL sono correlata linearmente alla frazione di colorante rimanendo in SUV. In secondo luogo, alte concentrazioni di etichette lipidi necessari per autotempra possono influenzare il processo di fusione.
Per polarizzata eccitazione TIRF, un impianto home-costruito come in figura 4 è costo-efficace e fornisce un eccellente controllo sulle proprietà di polarizzazione. Tuttavia, un impianto home-costruito non è necessario, come almeno alcuni microscopi TIRF commerciali usano di eccitazione polarizzata e off-the-shelfI moduli scissione emissione di due colori sono disponibili. Un microscopio commerciale che utilizza un (PM) di fibra a mantenimento di polarizzazione per accoppiare la luce di eccitazione polarizzata emergente dal laser nel microscopio è stato usato con successo in precedenza 25-27. Su un tale strumento, la polarizzazione può essere ruotata semplicemente ruotando l'ingresso in fibra PM all'unità TIRF del microscopio. Al contrario, altre configurazioni commerciali possono impiegare una polarizzazione beam splitter nell'unità TIRF, le regolazioni di polarizzazione più difficile o impossibile. Purtroppo, proprietà di polarizzazione spesso non sono specificati per sistemi commerciali. Pertanto, è fondamentale per gli utenti che non vogliono costruire un sistema home-costruito per discutere di proprietà di polarizzazione e di provare una demo prima di investire in una particolare opzione commerciale.
il monitoraggio simultaneo di lipidi e il rilascio del carico solubile fornisce ulteriori informazioni sul processo di fusione. Molti i pori sembrano sigillare dopo releasing solo una frazione dell'etichetta solubile incapsulato in un SUV 27. Di solito tutte le etichette di lipidi vengono rilasciati per il momento il poro risigilla, in modo da pori nuova chiusura non era sospettato in precedenza dal monitoraggio di rilascio dei lipidi da soli. Trasferimento, infatti, il monitoraggio rilascio di lipidi da soli, Kiessling et al. 29 avevano suggerito di lipidi etichettati nella SBL si è verificato tramite estremamente rapido collasso di tutta la membrana di SUV nel SBL.
Richiusura della fusione pori dopo il rilascio parziale dei contenuti è stato precedentemente osservato in un saggio di fusione cerotto doppio strato liposomi-tethered. Rawle et al. 54 patch doppio strato freestanding preparate che erano legati su un vetrino da ~ 8 nm lunghi distanziatori DNA. Hanno poi introdotto liposomi cui la fusione per la patch doppio strato è stato guidato da ibridazione di complementari coniugati a singolo filamento di DNA-lipidi ancorate alle membrane liposomi e patch. 12% di tutti gli eventi erano fusioni transitori, con conseguente parterilascio ial dei contenuti liposomi. Liposomi eventi di rottura, visto in alcuni precedenti saggi di fusione SUV-SBL 44, erano molto rari. Gli autori hanno suggerito il ~ 8 nm spazio sotto il cerotto doppio strato, prevenendo interazioni doppio strato-substrato, possono hanno permesso di trasferimento non perde dei contenuti liposomi allo spazio sotto il doppio strato. Inoltre, la diffusione delle etichette solubili rilasciati non era significativamente ostacolata nello spazio tra il doppio strato planare e il substrato di vetro. Nel saggio qui descritto, il doppio strato planare è analogamente mantenuto ~ 5 nm dal vetrino coprioggetti dalla presenza di poli (glicole etilenico) coniugati -lipid.
Le informazioni aggiuntive fornite dal monitoraggio a due colori di merci solubile e lipidi viene fornito con alcuni compromessi. In primo luogo, la preparazione dei SUV con le due etichette è un po 'più complessa di etichette tra cui lipidi da soli. In secondo luogo, vi è una certa inevitabile perdita di segnale-rumore quando il monitoraggio dei segnali dalle etichette lipidi in tegli stesso tasso di acquisizione come negli esperimenti di rilascio di lipidi monocolore (15-20 msec per frame). Alcune di questa perdita è dovuta al fatto che DID, usato come etichetta lipidi insieme con il marcatore SRB solubili, non è brillante come LR-PE usato in esperimenti solo colore. Ulteriori perdite derivano da sfondo superiore a causa simultanea eccitazione due laser e componenti aggiuntivi nel cammino ottico (Figura 4). Finora, il rumore del segnale-a-basso ci ha impedito di tracciamento affidabile del singolo DID etichette in esperimenti di due colori. Questo a sua volta impedito un'analisi approfondita per estrarre le informazioni che possono essere con misurazioni LR-PE monocolore. Questa sfida può essere superata in futuro ottimizzando condizioni di acquisizione e testare altre coppie di etichette compatibili con proprietà migliorate.
il monitoraggio simultaneo di rilascio del carico solubili e dei lipidi può anche rilevare hemifusion, uno stato in cui i volantini prossimali sono fusi, ma distalevolantini non hanno. Infatti, tali esperimenti due colori possono fornire l'unica valutazione conclusiva della presenza di hemifusion in altri saggi in vitro fusione 18,20. Tuttavia, hemifusion può essere rilevato in modo affidabile in monocolore SUV-SBL solo 22,44 rilascio monitoraggio esperimenti lipidico. In tali esperimenti, circa 1/2 l'intensità iniziale label lipidico di un SUV rimane nel punto di fusione. L'intensità rimanente può scomparire quando i volantini distali fondono qualche tempo dopo, se la macchia non ha ancora sbiancato.
L'utilizzo di lipidi PEG di introdurre un morbido cuscino tra la SBL ed il supporto di vetro è stato fondamentale nel riprodurre il requisito SNAP25 di esocitosi. I risultati di protocollo in un PEG pennello copre il lato della SBL che si affaccia sul canale di flusso pure. Questo ha alcuni vantaggi, come la riduzione delle interazioni non specifiche tra SUV e la SBL, come spiegato nella Introduzione. Se proteine che interagiscono con i fosfolipidi dellaSBL vengono introdotti nel dosaggio, tuttavia, la spazzola PEG presenterà una barriera sterica contro tali interazioni. Così, per alcune applicazioni, sarebbe opportuno includere lo strato PEG asimmetricamente, solo tra il SBL ed il vetrino di vetro. Il lato del SBL fronte al canale di flusso sarebbe quindi in grado di interagire con il legame fosfolipide proteine, come synaptotagmin. Ciò può essere realizzato utilizzando catene PEG bi-funzionali che possono essere covalentemente legati al coprioggetto ad una estremità e che trasporta un ancoraggio lipidico all'altra. Metodi Langmuir-Blodgett possono poi essere utilizzati per depositare un monostrato lipidico sullo strato PEG 55. Liposomi introdotte sopra questo fusibile superficie idrofoba con esso, formando un doppio strato 56. Si spera che questo approccio sarà combinato con microfluidica in futuro.
Si prevede che il saggio SUV-SBL presentato qui sarà utile per esplorare il ruolo delle proteine addizionali che interagiscono con Snares,come la famiglia Munc18 / Sec1, il sensore di calcio synaptotagmin-1, e complexin.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Gli autori dichiarano di non avere interessi finanziari concorrenti.
Materials
| Name | Company | Catalog Number | Comments |
| Reagents | |||
| Milli-Q (MQ) water | Millipore | ||
| KOH | J.T. Baker | 3040-05 | |
| Ethanol 190 Proof | Decon | ||
| Isopropanol | Fisher Chemical | A416P4 | |
| HEPES | AmericanBio | AB00892 | |
| Sodium Cholride (KCl) | AmericanBio | AB01915 | |
| Dithiothreitol | AmericanBio | AB00490 | |
| N-[2-hydroxyethyl] piperazine-N'-[2-ethanesulfonic acid] (HEPES) | AmericanBio | AB00892 | |
| EGTA | Acros Organics | 409911000 | |
| Buffers | |||
| HEPES-KOH buffer (pH 7.4) | 25 mM HEPES-KOH, 140 mM KCl, 100 μM EGTA, 1 mM DTT | ||
| Solvents | |||
| Chloroform | J.T. Baker | 9180-01 | in glass bottle, CAUTION, wear PPE |
| Methanol | J.T. Baker | 9070-03 | in glass bottle, CAUTION, wear PPE |
| Liposome preparation | |||
| Gastight Hamilton syringe | Hamilton | var. sizes | only use glass sringe with solents (Chlorophorm/ Methanon, 2:1, v/v) |
| Glass tubes Pyrex Vista 11 ml, 16x100 mm screw cap culture tube | Pyrex | 70825-16 | clean thoroughly, rinse with chloroform |
| 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine, 16:0-18:1 PC (POPC) | Avanti Polar Lipids | 850457 | Lipids come dissolved in CHCl3 or as lyphilized powder in sealed vials. Aliquot upon opening. Store extra as dried lipid films under inert atmosphere at -20 °C. Keep stocks in CHCl3/MeOH (2:1, v/v) at -20 °C. let come to RT before opening |
| 1,2-dioleoyl-sn-glycero-3-phospho-L-serine (sodium salt), 18:1 PS (DOPS) | Avanti Polar Lipids | 840035 | Same as above. |
| 1-stearoyl-2-arachidonoyl-sn-glycero-3-phosphoethanolamine, 18:0-20:4 PE (SAPE) | Avanti Polar Lipids | 850804 | Same as above. |
| L-α-phosphatidylinositol-4,5-bisphosphate (Brain, Porcine) (ammonium salt), Brain PI(4,5)P2 | Avanti Polar Lipids | 840046 | Same as above. |
| 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(7-nitro-2-1,3-benzoxadiazol-4-yl) (ammonium salt), 18:1 NBD PE | Avanti Polar Lipids | 810145 | Same as above. |
| 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (ammonium salt), 18:1 PEG2000 PE | Avanti Polar Lipids | 880130 | Same as above. |
| cholesterol (ovine wool, >98%) | Avanti Polar Lipids | 700000 | Same as above. |
| DiD' oil; DiIC18(5) oil (1,1'-Dioctadecyl-3,3,3',3'-Tetramethylindodicarbocyanine Perchlorate) | Molecular Probes | D-307 | |
| Rotavapor R-210 | Buchi | R-210 | heat bath above Tm of lipids used |
| OG n-Octyl-β-D-Glucopyranoside | Affymetrix | 0311 | store at -20 °C, let come to RT before opening |
| Shaker - Eppendorf Thermomixer R | Eppendorf | ||
| Slide-A-Lyze Dialysis Cassettes, 20k MWCO, 3 ml | life technologies | 66003 | |
| Bio-Beads SM-2 Adsorbents | Bio-Rad | 1523920 | |
| OptiPrep Density Gradient Medium | Sigma-Aldrich | D1556 | |
| Ultracentrifugation tube, Thinwall, Ultra-Clear, 13.2 ml, 14 x 89 mm | Beckman Coulter | 41121703 | |
| Beckman SW41 Ti rotor | |||
| SuflorhodamineB | Molecular Probes | S-1307 | |
| Econo-Column Chromatography Columns, 2.5 × 10 cm | Bio-Rad | 7372512 | |
| Sepharose CL-4B | GE Healthcare | 17-0150-01 | |
| SYPRO Orange Protein Gel Stain | Molecular Probes | S-6650 | 5,000x Concentrate in DMSO |
| PDMS block | |||
| Sylgard 184 Silicone elastomer kit, PDMS | Dow Corning | 3097358-1004 | |
| Pyrex glass petri dish, 150 x 20 mm, complete with cover | Corning | 3160-152 | |
| Hole puncher - Reusable Biopsy Punch, 0.75 mm | World Precision Instruments | 504529 | |
| Manual Hole Punching Machine | SYNEO | MHPM-UNV | |
| Drill .035 x .026 x 1.5 304 SS TiN coated round punch | SYNEO | CR0350265N20R4 | drill diameter: 0.9 mm, punch bore size: 0.74 mm |
| Tygon Microbore tubing, 0.25 mm ID, 0.76 mm OD | Cole-Parmer | 06419-00 | 0.010" ID, 0.030" OD |
| Silicone Tubing (0.51 mm ID, 2.1 mm OD | Cole-Parmer | 95802-00 | 0.020" ID, 0.083" OD |
| Cover glass - cleanroom cleaned | |||
| Schott Nexterion cover slip glass D | Schott | 1472305 | |
| plasma cleaner | Harrick | PDC-32G | |
| pTIRF setup and accessories | |||
| IX81 microscope body | Olympus | IX81 | |
| EM CCD camera | Andor | ixon-ultra-897 | |
| Thermo Plate, heated microscope stage | Tokai Hit | MATS-U52RA26 | |
| 1 ml hamilton glass syringes (4x) | Hamilton | 81365 | |
| syringe pump | kd Scientific | KDS-230 | |
References
- Sudhof, T. C., Rothman, J. E. Membrane fusion: grappling with SNARE and SM proteins. Science. 323, 474-477 (2009).
- Wickner, W., Schekman, R.
- Harrison, S. C.
- Jahn, R., Scheller, R. H.
- Lindau, M., Alvarez de Toledo, G.
- Staal, R. G., Mosharov, E. V., Sulzer, D. Dopamine neurons release transmitter via a flickering fusion pore. Nat Neurosci. 7, 341-346 (2004).
- Wu, Z., et al. Nanodisc-cell fusion: Control of fusion pore nucleation and lifetimes by SNARE protein transmembrane domains. Sci. Rep. 6, 27287 (2016).
- Alabi, A. A., Tsien, R. W. Perspectives on kiss-and-run: role in exocytosis, endocytosis, and neurotransmission. Ann Rev Physiol. 75, 393-422 (2013).
- Rossetto, O., Pirazzini, M., Montecucco, C. Botulinum neurotoxins: genetic, structural and mechanistic insights. Nature Rev Microbiol. 12, 535-549 (2014).
- Weber, T., et al. SNAREpins: minimal machinery for membrane fusion. Cell. 92, 759-772 (1998).
- Nickel, W., et al. Content mixing and membrane integrity during membrane fusion driven by pairing of isolated v-SNAREs and t-SNAREs. Proc Natl Acad Sci U S A. 96, 12571-12576 (1999).
- McNew, J. A., et al. Compartmental specificity of cellular membrane fusion encoded in SNARE proteins. Nature. 407, 153-159 (2000).
- Melia, T. J., You, D. Q., Tareste, D. C., Rothman, J. E. Lipidic antagonists to SNARE-mediated fusion. J Biol Chem. 281, 29597-29605 (2006).
- Hernandez, J. M., et al. Membrane fusion intermediates via directional and full assembly of the SNARE complex. Science. 336, 1581-1584 (2012).
- Fix, M., et al. Imaging single membrane fusion events mediated by SNARE proteins. Proc Natl Acad Sci U S A. 101, 7311-7316 (2004).
- Bowen, M. E., Weninger, K., Brunger, A. T., Chu, S. Single molecule observation of liposome-bilayer fusion thermally induced by soluble N-ethyl maleimide sensitive-factor attachment protein receptors (SNAREs). Biophys J. 87, 3569-3584 (2004).
- Liu, T., Tucker, W. C., Bhalla, A., Chapman, E. R., Weisshaar, J. C. SNARE-driven, 25-millisecond vesicle fusion in vitro. Biophys J. 89, 2458-2472 (2005).
- Yoon, T. Y., Okumus, B., Zhang, F., Shin, Y. K., Ha, T. Multiple intermediates in SNARE-induced membrane fusion. Proc Natl Acad Sci U S A. 103, 19731-19736 (2006).
- Diao, J., et al. A single-vesicle content mixing assay for SNARE-mediated membrane fusion. Nat Commun. 1, 1-6 (2010).
- Kyoung, M., et al. In vitro system capable of differentiating fast Ca2+-triggered content mixing from lipid exchange for mechanistic studies of neurotransmitter release. Proc Natl Acad Sci U S A. 108, E304-E313 (2011).
- Domanska, M. K., Kiessling, V., Stein, A., Fasshauer, D., Tamm, L. K. Single vesicle millisecond fusion kinetics reveals number of SNARE complexes optimal for fast SNARE-mediated membrane fusion. J Biol Chem. 284, 32158-32166 (2009).
- Kreutzberger, A. J., Kiessling, V., Tamm, L. K. High Cholesterol Obviates a Prolonged Hemifusion Intermediate in Fast SNARE-Mediated Membrane Fusion. Biophys J. 109, 319-329 (2015).
- Schwenen, L. L., et al. Resolving single membrane fusion events on planar pore-spanning membranes. Sci Rep. 5, 12006 (2015).
- Karatekin, E., et al. A fast, single-vesicle fusion assay mimics physiological SNARE requirements. Proc Natl Acad Sci U S A. 107, 3517-3521 (2010).
- Karatekin, E., Rothman, J. E. Fusion of single proteoliposomes with planar, cushioned bilayers in microfluidic flow cells. Nat Protoc. 7, 903-920 (2012).
- Smith, M. B., et al. Interactive, computer-assisted tracking of speckle trajectories in fluorescence microscopy: application to actin polymerization and membrane fusion. Biophys J. 101, 1794-1804 (2011).
- Stratton, B. S., et al. Cholesterol Increases the Openness of SNARE-mediated Flickering Fusion Pores. Biophysical journal. 110, (2016).
- Diao, J., et al. Synaptic proteins promote calcium-triggered fast transition from point contact to full fusion. Elife. 1, e00109 (2012).
- Kiessling, V., Domanska, M. K., Tamm, L. K. Single SNARE-mediated vesicle fusion observed in vitro by polarized TIRFM. Biophys J. 99, 4047-4055 (2010).
- Blasi, J., et al. Botulinum neurotoxin A selectively cleaves the synaptic protein SNAP-25. Nature. 365, 160-163 (1993).
- Washbourne, P., et al. Genetic ablation of the t-SNARE SNAP-25 distinguishes mechanisms of neuroexocytosis. Nat Neurosci. 5, 19-26 (2002).
- Diaz, A. J., Albertorio, F., Daniel, S., Cremer, P. S. Double cushions preserve transmembrane protein mobility in supported bilayer systems. Langmuir. 24, 6820-6826 (2008).
- Floyd, D. L., Ragains, J. R., Skehel, J. J., Harrison, S. C., van Oijen, A. M. Single-particle kinetics of influenza virus membrane fusion. Proc Natl Acad Sci U S A. 105, 15382-15387 (2008).
- Albertorio, F., et al. Fluid and air-stable lipopolymer membranes for biosensor applications. Langmuir. 21, 7476-7482 (2005).
- Daniel, S., Albertorio, F., Cremer, P. S. Making lipid membranes rough, tough, and ready to hit the road. Mrs Bulletin. 31, 536-540 (2006).
- Gao, Y., et al. Single reconstituted neuronal SNARE complexes zipper in three distinct stages. Science. 337, 1340-1343 (2012).
- Kenworthy, A. K., Hristova, K., Needham, D., Mcintosh, T. J. Range and Magnitude of the Steric Pressure between Bilayers Containing Phospholipids with Covalently Attached Poly(Ethylene Glycol). Biophys J. 68, 1921-1936 (1995).
- Knoll, W., et al. Solid supported lipid membranes: New concepts for the biomimetic functionalization of solid surfaces. Biointerphases. 3, Fa125-Fa135 (2008).
- Quinn, P., Griffiths, G., Warren, G. Density of newly synthesized plasma membrane proteins in intracellular membranes II. Biochemical studies. J Cell Biol. 98, 2142-2147 (1984).
- Sund, S. E., Swanson, J. A., Axelrod, D. Cell membrane orientation visualized by polarized total internal reflection fluorescence. Biophys J. 77, 2266-2283 (1999).
- Johnson, D. S., Toledo-Crow, R., Mattheyses, A. L., Simon, S. M. Polarization-controlled TIRFM with focal drift and spatial field intensity correction. Biophys J. 106, 1008-1019 (2014).
- Anantharam, A., Onoa, B., Edwards, R. H., Holz, R. W., Axelrod, D. Localized topological changes of the plasma membrane upon exocytosis visualized by polarized TIRFM. J Cell Biol. 188, 415-428 (2010).
- Axelrod, D. Carbocyanine dye orientation in red cell membrane studied by microscopic fluorescence polarization. Biophys J. 26, 557-573 (1979).
- Wang, T., Smith, E. A., Chapman, E. R., Weisshaar, J. C. Lipid mixing and content release in single-vesicle, SNARE-driven fusion assay with 1-5 msec resolution. Biophys J. 96, 4122-4131 (2009).
- Chernomordik, L. V., Frolov, V. A., Leikina, E., Bronk, P., Zimmerberg, J. The pathway of membrane fusion catalyzed by influenza hemagglutinin: restriction of lipids, hemifusion, and lipidic fusion pore formation. J Cell Biol. 140, 1369-1382 (1998).
- Takamori, S., et al. Molecular anatomy of a trafficking organelle. Cell. 127, 831-846 (2006).
- Wilhelm, B. G., et al. Composition of isolated synaptic boutons reveals the amounts of vesicle trafficking proteins. Science. 344, 1023-1028 (2014).
- Scott, B. L., et al. Liposome fusion assay to monitor intracellular membrane fusion machines. Methods Enzymol. 372, 274-300 (2003).
- Linkert, M., et al. Metadata matters: access to image data in the real world. J Cell Biol. 189, 777-782 (2010).
- Soumpasis, D. M. Theoretical analysis of fluorescence photobleaching recovery experiments. Biophys J. 41, 95-97 (1983).
- Ohki, S. A mechanism of divalent ion-induced phosphatidylserine membrane fusion. Biochim Biophys Acta. 689, 1-11 (1982).
- Berquand, A., et al. Two-step formation of streptavidin-supported lipid bilayers by PEG-triggered vesicle fusion. Fluorescence and atomic force microscopy characterization. Langmuir. 19, 1700-1707 (2003).
- Tamm, L. K., McConnell, H. M.
- Rawle, R. J., van Lengerich, B., Chung, M., Bendix, P. M., Boxer, S. G. Vesicle fusion observed by content transfer across a tethered lipid bilayer. Biophys J. 101, L37-L39 (2011).
- Wagner, M. L., Tamm, L. K. Reconstituted syntaxin1a/SNAP25 interacts with negatively charged lipids as measured by lateral diffusion in planar supported bilayers. Biophys J. 81, 266-275 (2001).
- Kalb, E., Frey, S., Tamm, L. K. Formation of Supported Planar Bilayers by Fusion of Vesicles to Supported Phospholipid Monolayers. Biochimica Et Biophysica Acta. 1103, 307-316 (1992).
