

Summary
A continuación, se presenta un protocolo para detectar eventos individuales, SNARE mediada por liposomas y de fusión entre bicapas soportadas en los canales de microfluidos utilizando TIRFM polarizado, con sensibilidad única molécula y ~ 15 ms tiempo de resolución. Lípidos y liberación de carga solubles se pueden detectar de forma simultánea. tamaño de los liposomas, de difusividad de los lípidos, y poro de fusión propiedades se miden.
Abstract
En el proceso omnipresente de fusión de la membrana de la apertura de un poro de fusión establece la primera conexión entre dos compartimentos separados anteriormente. Durante neurotransmisor o la liberación de hormonas a través de la exocitosis, el poro de fusión puede abrir y cerrar transitoriamente en varias ocasiones, la regulación de la cinética de liberación de carga. la dinámica de los poros también determinan el modo de reciclaje de vesículas; resellado resultados irreversibles en la fusión transitoria, "kiss-and-run", mientras que la dilatación conduce a la fusión completa. Para entender mejor los factores que gobiernan la dinámica de los poros, hemos desarrollado un ensayo para monitorizar la fusión de membranas mediante microscopía de fluorescencia polarizada de reflexión interna total (TIRF) con sensibilidad única molécula y ~ 15 ms tiempo de resolución en un bioquímicamente bien definidas sistema in vitro. La fusión etiquetada de manera fluorescente pequeñas vesículas unilamelares que contienen proteínas v-SNARE (V-SUV) con una doble capa de apoyo plana t-SNARE, apoyado en un cojín de polímero blando (t-SBL, bicapa t-apoyado), Se controla. El ensayo utiliza canales de flujo de microfluidos que garantizan un mínimo consumo de muestra, mientras que el suministro de una densidad constante de los SUV. La explotación de la mejora rápida de la señal después de la transferencia de las etiquetas de lípidos de la SUV de la LPS durante la fusión, la cinética de la transferencia de colorantes de lípidos se controla. La sensibilidad de la microscopía TIRF permite el seguimiento de las etiquetas de lípidos fluorescentes individuales, de los cuales difusividad de lípidos y tamaño SUV se puede deducir por cada evento de fusión. Los tiempos de liberación de colorante de lípidos pueden ser mucho más de lo esperado para el paso sin obstáculos a través de los poros abiertos de forma permanente. El uso de un modelo que asume el retraso de la liberación de lípidos se debe a poros parpadeo, un poro "apertura", la fracción de tiempo que el poro se mantiene abierto durante la fusión, puede ser estimado. Un marcador soluble puede ser encapsulada en los terrenos para el seguimiento simultáneo de los lípidos y la liberación de la carga soluble. Estas mediciones indican algunos poros pueden volver a cerrar después de perder una fracción de la carga soluble.
Introduction
La fusión de membranas es un proceso biológico universal, necesaria para el tráfico intracelular de lípidos y proteínas, la secreción, la fertilización, el desarrollo y la envolvió entrada del virus en organismos huésped 1-3. Para la mayoría de las reacciones de fusión intracelular, incluida la liberación de las hormonas y los neurotransmisores a través de la exocitosis, la energía para fusionar dos bicapas lipídicas se proporciona por formación de un haz de cuatro hélices entre las proteínas N-etilmaleimida sensible receptor de la proteína factor de unión soluble cognado (SNARE), anclada en la vesícula (v-SNARE) y la membrana diana (t-SNARE) 4, respectivamente. Sináptica exocitosis de vesículas es la reacción de fusión más estrechamente regulado y se produce dentro de una milésima de segundo después de la llegada de un potencial de acción 1,4,5. El poro de fusión, la conexión inicial entre los dos compartimientos de fusión, puede parpadear abierto y cerrado varias veces antes de volver a sellar o expansión irreversible 5-7. Los resultados anterioresen transitoria, "Kiss & run" de fusión, mientras que la segunda conduce a la fusión completa. Los factores que regulan el equilibrio entre estos dos modos de fusión y mecanismos que regulan el parpadeo de los poros no son bien entendidos 5,8.
Se requieren proteínas SNARE de exocitosis; la fusión de vesículas sinápticas se suprimirán a partir de la escisión de trampas por neurotoxinas 9. Los experimentos de fusión a granel mediante pequeñas vesículas unilaminares (SUV) mostraron que SNAREs no sólo son necesarios, sino también suficiente para impulsar la fusión de membranas 10. En este ensayo mayor, SUVs reconstituyeron con v-SNARE (v-SUV) fueron dopados con fosfolípidos fluorescentes (N - (7-nitro-2-1,3-benzoxadiazol-4-il) -phosphoethanolamine (NBD-PE) y ( N -. (lisamina rodamina B sulfonil) -phosphoethanolamine (LR-PE) y se mezcló con vesículas que contienen no marcados t-SNARE (t-SUV) Inicialmente la fluorescencia de NBD-PE en v-SUV se extinguió mediante la transferencia de energía de resonancia de Förster (FRET ) a LR-PE. Como laboratorioELED v-SUVs se fusionan con no marcados camisetas SUVs, la densidad superficial fluoróforo en la membrana ahora combinada se reduce y el incremento resultante en la fluorescencia NBD-PE informa de la extensión de los lípidos de mezcla 10. A medida que el ensayo mayor es fácil de configurar y analizar, se ha utilizado ampliamente para estudiar los mecanismos de fusión SNARE mediada 10-14. Sin embargo, tiene varias limitaciones, tales como una baja sensibilidad y una pobre resolución de tiempo. Lo más importante, como una medición de conjunto, el promedio es los resultados de todos los acontecimientos que hacen discriminación entre acoplamiento y de fusión, así como la detección de intermedios hemifusion difíciles.
La última década varios grupos, incluido el nuestro, se han desarrollado nuevos ensayos para monitorizar los eventos de fusión a nivel de la vesícula única 15-27. Ha y sus colegas utilizaron en V SUV atados sobre una superficie y un seguimiento de su fusión con T-SUV gratuitas 18,19. la mezcla de lípidos se controló utilizando FRET entre un par de fluoróforos unidos a lípidos emcamas en el V y T-SUV, respectivamente, utilizando microscopía de fluorescencia de reflexión total interna (TIRF) 18. Más tarde, el laboratorio de Brunger utiliza una sola especie de lípidos de la etiqueta junto con un marcador de contenidos para la detección simultánea de lípidos y mezclando 20,28. Tanto el lipídica y los marcadores de contenidos se incluyeron en altas concentraciones, auto-enfriamiento rápido; fusión con SUVs no marcados como resultado de la fluorescencia desatenuación 20,28.
Otros han fusionado en V SUV de bicapas planas reconstituidos con t-SNARE 15-17,21-27,29. La geometría planar del objetivo (t-SNARE que contiene) bicapa imita mejor el proceso de fusión fisiológica de vesículas pequeñas, muy curvadas con una membrana de plasma plana. El grupo Steinem emplea membranas de poros que abarca reconstituidos con t-SNARE, suspendidos sobre un sustrato de nitruro de silicio poroso y detecta la fusión con la V-SUV individuales mediante microscopia láser confocal de barrido 23. otros fusadas en V SUV a bicapas planas reconstituidos con t-SNARE, soportados sobre un sustrato de vidrio 15-17,21,22,24-27,29. La gran ventaja de utilizar bicapas soportadas (SBLS) es que la microscopía TIRF se puede utilizar para detectar eventos de conexión y de fusión con una excelente relación señal-ruido y sin interferencia de envío V-SUV, aunque el uso de microfluidos también proporciona una resolución de un solo evento usando -campo lejano estándar microscopía de epifluorescencia 24.
Una preocupación importante es si y cómo las interacciones sustrato bicapa afectar a la calidad apoyados bicapa y el proceso de fusión. Los primeros trabajos hizo uso de SBLS planas que fueron apoyados directamente sobre un sustrato de vidrio o cuarzo 15-17. Estos SBLS se hicieron por adsorción, el estallar, la difusión y la fusión de las membranas t-SUV en el sustrato. Pronto se advirtió, sin embargo, que la omisión de un componente clave t-SNARE, SNAP25, desde SBLS preparados de esta manera resultó en v-SUV de atraque y fusión de cinética indistinguisHable de los obtenidos utilizando la completa t-SNARE 17. Debido SNAP25 es absolutamente necesaria para la fusión in vivo 30,31, la relevancia fisiológica de estos primeros intentos se puso en tela de juicio. El grupo de Tamm superó este desafío mediante la formación de bicapa controla mejor el apoyo 21. Se utiliza la deposición de Langmuir-Blodgett por primera valva libre de proteínas de la SBL, seguido de la fusión de esa monocapa con t-SUVs 21. Esto dio lugar a la fusión SNAP25-dependiente.
Para evitar artefactos potenciales asociados con una bicapa soportada directamente sobre un sustrato de vidrio sin necesidad de utilizar métodos de Langmuir-Blodgett, Karatekin et al. Introdujo un poli hidratado (etilenglicol) (PEG) amortiguador suave, entre la bicapa y el sustrato 24. Esta modificación también resultó en la fusión SNAP25 dependiente 24. Bicapas acolchados en una capa de polímero blando habían sido conocidos para conservar mejor transmembranala movilidad y función de proteínas de 32 años, y habían sido utilizados en estudios de fusión con virus 33. Además, bicapas PEGilados parecen conservar cierta capacidad de auto-sanar y son muy robustos 34,35. En primer lugar, una fracción de cadenas de PEG, de lípidos ligado disponibles comercialmente están incluidos en la membrana de t-SUV. Cuando estas camisetas SUVs se rompen y forman una bicapa planar sobre un sustrato de vidrio, un cepillo de PEG cubre ambas valvas de la bicapa planar. Debido a la formación de bicapa planar es impulsado por la adhesión de las cadenas de PEG que rodean camisetas SUVs sobre la superficie de cristal hidrófilo, estallido liposoma y formación de bicapa planar son relativamente insensibles a la composición lipídica utilizada. Sin embargo, cuando se incluyen grandes cantidades de colesterol, aumentando las propiedades de cohesión de los SUV, las SUVs no pueden estallar de forma espontánea. Si este es el caso, los iones de choque o divalentes osmóticos pueden ser empleados para ayudar a la formación de bicapa planar 25.
Como se mencionó anteriormente, en este apabordaje un cepillo de PEG cubre ambos lados de la planar, bicapa compatible. El cepillo de cara al canal de flujo de microfluidos ayuda a evitar la adhesión no específica de entrantes v-SUVs que también suelen estar cubiertas de una capa de PEG. La formación de complejos v-t-SNARE y comienza a partir de la membrana distal-N-terminales y procede por etapas hacia los dominios de la membrana proximal-36. Para el V-SUV para interactuar con el t-SBL, horizontal y vertical necesidad t-SNARE N-terminales para sobresalir por encima de los cepillos de PEG, que parece ser el caso en las condiciones del ensayo. Altura del cepillo puede adaptarse para estudiar las proteínas distintas de SNAREs variando la densidad de los lípidos PEGilados y la longitud de la cadena PEG 37,38. Otro de los beneficios de los cepillos de PEG que cubren las superficies proximales de las bicapas de fusión es que se mimetizan con el entorno lleno de las membranas biológicas que están llenas de 30.000-40.000 proteínas integrales de membrana por cuadrado micras 39. Al igual que las cadenas de PEG en este ensayo, la repucapa de proteína lsive que cubre las membranas biológicas necesita ser empujado a un lado para permitir el contacto entre las dos bicapas de fosfolípidos para la fusión que se produzca.
canales de flujo de microfluidos se utilizan en este ensayo, ya que ofrecen ventajas únicas. En primer lugar, el flujo de microfluidos permite una mayor deposición uniforme de t-SUV a extenderse y se fusionan para formar el t-SBL. En segundo lugar, el volumen del canal pequeña (<1 l), se reduce el consumo de muestra. En tercer lugar, los pequeños volúmenes requeridos permiten todo el experimento que se llevó a cabo bajo un flujo constante. Flujo elimina débilmente, presumiblemente de forma no específica, se adhirieron en V SUVs de la SBL 16. También mantiene una densidad constante de v-SUV por encima de la t-SBL, lo que simplifica el análisis cinético 17. Por último, vesículas se acopló se distinguen fácilmente de los libres llevadas por el flujo 25. En cuarto lugar, varios canales de microfluidos pueden ser utilizados en el mismo cubreobjetos, cada sondeo de una condición diferente. Esto permite la comparación de las condiciones Duriendo la misma corrida experimental. Un enfoque similar ha sido utilizado por el grupo van Oijen para estudiar la fusión entre el virus de la gripe y SBLS acolchados 33.
En la microscopía TIRF, el decaimiento exponencial del campo evanescente (con una constante de ~ 100 nm) descomposición confines de fluorescencia de excitación a aquellas moléculas que se encuentran en una proximidad muy cercana de la interfaz de la memoria intermedia de cristal. Esto minimiza la contribución de moléculas fluorescentes que están más lejos, aumenta la relación señal-ruido, y permite que la sensibilidad de una sola molécula con tiempos de exposición marco de 10 a 40 mseg. El campo evanescente también conduce a un aumento de la señal en fusión: como la transferencia de lípidos marcados desde el SUV en el SBL, se encuentran, en promedio, en un campo de excitación más fuerte. Este aumento de la fluorescencia es más fuerte para los liposomas más grandes.
Si la luz polarizada se usa para generar el campo evanescente, efectos adicionales contribuyen a los cambios en la fluorescencia tras la tRANSFERENCIA de las etiquetas de los SUV en el SBL. Algunos tintes de lípidos tienen un dipolo de transición orientada con un ángulo medio preferido con respecto a la bicapa en el que están inmersos. Esto crea una diferencia en la cantidad de fluorescencia emitida por los fluoróforos cuando están en la SUV frente a la SBL, ya que el haz polarizado excitará colorantes en las dos membranas de manera diferente. En el primer caso, el haz de excitación va a interactuar con dipolos de transición orientadas alrededor de la camioneta esférica, mientras que para este último, las orientaciones del dipolo se limitarán por la geometría plana SBL. Por ejemplo, cuando se utiliza luz polarizada s incidente (polarizado normal al plano de incidencia), la excitación es más eficiente cuando el colorante se encuentra en el SBL que en el SUV para un orientado paralelo colorante lípidos transición de dipolo a la membrana 29,40 (como la de Dil o DiD 41-43). Un SUV dopado con un fluoróforo tal está atenuada cuando se acopla sobre el SBL (Figura 7, representat ive Resultados). Como un poro de fusión se abre y conecta las membranas SUV y SBL, sondas fluorescentes se difunden en la SBL y se vuelven más probabilidades de ser excitada por el campo evanescente polarizada s 25,27,29. En consecuencia, la señal de fluorescencia integrado alrededor del sitio de fusión aumenta bruscamente durante la transferencia de colorantes de la SUV en el SBL 27 (Figura 3 y Figura 7). Un factor adicional que contribuye a indicar los cambios que acompañan a la fusión se desatenuación de marcadores fluorescentes a medida que se diluyen cuando se transfiere en el SBL. La contribución de desatenuación suele ser menor en comparación con deterioro del campo de polarización y efectos evanescentes en el ensayo descrito aquí, ya que sólo una pequeña fracción (  ) De los lípidos están etiquetados.
) De los lípidos están etiquetados.
El aumento de la señal en fusión puede ser explotado para deducir las propiedades de poro de fusión mediante la comparación del tiempo,1 "src =" / files / ftp_upload / 54349 / 54349eq2.jpg "/>, requerido para un lípido escape a través de un poro que es completamente permeable a los lípidos al tiempo real de la liberación,  . Si las dos escalas de tiempo son comparables, sería concluir que el poro presenta poca resistencia al flujo de los lípidos. Sin embargo, si el tiempo de liberación real es significativamente más largo que el tiempo para la liberación de difusión limitada, esto indicaría un proceso, tales como el parpadeo de los poros, que retarda la liberación de los lípidos. El tiempo de liberación de difusión limitada,
. Si las dos escalas de tiempo son comparables, sería concluir que el poro presenta poca resistencia al flujo de los lípidos. Sin embargo, si el tiempo de liberación real es significativamente más largo que el tiempo para la liberación de difusión limitada, esto indicaría un proceso, tales como el parpadeo de los poros, que retarda la liberación de los lípidos. El tiempo de liberación de difusión limitada,  , Depende del tamaño del liposoma de fusión y lípidos difusividad; su estimación requiere que estos dos parámetros que se deben cuantificar. La sensibilidad única molécula del ensayo permite la difusividad de lípidos que se mide mediante el seguimiento de varios fluoróforos única de lípidos después de su liberación en el SBL para cada evento de fusión 26. El tamaño de cada vesícula fusionandopuede estimarse a 27 mediante la combinación de (i) la intensidad de un solo colorante de los lípidos, (ii) el cambio en la fluorescencia total alrededor de un sitio de acoplamiento después de que todos los fluoróforos se transfieren a la SBL en fusión, (iii) el conocido densidad etiquetado de SUV lípidos, y (iv) la superficie por los lípidos. Para muchos eventos de fusión, se encontró que los tiempos reales de liberación de lípidos a ser mucho más lento de lo esperado por la liberación controlada por difusión 27, como se ha señalado anteriormente suponiendo tamaño SUV uniforme 44. Suponiendo retraso de la liberación de lípidos se debe a poros parpadeo, un modelo cuantitativo permite la estimación de la "apertura de poros", la fracción de tiempo que el poro se mantiene abierto durante la fusión 27.
, Depende del tamaño del liposoma de fusión y lípidos difusividad; su estimación requiere que estos dos parámetros que se deben cuantificar. La sensibilidad única molécula del ensayo permite la difusividad de lípidos que se mide mediante el seguimiento de varios fluoróforos única de lípidos después de su liberación en el SBL para cada evento de fusión 26. El tamaño de cada vesícula fusionandopuede estimarse a 27 mediante la combinación de (i) la intensidad de un solo colorante de los lípidos, (ii) el cambio en la fluorescencia total alrededor de un sitio de acoplamiento después de que todos los fluoróforos se transfieren a la SBL en fusión, (iii) el conocido densidad etiquetado de SUV lípidos, y (iv) la superficie por los lípidos. Para muchos eventos de fusión, se encontró que los tiempos reales de liberación de lípidos a ser mucho más lento de lo esperado por la liberación controlada por difusión 27, como se ha señalado anteriormente suponiendo tamaño SUV uniforme 44. Suponiendo retraso de la liberación de lípidos se debe a poros parpadeo, un modelo cuantitativo permite la estimación de la "apertura de poros", la fracción de tiempo que el poro se mantiene abierto durante la fusión 27.
Siempre que sea posible, es importante para poner a prueba los mecanismos de fusión utilizando tanto los lípidos y las etiquetas de contenidos solubles. Por ejemplo, la liberación de lípidos podría retardarse por procedimientos distintos de parpadeo de poros, tales como la restricción de la difusión de lípidos por las proteínas SNARE que surround camisetasél poro. Si este fuera el caso, entonces la liberación del contenido precedería retira de las etiquetas de lípidos, siempre y cuando el poro es lo suficientemente grande como para permitir el paso de sondas solubles. Un defecto más fundamental en el enfoque podría ser en el supuesto de que la transferencia de lípidos marcados a la SBL se produce a través de un poro de fusión estrecho que conecta el SBL a una vesícula que se ha conservado en gran medida su forma pre-fusión. Transferencia de lípidos en el SBL también podría ser el resultado de una rápida dilatación del poro de fusión con una concomitante, extremadamente rápido colapso de la camioneta en la membrana SBL, como sugerido anteriormente sobre la base de datos de liberación de lípidos solo 29. Seguimiento tanto de lípidos y contenido liberar de forma simultánea, se encontró que muchos poros volvieron a sellar después de soltar todas sus etiquetas de lípidos, pero conserva algunos de su carga soluble 27. Esto indica que al menos algunos liposomas no se colapsan en el SBL después de la fusión, y que la transferencia de colorante de los lípidos en el SBL se produce a través de un poro de fusión. Además, lIPID y contenidos de liberación se produjo de forma simultánea 27, por lo que es poco probable que el retraso de la liberación de lípidos era debido al impedimento de la difusión de lípidos por las proteínas SNARE que rodean el poro 45.
Un protocolo de fusión SUV-SBL que no Liberar la monitorización de contenidos solubles se publicó anteriormente por Karatekin y Rothman 25. Aquí, los desarrollos más recientes se incluyen, seguimiento a saber simultánea de lípidos y liberan los contenidos y la estimación de las propiedades de SUV, lípidos, y poro de fusión 27. El protocolo comienza con instrucciones para la preparación de las células de microfluidos, hechos por la unión de un poli (dimetil siloxano) ranuras (PDMS) de elastómero de bloques que contienen con un cubreobjetos de vidrio 25. A continuación, se explica la preparación de V-SUVs tanto con lípidos y marcadores de contenido. Las secciones 4 y 5 proporcionan instrucciones para el montaje de las células de microfluidos, formando los SBLS in situ y la comprobación de defectos y la fluidez, la introducción de los Servicios VeterinariosUVS en las células de flujo y la detección de eventos de fusión. Sección 6 proporciona instrucciones para el análisis de datos.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. Preparación de un bloque de PDMS para formar el canal de microfluidos

Figura 1. La microfabricación de la plantilla de celda de flujo y la preparación de bloques PDMS. (A) Diseño de una celda de flujo de cuatro canales que se ajusta sobre un cubreobjetos de vidrio 24 x 60 mm (parte inferior). Seis diseños idénticos están dispuestas para que quepan en una oblea de silicio de 10 cm (superior). (B) Cortar el bloque de PDMS espesor de aproximadamente 5-8 mm en una perforadora. (C) La inserción del tubo en el agujero perforado con un par de pinzas. Por favor, haga clic aquí para ver una versión más grande de esta figura.
- Obtener una plantilla de celda de flujo como la de la Figura 1A. canales típicos son 0.3-1 mm de ancho, 70-100 m de altura, y 1-2 cm de largo. Fabricar una plantilla de nosotrosing técnicas estándar de fotolitografía en una sala limpia 25 o si el acceso para sala blanca no está disponible. Alternativamente, la máquina de una plantilla con las dimensiones más grandes de un material adecuado.
Nota: El personal de sala limpia puede entrenar y guiar a los usuarios inexpertos (acceso a una instalación de salas blancas siempre está restringida a los usuarios con una formación adecuada) en el diseño y el orden de una máscara, la limpieza de la oblea, y la fotolitografía. Una vez que se obtiene una plantilla, que puede ser utilizado fuera de la sala limpia varias veces, siempre que se mantiene en un plato cerrado y se tiene cuidado de excluir polvo. - Preparar una mezcla de ~ 100 ml PDMS (base de elastómero de silicona) y ~ 10 ml de agente de reticulación (agente de curado) en un vaso de plástico desechable. Rompa la punta de una pipeta desechable para manejar el PDMS viscosos con mayor facilidad. Pesar el PDMS en la taza plástica como pipeteo no es exacta.
- Se agita bien la mezcla. Retire las muchas burbujas de aire por desgasificación en un desecador de vacío (alrededor de 20min). A medida que la copa se coloca bajo vacío, las burbujas inicialmente crecer en tamaño, aumentando el volumen de la mezcla en gran medida. Controlar el vacío que se aplica y asegurarse de que la copa es lo suficientemente profundo para evitar un derrame.
- Vierta una gota grande de la mezcla de PDMS desgasificado en una placa de Petri de vidrio (150 mm x 20 mm) y presione la oblea en el PDMS con la plantilla hacia arriba. Esto evita las burbujas queden atrapados debajo de la oblea. burbujas atrapadas se expandirán y pueden inclinar la oblea cuando el plato se coloca en el horno. Ahora vierta PDMS desgasificados en la plantilla en la parte superior de la oblea hasta que está cubierto por aproximadamente 5-8 mm de PMDS. Si se produce una burbuja de aire retire suavemente con una punta de pipeta.
- Hornear los PMDS en un horno a 60 ° C durante 3 horas. Asegúrese de que el plato esté nivelada.
- Utilice una nueva hoja de bisturí para cortar un bloque de PDMS que contiene las estructuras de canales moldeados. El bloque de cortar necesita para que quepan en un cubreobjetos (véase el paso 4.1.9).
- Pelar el bloque cortado de la cápsula y plas sobre un pedazo de papel de aluminio limpio.
Nota: Los PMDS bloques reticulados se pueden mantener durante unos meses. En una sola oblea, hay 6 conjuntos de canales de flujo (Figura 1A), por lo 6 PDMS bloques se pueden producir en un solo moldeo PDMS. Después de cortar todos los 6 de los bloques de PDMS, piezas sueltas limpias de PDMS, pero por lo demás no retire el PDMS restantes antes de verter y reticular el siguiente lote, lo que requerirá sólo la mitad de los PDMS utilizados para el primer lote. Una buena plantilla puede durar unos pocos años. - Utilice una perforadora para perforar a través del bloque de PDMS en un movimiento recto (Figura 1B). Comience en el lado de las ranuras de canal. Asegúrese de retirar la pieza troquelada de PDMS. Repita este procedimiento para todos los ocho orificios del diseño de cuatro canales.
- Coloque el lado del canal de bloque PDMS abajo sobre un nuevo y sin arrugas trozo de papel de aluminio. Almacenar el bloque de hasta unos pocos meses en una caja seca, idealmente en un desecador.
- Empuje la tubería (0,25 mm ID, 0,76 mm OD) alrededor de un tercio en el agujero perforado usando un par de pinzas (Figura 1C). Corte el tubo inclinado para la inserción más fácil. Deja tubos de tiempo suficiente para alcanzar el depósito de SUV y la bomba de jeringa, respectivamente. Después de colocar el chip montado en el microscopio, cortar los tubos de nuevo si son demasiado largos.
- Para conectarse a las jeringas de la bomba, cortar un trozo de tubo de silicona más grande (0,51 mm de diámetro, 2,1 mm de diámetro exterior) e insertar el tubo de disolvente en un lado. Este bloque listo-a-uso de PDMS se puede mantener durante unos meses.
2. Limpieza Cubreobjetos
- Cubreobjetos limpias de acuerdo con el protocolo descrito en Karatekin y Rothman 25 utilizando una mezcla fuerte oxidante de ácido sulfúrico (H 2 SO 4) y peróxido de hidrógeno (H 2 O 2). Realizar este procedimiento en una sala limpia, y observar las precauciones de seguridad apropiadas. Como alternativa, utilice una sala limpia limpiado coverslip que está disponible comercialmente (ver Lista de Materiales).
3. Preparación de las v-SUV que contienen tanto lípidos y etiquetas de contenido

Figura 2. Esquema de la preparación de SUV. Los lípidos se mezclan en un tubo de vidrio (1) y el disolvente se evapora para formar una película lipídica mediante la rotación del tubo en un baño de agua (2). Que quedan trazas de disolvente se eliminaron bajo alto vacío (3). La película lipídica se hidrata en un tampón de reconstitución que contiene el detergente y la proteína mientras que se agita vorticialmente (4). Si el contenido de colorante se ha de encapsular, se incluye en este paso, así como en la etapa de dilución (5). La dilución de la concentración de detergente por debajo de su concentración micelar crítica conduce a la formación de liposomas. El detergente se dializa noche fuera de casa (6). Para t-SUV para la formación de SBL incluyendo NBD-PE (verde), las vesículas se hacen flotar en un gradiente de densidad y se recogen enla interfase entre dos capas (7a). Para separar las v-SUV con marcador contenido encapsulado del colorante libre la muestra se ejecuta sobre una columna de exclusión por tamaño y se recogió en fracciones de 0,5 ml (7b). Haga clic aquí para ver una versión más grande de esta figura.
- Preparar 4 l de tampón de reconstitución, que contiene 25 mM HEPES-KOH, KCl 140 mM, 100 mM EGTA y DTT 1 mM, pH 7,4. Utilizar la mayor parte del tampón de diálisis y 100 ml para los otros pasos.
Nota: Otros tampones pueden ser utilizados, pero es importante para mantener la osmolaridad tampón consistente en todo el experimento y elegir condiciones en las que las proteínas son funcionales. Para la preparación de las v-SUV que contienen sólo las etiquetas de lípidos y t-SUV, siga Karatekin y Rothman 25. - Dado que las poblaciones de lípidos se almacenan a -20 ° C en cloroformo o cloroformo: metanol (2: 1, v / v), deje viales alcancen la temperatura ambiente antes de abrir paraevitar la condensación.
- Enjuague un tubo de vidrio con cloroformo para eliminar trazas de detergente y mezclar los lípidos en la relación deseada con una cantidad final de 1 mol de una mezcla de cloroformo y metanol (2: 1, v / v). Sólo use jeringas de vidrio / tubos de manejar disolventes / lípidos orgánicos disueltos.
Nota: Los lípidos utilizados aquí son POPC: SAPE: DOPS: Colesterol: PEG2000-DOPE: hizo en una relación molar de 57,4: 15: 12: 10: 4,6: 1 para que contienen vesículas v-SNARE y POPC: SAPE: DOPS: Colesterol : PI cerebro (4,5) P2: PEG2000-DOPE: NBD-PE (54,9: 15: 12: 10: 3: 4,6: 0,5) para t-SNARE SUVs. Ver Material para más detalles. Otras composiciones lipídicas se pueden usar. - Evaporar el disolvente bajo una corriente suave de nitrógeno, o en un evaporador rotatorio. Sumergir la punta del tubo en un baño de agua (37 ° C) se calienta por encima de la temperatura de fusión más elevado de los lípidos para producir una película de lípido homogénea y para evitar grandes cambios en la temperatura mientras se evapora el disolvente. Comience vacío a alrededor300 mbar hasta que se forma la película de lípido, y luego continúan por ~ 2 min a alta posible de vacío en el evaporador rotatorio.
- Retire el tubo de vidrio y se envuelve en papel de aluminio para evitar la exposición a la luz y eliminar las trazas restantes de disolvente colocando el tubo en un desecador a alto vacío durante al menos 2 horas. Este paso también se puede ejecutar durante la noche.
- Para rehidratar la película lipídica seca, preparar una mezcla de detergente (n-octil-β-D-glucopiranósido, OG), proteína (v-SNARE), y SRB en tampón de reconstitución (500 l de volumen final). Mantener la concentración de detergente final ~ 2 veces superior a la concentración micelar crítica (CMC, 20-25 mM durante OG) y ajustar el volumen final de tal manera que el detergente a la proporción de lípidos es> 10. Disolver sulforodamina B en polvo (SRB) en la solución de proteína-detergente para obtener una concentración final de SRB 10-50 mM agitando suavemente la solución. Ajustar la osmolaridad del tampón de reconstitución cuando se añade una alta concentración de SRB al reducir el concentration de cloruro de potasio en consecuencia.
Nota: lípido-proteína-a (LP) para t-SNARE es alta (LP ~ 20.000, ~ 70 t-SNARE por metro micras 25.). Significativamente mayores densidades t-SNARE en el SBL pueden conducir a agregados de proteínas inactivas con redujo significativamente las tasas de fusión 17,24. LP para v-SNARE es mucho más bajo (LP ~ 200 ~ 7.000 v-SNARE por m 2), cerca de las densidades v-SNARE que se encuentran en las vesículas sinápticas 46,47. Para garantizar la función de la proteína después de la expresión recombinante y la purificación de la V y t-SNARE es útil para llevar a cabo un ensayo de fusión a granel sim 48 antes de pasar por todas las etapas del ensayo fusión de vesículas sola. - Agitar suavemente el tubo de vidrio precalentada que contiene la película lipídica seca, mientras que la adición de la solución de proteína-detergente-SRB desde el paso 3.6. Trate de evitar la creación de burbujas. Continuarán sacudiendo durante 15 minutos a 37 ° C.
- Diluir el detergente cinco veces mediante la adición de reconstitucióntampón que contiene SRB (añadir 2 ml, 2,5 ml de volumen final de vórtex), mientras que rápidamente para evitar gradientes de concentración. Continuarán sacudiendo durante 15 minutos a 1 hora a 37 ° C.
Nota: Longer eficacia de reconstitución de proteínas aumento de incubación. La dilución rápida disminuye la concentración de detergente por debajo de la CMC y conduce a la formación de pequeños liposomas. - Se dializa la suspensión de vesículas primero contra ~ 1 L de tampón de reconstitución durante 1-2 horas a temperatura ambiente y luego contra 3 L de tampón de reconstitución durante la noche a 4 ° C con 4 g de adsorbente de poliestireno, usando un tubo de diálisis de MWCO de 20.000 o cassette. Utilizar diferentes vasos para la diálisis de vesículas con y sin SRB para evitar la contaminación cruzada.
- Equilibrar una columna de filtración de gel con tampón de reconstitución. Ejecutar la suspensión de vesículas a través de la columna para separar SRB libre de los v-SUV con SRB encapsulado. Utilice tampón de reconstitución sin SRB como eluyente una vez que toda la muestra tieneentrado en la columna. Recoger en V SUV en fracciones de 0,5 ml.
- Verificar éxito encapsulación SRB en concentraciones de auto-inactivó mediante la medición de la fluorescencia SRB antes y después de la adición de detergente a una alícuota de 16 vesículas. El uso de un espectrómetro de fluorescencia, excitar la muestra a 550 nm y escanear la emisión SRB entre 570 nm y 630 nm. Esperar un aumento en la fluorescencia pliegue 4-8 SRB dependiendo de la cantidad encapsulada después de la adición de detergente como la membrana se solubiliza y se diluye el SRB en libertad.
- Caracterizar la recuperación de lípidos y de proteínas utilizando la espectroscopia de fluorescencia 24 y electroforesis en gel de SDS-PAGE, respectivamente. Use un método de tinción sensible, especialmente para las muestras t-SUV alta LP (véase la Lista de Materiales). Normalmente alrededor del 50% de ambos de entrada de lípidos y proteína se pierde durante la preparación resultante en un LP cerca del valor nominal.
- Caracterizar tamaños SUV utilizando dispersión dinámica de la luz 24 o electrones microscopy 48. SRB tienda que contiene V-SUV a 4 ° C hasta ~ 3-4 días. No congele, como la congelación y descongelación se rompe la membrana y libera SRB encapsulado.
4. SUV-SBL Fusión Ensayo de Seguimiento de los lípidos de estreno Sólo
- La formación de la bicapa soportada tethered dentro de los canales de flujo de microfluidos
- Coloque el bloque PDMS (paso 1,11) a alto vacío durante al menos 20 min antes del experimento para eliminar gases disueltos. Esto reduce enormemente el riesgo de burbujas de aire en los canales de microfluidos durante el experimento de fusión.
- Activar la configuración de microscopio y se calienta la fase y soporte de muestra (Figuras 3 y 4) a la temperatura deseada.
- Filtrar el tampón de reconstitución usado para diluir las vesículas a través de un filtro con 0,45 micras o menor tamaño de poro.
- Diluir ~ 30 l de la NBD-PE etiquetados camisetas SUVs o sin proteínas (PF-SUV) liposomas de control (accionessolución de lípidos 0,5-1 mM) con ~ 60 l de tampón. La concentración final no es crítica aquí.
- Degas esta mezcla usando una jeringa de 3 ml. Presione a cabo la mayor parte del aire por encima de la muestra, mientras que manteniendo la jeringa vertical. Sellar la punta (sin aguja) utilizando una película de parafina y crear un vacío tirando hacia abajo el émbolo. Toca el cilindro de la jeringa para acelerar la desgasificación de la solución. Repita este proceso varias veces hasta que no se produzcan más burbujas cuando se aplica vacío.
- Use una aguja hipodérmica con un diámetro ligeramente más grande que el tubo que está conectado al bloque de PDMS para perforar un agujero en la tapa de un tubo de microcentrífuga. Asegúrese de que la pieza troquelada de plástico no está dentro del tubo, ya que podría obstruir el tubo conectado a la entrada del canal de microfluidos más tarde.
- Llenar la solución SUV desgasificado en el tubo de microcentrífuga que tiene un agujero en la tapa y colocarla en el soporte de la platina del microscopio que se equilibre a la temperatura establecida.
- place un cubreobjetos previamente lavado (sección 2) en un limpiador de plasma y ejecutar plasma de aire durante unos 5 minutos. Ponga el cubreobjetos tratada con plasma (cara tratada hacia arriba) en la parte superior de unos pocos tejidos sin pelusa que actúa como un cojín.
- Retire el papel de aluminio del bloque PDMS desgasificado y colocar el bloque en la parte superior del cubreobjetos. Cuando se reutilice el bloque PDMS poner y separar un trozo de cinta adhesiva a un lado del canal para limpiarlo. Presione hacia abajo el PDMS bloquear en el cubreobjetos usando un par de pinzas para que se pegue, pero no presione demasiado duro como el vidrio podría romperse.
- Coloque la celda de flujo montado sobre la platina del microscopio y conectar el tubo al depósito SUV y la bomba de jeringa, respectivamente. Tape el cubreobjetos a la etapa (Figura 3B).
- Iniciar la aspiración de SUVs a 3 l / min hasta que la solución llena los canales por completo (~ 2,25 mm / seg para la sección transversal del canal x 300 micras 75 micras). Cuando las soluciones para todos los canales empiezan a moverse hasta THe tubería en el lado de salida, reducir el flujo de 0.5 l / min y se incuba durante 30 a 45 min.
Nota: El tiempo entre el plasma el tratamiento de la lámina de vidrio y fluyendo los SUVs en el canal no debe exceder de 10 a 20 min como el efecto del tratamiento de plasma es transitorio. - Compruebe los canales para cualquier fuga. Utilice un objetivo de aire 10-20x para observar la fluorescencia de NBD-PE de la NBD-PE que contiene T o PF-SUV. Excite NBD fluoróforos usando el láser de 488 nm. Las fugas son difíciles de detectar usando iluminación de campo brillante.
- Poner un tubo con tampón de reconstitución desgasificado en el soporte y dejar que la temperatura se equilibre cambios tan rápidos en la temperatura podría causar defectos en el SBL. Detener el flujo y esperar ~ 1 min para asegurarse de que el flujo se ha detenido por completo y ninguna burbuja de aire será aspirado en el tubo antes de cambiar los tubos de entrada para el tampón desgasificado.
- Enjuague todos los canales con tampón desgasificado para lavar todo terrenos no consolidados.
- Cambiar a un higher objetivo ampliación TIRF (60X, aceite, NA 1,45 a 1,49) y verifique que la bicapa parece homogénea y está libre de cualquier defecto evidente a gran escala, tales como manchas oscuras o túbulos de lípidos que se extienden hacia fuera de la LPS.
- Comprobar la fluidez de la bicapa
- Si una unidad dedicada FRAP no está disponible, o si una secuencia FRAP no se puede programar, a continuación, cualitativamente fluidez de la membrana de prueba de la siguiente manera.
- Cierre el diafragma de campo a un tamaño pequeño (~ 40 m de diámetro) y ajustar la intensidad de la luz de excitación de 488 nm a través del software (20-80 mW, o 15-60 NW / m 2) para blanquear la fluorescencia NBD-PE en los expuestos área de manera significativa, pero no completamente. Para una bicapa de líquidos, en estado estacionario, la intensidad de fluorescencia en el medio de la zona expuesta debe ser más bajo que en los bordes, como moléculas de NBD-PE intactos entran en el área expuesta y difunden una cierta distancia antes del blanqueo. Por el contrario, si el adherido a la superficie SUV no pudo estallar, o por alguna otra razón la bicapa soportado no es fluida, todos los fluoróforos en el área expuesta debe blanquear.
Nota: Los valores de intensidad de láser en este y posteriores pasos se dan como punto de partida en bruto y deben ser optimizados para un conjunto dado de condiciones. - Detener la iluminación y empezar de nuevo unos minutos más tarde para verificar los resultados de las mediciones de estado estacionario
- Cierre el diafragma de campo a un tamaño pequeño (~ 40 m de diámetro) y ajustar la intensidad de la luz de excitación de 488 nm a través del software (20-80 mW, o 15-60 NW / m 2) para blanquear la fluorescencia NBD-PE en los expuestos área de manera significativa, pero no completamente. Para una bicapa de líquidos, en estado estacionario, la intensidad de fluorescencia en el medio de la zona expuesta debe ser más bajo que en los bordes, como moléculas de NBD-PE intactos entran en el área expuesta y difunden una cierta distancia antes del blanqueo. Por el contrario, si el adherido a la superficie SUV no pudo estallar, o por alguna otra razón la bicapa soportado no es fluida, todos los fluoróforos en el área expuesta debe blanquear.
- Programar una secuencia FRAP para una medición más cuantitativa, si es posible. Ver archivos suplementarios y la leyenda correspondiente para más detalles.
Nota: A veces, todo terrenos se adherirán en el cubreobjetos de vidrio, pero no pueden romperse y formar una bicapa fluida. Si esto ocurre, enjuague los canales con tampón de reconstitución que contiene desgasificado mM Mg 2 + 10 para ayudar a la formación de bicapa soportada. Utilice blanqueo de fluorescencia para evaluar la fluidez bicapa como en 4.2. Una vez que se forma una doble capa de líquido, enjuague con Mg 2 + libre recoNSTITUCIÓN tampón.
- Si una unidad dedicada FRAP no está disponible, o si una secuencia FRAP no se puede programar, a continuación, cualitativamente fluidez de la membrana de prueba de la siguiente manera.
- La introducción de las v-SUV en los canales de flujo de microfluidos
- Degas tampón de reconstitución y lo utilizan para diluir la solución madre de v-SUV por un factor de aproximadamente 3 10-05 10 dependiendo de la concentración de la v-SUV. Objetivo para una dilución que resulta en alrededor de 10 a 100 eventos de fusión dentro de 60 seg en el campo de visión.
Nota: Demasiados fusiones aumentan la fluorescencia de fondo (ya que cada evento depósitos LR-PE o DiD etiquetas de lípidos en el SBL) y hacen que la detección y el análisis de los eventos de fusión difícil. Por el contrario, una tasa de fusión que es demasiado bajos resultados en las estadísticas pobres o requiere la adquisición de muchas más películas. Para una concentración v-SUV de lípidos 0,1 mM, empezar por dilución de 5 l SUV de valores en tampón de reconstitución 995 l y luego se diluye 5-50 l de esto en 950-995 l de tampón de reconstitución. - Deje que la temperatura se equilibre antes de detener el flujo y la inserting el tubo de entrada en la solución v-SUV diluida.
- Ajuste el ángulo TIRF y la polarización de la siguiente manera.
- Después de ajustar la polarización deseada girando el haz de excitación, ajustar la inclinación del espejo al mando de la posición del haz a través de un motor paso a paso a través del software. Mueva lentamente la posición del haz de láser desde el centro del objetivo en el plano focal de nuevo a un lado fuera del centro. Observe la luz del objetivo en el lado frontal emergen con un ángulo cada vez mayor con respecto al eje objetivo que la posición se desplaza más fuera del centro.
- Tenga en cuenta la posición del motor cuando el haz que sale desaparece primero en el objetivo, es decir, cuando se logra la primera TIR.
- Lentamente mantener en movimiento la posición del haz más fuera del centro mientras se monitoriza la fluorescencia de la superficie. Tenga en cuenta la posición del motor cuando la superficie de fluorescencia desaparece cuando el haz se ha movido demasiado lejos fuera del centro.
- Elegir un haz de positio n entre los dos límites determinado anteriormente. Para obtener la mejor relación señal-ruido y más mejora de la señal en la fusión, elegir una profundidad de penetración poco profunda (posición del haz más cerca del borde del objetivo) que todavía proporciona una iluminación uniforme de la viewfield.
Nota: Es mejor es mantener la misma posición del haz TIR (la misma profundidad de penetración) para todos los experimentos después que los ajustes están optimizados. Asegúrese de que la rotación de la polarización no da lugar a cambios en la posición del haz.
- De flujo V-SUVs en el canal a una velocidad de flujo de 2 l / min, que corresponde a una velocidad de flujo lineal promedio de ~ 1,5 mm / seg para una sección transversal de 75 micras x 300 micras. Cambiar a la configuración de excitación / emisión para supervisar la mezcla de lípidos (LR o sólo lo hizo).
- Degas tampón de reconstitución y lo utilizan para diluir la solución madre de v-SUV por un factor de aproximadamente 3 10-05 10 dependiendo de la concentración de la v-SUV. Objetivo para una dilución que resulta en alrededor de 10 a 100 eventos de fusión dentro de 60 seg en el campo de visión.
- La observación de la fusión entre individuales v-terrenos y el SBL
.jpg "/>
Figura 3. La configuración experimental pTIRF. (A) Representación esquemática de un v-SUV y un t-SBL en un sustrato de vidrio. TIRFM imágenes en falso color de evento de fusión única SUV-SBL muestran acoplamiento de lípidos (1) y la liberación de colorante de lípidos en el SBL (2) seguido por el blanqueamiento y la disminución de la intensidad de fluorescencia (3). Se muestra la intensidad de fluorescencia total (suma de los valores de los píxeles en el cuadro 5.3 micras x 5,3 m) de la señal. (B) El cubreobjetos unido al bloque de PDMS se graba en el escenario climatizada. tubo de entrada para los canales de microfluidos tomen muestras de tubo en el soporte de la muestra de metal (derecha) aspirado por la bomba de jeringa (izquierda). Por debajo de la bomba es la unidad de emisión dual. Haga clic aquí para ver una versión más grande de esta figura.
OAD / 54349 / 54349fig4.jpg "/>
Figura 4. Representación esquemática de la configuración experimental. La onda evanescente se crea en la interfaz de la memoria intermedia de vidrio en el canal microfluídico. El SBL se forma en el cristal y v-SUV se aspiran desde el soporte de la muestra de metal (parte superior derecha) a través del canal en la jeringa (parte superior izquierda). M, espejo; DM, espejo dicroico; L, la lente; F, filtro; P, polarizador. Por favor, haga clic aquí para ver una versión más grande de esta figura.
- Continuamente excitar la fluorescencia y supervisar v-SUV usando un 561 nm (para LR-PE) o 638 nm (para DID) de láser en función del fluoróforo incorporado en v-SUV. Como v-SUV llegan al canal de flujo y el muelle en y se fusionan con el SBL, la señal de fluorescencia de fondo empieza a acumularse.
- Ajustar la intensidad del láser de excitación a través del software para blanquear continuamente el fondofluorescencia de tal manera que al nuevos eventos de conexión y el estado de equilibrio de fusión puede ser fácilmente observado.
Nota: Si blanqueo es demasiado lento, la fluorescencia de fondo será demasiado alta. Si blanqueo es demasiado rápido, las señales de los SUV atracados y de fluoróforos liberados en el SBL se desvanecen rápidamente, acortando la ventana durante la cual la fusión de los SUV atracados puede ser detectado o un fluoróforo puede ser rastreado. Para LR-PE excitado a 561 nm, 2.5 a 7.5 mW de potencia iluminando un círculo 190 micras de diámetro (100 a 250 nW / m 2) es un valor razonable para empezar. Por 638 nm de excitación de DiD, 0.8-1.6 mW de potencia sobre un círculo de 190 m de diámetro (30-60 NW / m 2) se puede utilizar para las pruebas iniciales. - Adquirir varias películas en diferentes posiciones en un canal dado. Compruebe fluorescencia NBD-PE para verificar que la SBL no tiene defectos en estas posiciones. Adquirir películas de formato completo a la tasa máxima (~ 50 cuadros / seg) o una región recortada de interés a mayor velocidad de cuadro (hasta~ 100 Hz) durante 60 segundos.
- Mover a otro canal de microfluidos y repetir las grabaciones para otras condiciones. Incluir los controles negativos, tales como un SBL o SUVs libre de proteína, añadir el dominio citoplásmico soluble de la v-SNARE VAMP2 (CDV) como un inhibidor o tratar v-SUV con neurotoxina del tétanos (30 min a 37 ° C) en una o más de los canales en el mismo cubreobjetos.
- Limpiar y reciclar PDMS
- Con el fin de volver a utilizar el bloque de PDMS, enjuague los canales de microfluidos con ~ 200 l de etanol al 70% a un caudal de 5 l / min. Por último, aspirar aire a través de los tubos y canales.
- Separar los PDMS bloquean suavemente desde el cubreobjetos apretándolo ligeramente. Colocarlo en una hoja limpia de papel de aluminio. Almacenar en un desecador de vacío. Puede ser reutilizado varias veces.
- Para una limpieza más a fondo enjuagar los canales con una solución de hidróxido de detergente o de sodio antes de 70% de etanol. Alternativamente, quite todos los tubos lado a otro M El PDMS y sonicar el bloque durante 30 minutos en isopropanol, antes del secado y la inserción de un tubo nuevo.
5. SUV-SBL Fusión Ensayo de Seguimiento contenido en lípidos y de liberación simultáneamente
- Para el monitoreo de doble color de lípidos simultánea y contenidos solubles en libertad, sigue los mismos pasos que en el apartado 4, excepto liposomas uso rotuladas con tanto un lípido (DID) y un contenido de solubles (SRB) etiqueta, tal como se explica en la sección 3. SRB está encapsulado a 10 mM y es inicialmente muy auto-apagará.
- El uso de la configuración mostrada esquemáticamente en la Figura 4, excitar SRB y DiD fluorescencia simultáneamente usando 561 nm y 638 nm láser, respectivamente. Un espejo dicroico (640 nm) se divide la emisión en dos haces que se ejecutan a través de un corto (595/50 nm) y un filtro (700/75 nm) de longitud de onda larga para detectar la SRB y DiD emisiones, respectivamente. Los dos haces de emisión se proyectan de lado a lado en el chip EM-CCD.
- Análisis de los datos FRAP
- Utilice el programa MATLAB proporcionado en la información complementaria para estimar el coeficiente de difusión de lípidos, D. El programa lee una lista de archivos OME-TIFF 49, detecta el área de blanqueado, representa los valores medios de píxel en el área de blanqueado como una función del tiempo, y se ajusta a la curva de recuperación resultante a un modelo por Soumpasis 50 para extraer el tiempo de recuperación,
 , Donde w es el radio del círculo blanqueada.
, Donde w es el radio del círculo blanqueada.
Nota: Se proporcionó un programa MATLAB anterior 25 para el análisis de las películas FRAP utilizando archivos Nikon ND2. El actual programa lee los archivos TIFF OME-49, porque la mayoría de los formatos de archivo pueden ser fácilmente convertidos en OME-TIFF. Un análisis cuantitativo de los datos FRAP es más fácil y más precisa cuando se blanquea es instantánea, el bleacHED región es un círculo, y la decoloración durante la lectura de salida es insignificante. Aunque estos criterios no se cumplen estrictamente en las FRAP mediciones simples descritos aquí, se puede obtener una estimación razonable del coeficiente de difusión. Para una estimación más precisa, utilizar el seguimiento de los tintes de lípidos simples (sección 6.3).
- Utilice el programa MATLAB proporcionado en la información complementaria para estimar el coeficiente de difusión de lípidos, D. El programa lee una lista de archivos OME-TIFF 49, detecta el área de blanqueado, representa los valores medios de píxel en el área de blanqueado como una función del tiempo, y se ajusta a la curva de recuperación resultante a un modelo por Soumpasis 50 para extraer el tiempo de recuperación,
- Análisis de la tasa de acoplamiento, la tasa de fusión y los tiempos de retardo-docking-a la fusión
- ImageJ utilizar para abrir una película para analizar y ajustar el brillo y el contraste para identificar claramente acoplamiento de vesículas y eventos de fusión. Iniciar el plugin SpeckleTrackerJ 26. Véase Smith et al. 26 y en línea documentación para SpeckleTrackerJ para obtener instrucciones para su uso.
- Identificar todos los SUV de nueva atracados en SpeckleTrackerJ. Para distinguir los SUV que se acoplan fuertemente de las que rebotan en el SBL, imponer una duración de conexión mínima de unos cuantos fotogramas. Guardar las pistas, y repetir para todas las películas.
Nota: Para el doctasa de rey, lo único que importa es identificar cuando un SUV acoplado, por lo que sólo el primer fotograma en el que los SUVs atracado tiene que ser registrado en estas pistas. El resto de las pistas no se tendrá en cuenta en el análisis. Sin embargo, auto-seguimiento de cada SUV hasta que se blanquea o fusibles ayuda SUV marca que ya han sido realizados. - Identificar todas las vesículas se fusionan. Para estos, las pistas deben incluir todos los fotogramas de la primera trama de un SUV acoplado hasta que la primera trama en la que la fusión es evidente por un aumento súbito en la fluorescencia del punto de seguimiento. La duración de estas pistas se utilizan para calcular los retrasos-docking-a fusión. Guardar todas las pistas. Repita este procedimiento para todas las películas.
- Para el análisis de los datos de conexión por lotes o de fusión, compilar una lista de los archivos de la trayectoria de las películas relevantes y ejecutar los programas de MATLAB provistos de Karatekin y Rothman 25. Siga las instrucciones que se proporcionan junto con los programas.
Nota: Los programas de trazar el semeneventos de conexión y de fusión ulative como una función del tiempo, en base a información extraída de los archivos de trayectoria. las tasas de atraque y de fusión se estiman a partir de las pendientes de estas parcelas. Para los datos de fusión, los retrasos-docking-a de fusión también se calculan y su distribución representan como un gráfico de supervivencia, es decir, la probabilidad de que la fusión todavía no se ha producido por un retardo dado después de acoplamiento.
- difusividad de lípidos
- Para cada evento de fusión, realizar un seguimiento de los lípidos fluorescentes individuales a medida que se discernible cuando se han difundido suficientemente lejos del sitio de fusión (Figura 6). Utilice SpeckleTrackerJ para el seguimiento, y guardar las pistas para su posterior análisis utilizando MATLAB. Dependiendo del tamaño de la vesícula de fusión, típicamente 3-30 fluoróforos individuales pueden ser rastreados. Debido a que las pistas más largas son deseables para el cálculo de
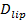 , Trata de realizar un seguimiento de las moléculas individuales como a largocomo sea posible, utilizando la corrección manual de las trayectorias si es necesario.
, Trata de realizar un seguimiento de las moléculas individuales como a largocomo sea posible, utilizando la corrección manual de las trayectorias si es necesario. - Calcular el cuadrado medio del desplazamiento (MSD) para el marcador trayectorias individuales de lípidos que duran> 40-50 marcos (alrededor de ~ 1,5 seg). Utilice el MSD para calcular el coeficiente de difusión de los lípidos, . Ver Smith et al. 26 y Stratton et al. 27 para más detalles.
- Para cada evento de fusión, realizar un seguimiento de los lípidos fluorescentes individuales a medida que se discernible cuando se han difundido suficientemente lejos del sitio de fusión (Figura 6). Utilice SpeckleTrackerJ para el seguimiento, y guardar las pistas para su posterior análisis utilizando MATLAB. Dependiendo del tamaño de la vesícula de fusión, típicamente 3-30 fluoróforos individuales pueden ser rastreados. Debido a que las pistas más largas son deseables para el cálculo de
- Intensidad colorante único de lípidos, el factor de reducción de la intensidad de SUV-SBL, y el tamaño de las vesículas
- Medir la suma de valores de píxel de un marcador de lípidos en un píxel 3 x 3 en la zona (0,8 m x 0,8 m) alrededor del marcador de ~ 15 cuadros antes y después de los blanqueadores de marcadores en un solo paso. Restar la intensidad de fondo promedio de más de los marcos de post-blanqueador, de la intensidad pre-blanqueador promedio antes del blanqueo para obtener la intensidad del marcador de lípidos de seguimiento. Repita la medición para tantos marcadores como práctico para un dado película.
- Trazar la distribución de intensidades de etiqueta única de lípidos,
 Y adaptarse a una gaussiana para estimar la media.
Y adaptarse a una gaussiana para estimar la media. - Calcular el retardo entre cuando se produjo la fusión y cuando la trayectoria de un solo marcador de lípidos lanzado en ese evento terminó en un blanqueo de un solo paso. Obtener el tiempo de blanqueo para un fluoróforo en el SBL,
 , Representando la función de supervivencia de los retrasos y la instalación de una exponencial.
, Representando la función de supervivencia de los retrasos y la instalación de una exponencial.
Nota: Debido a que las parejas polarizadas de campo de excitación más débilmente a los fluoróforos en un SUV, el tiempo de blanqueo de un SUV es por lo general mucho más lento 27. - Estimar
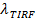 , El factor de reducción de intensidad para un colorante de lípidos cuando en el SUV con respecto a cuando está en la SBL, siguiendo Stratton 27 (tp_upload / 54349 / 54349eq8.jpg "/> es la intensidad de un solo tinte en el SUV).
, El factor de reducción de intensidad para un colorante de lípidos cuando en el SUV con respecto a cuando está en la SBL, siguiendo Stratton 27 (tp_upload / 54349 / 54349eq8.jpg "/> es la intensidad de un solo tinte en el SUV).
Nota: Si blanqueo eran insignificantes, sería igual a la intensidad atracado  (punto (1) en la Figura 3A, panel derecho), dividido por la intensidad total alcanzado después de que todos los fluoróforos se depositan en la LPS sobre la fusión (línea discontinua etiquetado
(punto (1) en la Figura 3A, panel derecho), dividido por la intensidad total alcanzado después de que todos los fluoróforos se depositan en la LPS sobre la fusión (línea discontinua etiquetado  en la Figura 3). Sin embargo, debido a la rápida de blanqueo en la SBL, normalmente no se alcanza y una estimación precisa de Requiere instalación la cinética de liberación a una expresión que incluye 27 udes tanto (Véase 6.4.3) y . Expresiones separados se proporcionan en Stratton et al 27 para los casos de la cinética de poros limitado y de liberación limitada de difusión.; el mejor de los casos en forma varía de un evento a otro (ver 6.5.1 para el caso de la cinética de poros limitada).
en la Figura 3). Sin embargo, debido a la rápida de blanqueo en la SBL, normalmente no se alcanza y una estimación precisa de Requiere instalación la cinética de liberación a una expresión que incluye 27 udes tanto (Véase 6.4.3) y . Expresiones separados se proporcionan en Stratton et al 27 para los casos de la cinética de poros limitado y de liberación limitada de difusión.; el mejor de los casos en forma varía de un evento a otro (ver 6.5.1 para el caso de la cinética de poros limitada). - Calcular el área de la vesícula para los eventos individuales de 27
 , dónde es la intensidad SUV atracado, es la intensidad de un solo colorante de lípidos significa en el SBL, es el factor de reducción de la intensidad de un colorante de los lípidos cuando se encuentra en la SUV con respecto a cuando está en la SBL, yación 1 "src =" / files / ftp_upload / 54349 / 54349eq12.jpg "/> es la densidad de área conocida de colorantes de lípidos.
, dónde es la intensidad SUV atracado, es la intensidad de un solo colorante de lípidos significa en el SBL, es el factor de reducción de la intensidad de un colorante de los lípidos cuando se encuentra en la SUV con respecto a cuando está en la SBL, yación 1 "src =" / files / ftp_upload / 54349 / 54349eq12.jpg "/> es la densidad de área conocida de colorantes de lípidos.
- Propiedades de fusión de poro
- Suponiendo que la liberación es de poros limitado, cabe la cinética de liberación etiqueta de lípidos al 27
 , dónde
, dónde  es la intensidad SUV atracado justo antes de la fusión y los otros parámetros son como se definen anteriormente. Utilice el valor de obtenido en el punto 6.4.3 como un parámetro fijo, y extraer las mejores estimaciones aptos para la y
es la intensidad SUV atracado justo antes de la fusión y los otros parámetros son como se definen anteriormente. Utilice el valor de obtenido en el punto 6.4.3 como un parámetro fijo, y extraer las mejores estimaciones aptos para la y  .
. - Estimar la fracción de tiempo que el poro está abierto, P 0, suponiendo que el retraso de la liberación de lípidos se debe a poros parpadeo 27:60;
 =
=  , Donde una ves es el área de la vesícula (sección 6.4.5), b es la altura de los poros (por lo general llevado a ser ~ 15 nm), r p ≈ 3 nm es el radio de poro efectivo como se ve por las etiquetas de lípidos que se difunden e incluye la mitad el espesor de dos capas (~ 2 nm), es la difusividad de lípidos (calculado en 6.3), y es el momento de que los lípidos para ser liberados de la camioneta en el SBL (desde 6.5.1).
, Donde una ves es el área de la vesícula (sección 6.4.5), b es la altura de los poros (por lo general llevado a ser ~ 15 nm), r p ≈ 3 nm es el radio de poro efectivo como se ve por las etiquetas de lípidos que se difunden e incluye la mitad el espesor de dos capas (~ 2 nm), es la difusividad de lípidos (calculado en 6.3), y es el momento de que los lípidos para ser liberados de la camioneta en el SBL (desde 6.5.1). - Para confirmar que un P nominal 0> 1 indica un poro completamente abierta, P 0 = 1, se ajusta a la evolución en el tiempo la intensidad a la ecuación 4 de Stratton et al. 27, la cinética predicho para un poro permanentemente abierta. Este ajuste debe ser mejor que fitting en la expresión 6.5.1 para un poro abierto permanentemente.
- Suponiendo que la liberación es de poros limitado, cabe la cinética de liberación etiqueta de lípidos al 27
 , Donde w es el radio del círculo blanqueada.
, Donde w es el radio del círculo blanqueada.  , Representando la función de supervivencia de los retrasos y la instalación de una exponencial.
, Representando la función de supervivencia de los retrasos y la instalación de una exponencial.  (punto (1) en la Figura 3A, panel derecho), dividido por la intensidad total alcanzado después de que todos los fluoróforos se depositan en la LPS sobre la fusión (línea discontinua etiquetado
(punto (1) en la Figura 3A, panel derecho), dividido por la intensidad total alcanzado después de que todos los fluoróforos se depositan en la LPS sobre la fusión (línea discontinua etiquetado  en la Figura 3). Sin embargo, debido a la rápida de blanqueo en la SBL,
en la Figura 3). Sin embargo, debido a la rápida de blanqueo en la SBL,  , dónde
, dónde  , dónde
, dónde  es la intensidad SUV atracado justo antes de la fusión y los otros parámetros son como se definen anteriormente. Utilice el valor de
es la intensidad SUV atracado justo antes de la fusión y los otros parámetros son como se definen anteriormente. Utilice el valor de  =
=  , Donde una ves es el área de la vesícula (sección 6.4.5), b es la altura de los poros (por lo general llevado a ser ~ 15 nm), r p ≈ 3 nm es el radio de poro efectivo como se ve por las etiquetas de lípidos que se difunden e incluye la mitad el espesor de dos capas (~ 2 nm),
, Donde una ves es el área de la vesícula (sección 6.4.5), b es la altura de los poros (por lo general llevado a ser ~ 15 nm), r p ≈ 3 nm es el radio de poro efectivo como se ve por las etiquetas de lípidos que se difunden e incluye la mitad el espesor de dos capas (~ 2 nm), Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Calidad SBL
Es crucial para verificar la calidad y fluidez de la SBL antes del experimento de fusión. La fluorescencia en el lado inferior, de vidrio de un canal microfluídico debe ser uniforme, sin defectos evidentes. Si una burbuja de aire pasa a través del canal, se suele dejar cicatrices visibles en el SBL. Si hay tan grandes cicatrices escala / defectos, no use ese canal. A veces los SUV puede adherirse al sustrato pero no pueden romperse y formar un SBL continua. Si ese es el caso, la fluorescencia todavía están muy uniforme, pero no se recuperará después de la decoloración de una pequeña región. La introducción mM Mg 2 + 10 menudo ayuda a resolver este problema. Otros agentes pueden ser introducidos para ayudar a reventar los SUV, como otros cationes divalentes 51, una solución de polímero 52, o choque osmótico 25.
Los cambios de temperatura provocan cambios en el área de SBL porque el área media ocupada por un lípido depende de la temperatura. El aumento de la temperatura o disminuyendo por 5 a 10 ° C o más puede resultar en exceso de área de entrar en tubos extruidos a partir de la superficie, o, manchas SBL-libres oscuras aparecen en la superficie, respectivamente 53. Por tanto, es importante mantener las variaciones de temperatura a un mínimo.
Figura 5 para una SBL que era relativamente libre de defectos y fluida.

Figura 5. FRAP para examinar SBL fluidez. Una secuencia de imágenes que muestran un área iluminada delimitada por el diafragma de campo cerrado (diámetro 49 micras) antes (primer cuadro) y después del blanqueo con un fuerte pulso de luz. La zona es proyectado en baja exposición a la luz para seguir la recuperación de fluorescencia por difusión de NBD marcado lípidos. El gráfico muestra la fluorescencia normalizada como una función del tiempo para varios canales de diferentes cubreobjetos que indican el alto nivel de reproducibilidad del método. La composición de los lípidos de la t-SBL era POPC: SAPE: DOPS: PI cerebro (4,5) P2:: Colesterol PEG2000-DOPE: NBD-PE (54,9: 15: 12: 10: 3: 4,6: 0,5% en moles ) y LP = 20.000. El área de blanqueo era 984.203 μm 3. El tiempo de recuperación y el coeficiente de difusión calculado a partir de un ajuste (véase 6.1) fueron τ = 67,3 seg y D = 1,99 m 2 / seg, respectivamente. Haga clic aquí para ver una versión más grande de esta figura.
Detección de un solo color de acoplamiento y fusión eventos usando una etiqueta de lípidos
recuento manual de los eventos de conexión y de fusión es una tarea tediosa ya que se necesitan 100-400 eventos por condición para buenas estadísticas. Desde SUVs pueden moverse en el SBL antes de fundirse, se necesita un software de seguimiento. SpeckleTrackerJ 26 plug-in para ImageJ tiene características que fueron desarrolladas específicamente para el seguimiento de los SUV para el análisis de los eventos de conexión y de fusión. A menudo, sólo una fracción de vesículas se acopló terminan de fusión;para calcular la tasa de acoplamiento muchos más SUVs necesitan ser rastreado de aquellas que se fusionan o sólo una parte de la película debe ser analizado se detectan hasta un número suficiente de eventos para una estimación fiable. Si el objetivo principal es la tasa de fusión y / o los retrasos en dique-a la fusión, solamente los todo terrenos de fusión necesitan ser identificados y rastreados.
Después de asegurar la calidad de la SBL, SUVs se introducen en los canales y su fluorescencia es de excitación continua bajo TIR usando luz polarizada s. Para los experimentos iniciales, puede ser necesario ajustar para sintonizar la profundidad de penetración de la onda evanescente que resulta el ángulo de incidencia del haz de excitación TIR. Un evento único representante de acoplamiento SUV y de fusión se muestra en la Figura 6A. La vesícula atracado durante la trama marcada D y la fusión se inició en el marco F. Acerca de 1,3 segundos más tarde (trama S) se convirtieron en los lípidos individuales discernible a medida que difunden fuera de la fusión sentarsemi. La intensidad de la fluorescencia total (suma de los valores de píxeles) en cada cuadro se representa como una función del tiempo en la Figura 6B, con las tramas en que se produjo indicados de acoplamiento y fusión. El número acumulado de eventos de fusión como una función del tiempo se representa en la figura 6C, durante cinco películas diferentes utilizando t-SNARE reconstituyó SBLS (t-SBL, LP = 20.000, aproximadamente 70 t-SNARE por metro cuadrado. Micras). La tasa de fusión de cada película se calculó a partir de un ajuste lineal a la pendiente. Esta tasa media ± SEM de fusión se muestra como un gráfico de barras en el panel D. El retraso en dique-a la fusión para el ejemplo que se muestra en la Figura 6A, B, fue de 0,07 seg. La distribución de los retardos calculados de manera similar para un total de 158 eventos se muestra en la Figura 6D como una parcela superviviente, es decir, la probabilidad de que una vesícula atracado aún no se ha fundido por un retardo dado. Alrededor de 1/2 de estas vesículas fusionado a menos de 100 milisegundos. Con el aumento de los niveles de colesterol, la distriblución de los retrasos en dique-a la fusión acorta, a pesar de que la difusión de lípidos ralentiza 27.
Es importante para ejecutar experimentos de control en paralelo. En el mismo cubreobjetos desde que se adquirieron los datos de la Figura 6B, D, las condiciones de control se pusieron a prueba en los canales vecinos. En un canal, el SBL no incluye ninguna t-SNARE. Fuera de 5 películas que duran 60 segundos cada uno, sólo 7 eventos de fusión se registraron en comparación con los 158 cuando el t-SNARE se incluyeron (Figura 6C). En otro canal, se incluyó el dominio citoplásmico soluble de la v-SNARE VAMP2 (CDV). CDV se une el t-SNARE en la SBL, evitando la unión del VAMP2 de longitud completa que se encuentra en los terrenos. En cinco películas 60 seg, no se detectaron eventos de fusión en presencia de CDV (Figura 6C).

La Figura 6. SUV-SBL atraque y fusión eventos. (A) Secuencia de imágenes de un evento de fusión v-SUV con el T-SBL imágenes de microscopía pTIRF siguientes a la difusión de la LR-PE. Las imágenes se toman cada 18 ms, sólo se muestra cada dos tramas. D y F marcan el inicio de acoplamiento y de fusión, respectivamente. etiquetas de lípidos individuales son identificables medida que se difunden fuera del sitio de fusión y otros (S). Dimensiones del marco: 16,7 micras x 16,7 m. (B) La intensidad de fluorescencia como una función de tiempo para el evento de fusión se muestra en (A). (C) Número acumulado de eventos de fusión como una función del tiempo para V-SUV marcadas con DiD de otro conjunto de experimentos. (D) la tasa de fusión normalizada (media ± SEM, sx micras x 2 pM SUV) -1, suponiendo 2 x 10 4 lípidos por SUV y (E) la probabilidad de supervivencia durante un tiempo determinado después del acoplamiento de la vesícula. La composición para todos SBLs fueron POPC: SAPE: DOPS: PI cerebro (4,5) P2:: Colesterol PEG2000-DOPE: NBD-PE (54,9: 15: 12: 10: 3: 4,6: 0,5% en moles) y LP = 20.000. V-SUVs estaban compuestos de POPC: SAPE: DOPS: Colesterol: PEG2000-DOPE: LR-PE (57,4: 15: 12: 10: 4,6: 1% en moles) y LP = 200 para A y B. Para el CE, la v -SUV composición fue similar, excepto que hice fue utilizado en lugar de LR-PE. Por favor, haga clic aquí para ver una versión más grande de esta figura.
Difusividad lípidos, vesículas tamaño, y la fusión de los poros Propiedades
La sensibilidad de una sola molécula del ensayo permite la extracción de varios parámetros, además de los tipos de conexión y de fusión y las distribuciones de retardo-docking-a fusión. Individuales, lípidos marcados con fluorescencia se vuelven discernible medida que se difunden fuera del sitio de fusión (Figura 6A). Seguimiento de tstos proporciona directamente difusividad lipídico 26, 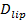 Y una medición digital de la tasa de blanqueo para fluoróforos en el SBL 27. Como se mencionó en la introducción, el de emisión de fluorescencia de las etiquetas difieren en la SUV frente en el SBL, debido a tres factores que son en general difíciles de separar: (i) desatenuación de etiquetas en fusión, debido a su dilución cuando se transfiere en el SBL , (ii) una intensidad de excitación más alta en el SBL debido a la descomposición del campo evanescente generado por TIR, y (iii) la orientación de los dipolos de transición fluoróforo con respecto a la polarización del haz de excitación. La magnitud del factor de (ii) depende de los tamaños relativos de la longitud de decadencia de campo evanescente y el tamaño de vesícula y es diferente para cada evento de fusión debido a la dispersidad de tamaños de SUV. Factor (iii) depende del fluoróforo y la polarización utilizada. En lugar de tratar de di sentangle estos efectos, se puede medir la magnitud del cambio de fluorescencia directamente mediante la comparación de los valores totales de píxeles en una región que rodea a un SUV antes de la fusión y después de todos los fluoróforos se han depositado en el SBL después de la fusión (Figura 3A, (1) vs. ( 2)). A diferencia de los trabajos anteriores 29,44, la región debe elegirse suficientemente grande como para que no fluoróforos se han dejado a través de la difusión de los tiempos considerados después de la fusión. La elección de una zona de pixel de 30 x 30 (8 x 8 micras micras) para el análisis de hasta 1,6 segundos después de la fusión es generalmente adecuada 27. Haciendo caso omiso de blanqueo por el momento, la relación de
Y una medición digital de la tasa de blanqueo para fluoróforos en el SBL 27. Como se mencionó en la introducción, el de emisión de fluorescencia de las etiquetas difieren en la SUV frente en el SBL, debido a tres factores que son en general difíciles de separar: (i) desatenuación de etiquetas en fusión, debido a su dilución cuando se transfiere en el SBL , (ii) una intensidad de excitación más alta en el SBL debido a la descomposición del campo evanescente generado por TIR, y (iii) la orientación de los dipolos de transición fluoróforo con respecto a la polarización del haz de excitación. La magnitud del factor de (ii) depende de los tamaños relativos de la longitud de decadencia de campo evanescente y el tamaño de vesícula y es diferente para cada evento de fusión debido a la dispersidad de tamaños de SUV. Factor (iii) depende del fluoróforo y la polarización utilizada. En lugar de tratar de di sentangle estos efectos, se puede medir la magnitud del cambio de fluorescencia directamente mediante la comparación de los valores totales de píxeles en una región que rodea a un SUV antes de la fusión y después de todos los fluoróforos se han depositado en el SBL después de la fusión (Figura 3A, (1) vs. ( 2)). A diferencia de los trabajos anteriores 29,44, la región debe elegirse suficientemente grande como para que no fluoróforos se han dejado a través de la difusión de los tiempos considerados después de la fusión. La elección de una zona de pixel de 30 x 30 (8 x 8 micras micras) para el análisis de hasta 1,6 segundos después de la fusión es generalmente adecuada 27. Haciendo caso omiso de blanqueo por el momento, la relación de  antes y después de la fusión proporciona el factor de reducción de la intensidad de fluorescencia,
antes y después de la fusión proporciona el factor de reducción de la intensidad de fluorescencia, 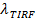 . El tamaño de las vesículas se puede estimar debido a la intensidad de fluorescencia de una sola etiqueta en el SBL (iles / ftp_upload / 54349 / 54349eq6.jpg "/>), el factor por el cual su intensidad se reduciría si se tratara de un SUV ( ), La densidad conocida de etiquetas en el SUV, y el área ocupada por un lípido. En la práctica, el blanqueo es generalmente lo suficientemente rápido que en el momento en todos los marcadores se depositan en la SBL, algunos ya han blanqueado. Por esta razón, para una estimación precisa, el factor de reducción de la intensidad de fluorescencia, , Y el tiempo de la liberación de lípidos,
. El tamaño de las vesículas se puede estimar debido a la intensidad de fluorescencia de una sola etiqueta en el SBL (iles / ftp_upload / 54349 / 54349eq6.jpg "/>), el factor por el cual su intensidad se reduciría si se tratara de un SUV ( ), La densidad conocida de etiquetas en el SUV, y el área ocupada por un lípido. En la práctica, el blanqueo es generalmente lo suficientemente rápido que en el momento en todos los marcadores se depositan en la SBL, algunos ya han blanqueado. Por esta razón, para una estimación precisa, el factor de reducción de la intensidad de fluorescencia, , Y el tiempo de la liberación de lípidos,  , Están mejor extraída de un ajuste a la evolución temporal de los valores totales de píxeles que tiene en cuenta de blanqueo 27. La tasa de blanqueo de etiquetas en el SBL se estima independientemente de seguimiento único tinte de lípidos, o mediante la instalación de una exponencial con el perfil total de la intensidad,
, Están mejor extraída de un ajuste a la evolución temporal de los valores totales de píxeles que tiene en cuenta de blanqueo 27. La tasa de blanqueo de etiquetas en el SBL se estima independientemente de seguimiento único tinte de lípidos, o mediante la instalación de una exponencial con el perfil total de la intensidad,  , A veces largas (por ejemplo, el régimen de (3) en la figura 3). Conociendo el tamaño de una vesícula de fusión y la difusividad de lípidos permite la comparación del tiempo real la liberación de lípidos, , Para el tiempo de difusión de un lípido alrededor de la vesícula
, A veces largas (por ejemplo, el régimen de (3) en la figura 3). Conociendo el tamaño de una vesícula de fusión y la difusividad de lípidos permite la comparación del tiempo real la liberación de lípidos, , Para el tiempo de difusión de un lípido alrededor de la vesícula  , Donde A es el área VES vesículas y es la difusividad de los lípidos. Si las dos escalas de tiempo son comparables, el poro presenta poca resistencia a la liberación de lípidos, y la liberación es de difusión limitada. Por el contrario, si
, Donde A es el área VES vesículas y es la difusividad de los lípidos. Si las dos escalas de tiempo son comparables, el poro presenta poca resistencia a la liberación de lípidos, y la liberación es de difusión limitada. Por el contrario, si  , Entonces el poro retarda significativamente el flujo de lípidos. Una medida cuantitativa de la retraso es la "apertura de poros", P 0, igual a la relación
, Entonces el poro retarda significativamente el flujo de lípidos. Una medida cuantitativa de la retraso es la "apertura de poros", P 0, igual a la relación  veces un factor oPara f unidad relacionado con geometría de los poros 27. Para una de dos estados, poro abierto / cerrado, P 0 es la fracción de tiempo en el estado abierto. Para un poro parpadeo cuyo tamaño varía de forma continua, P 0 representa el radio promediada en el tiempo con relación al radio completamente abierta.
veces un factor oPara f unidad relacionado con geometría de los poros 27. Para una de dos estados, poro abierto / cerrado, P 0 es la fracción de tiempo en el estado abierto. Para un poro parpadeo cuyo tamaño varía de forma continua, P 0 representa el radio promediada en el tiempo con relación al radio completamente abierta.
La relación señal-ruido tiene que ser lo suficientemente bueno para el seguimiento de los fluoróforos de lípidos individuales. Esto se logra más fácilmente en los experimentos de un solo color, usando un marcador brillante como LR-PE.
Detección simultánea de los lípidos y soluble de carga de lanzamiento
Un evento representativo se muestra en la Figura 7. A las altas concentraciones utilizadas para la encapsulación, la fluorescencia SRB es inicialmente auto-apagado. Por lo tanto, la mayor parte atracados en V SUV sólo son detectables por su señal DiD 27. Un fuerte aumentoEn el TID fluorescencia marca la mezcla de lípidos, como en los experimentos de un solo color en lípidos de la etiqueta. El inicio de la liberación de lípidos coincidió con el inicio de un aumento de la fluorescencia SRB. Este aumento era más probable debido a desatenuación SRB como las moléculas de SRB escaparon de la SUV y el SRB restante se diluyó a concentraciones por debajo de auto-enfriamiento. Curiosamente, la fluorescencia SRB alcanzó una meseta estable. Si el poro de fusión se mantuvo abierta, uno esperaría que la señal de SRB para aumentar a un máximo debido a desatenuación, seguido de una disminución de fondo como el SRB restante salió de la camioneta, o si el SUV se desplomó en el SBL. La meseta estable más probable indica el poro volverse a cerrar después de soltar todas las etiquetas de lípidos y una fracción de la carga soluble.
En algunos casos, la señal de la liberación de lípidos no se acompañó de cualquier contenido detectables liberar señales. Esto podría ser, ya sea porque los poros eran demasiado pequeños para permitir el paso de SRB, o algunala forma en la camioneta ya había perdido su carga soluble. Para distinguir entre estas dos posibilidades, el blanqueado de SUVs atracados puede ser analizado. El blanqueo de la etiqueta de lípidos DiD reduce su señal de fluorescencia en cuestión de segundos, mientras que la decoloración de los resultados SRB inicialmente independientes se extinguió en un aumento de la señal SRB como una fracción de la SRB son foto-convertidos a otros productos y auto-extinción se alivia. Aunque la mayoría de los SUV atracados (48 de los 58 analizados eventos) muestran el aumento esperado en la fluorescencia SRB durante largos períodos de iluminación, una pequeña fracción (10/58) no mostró ninguna señal de SRB en absoluto. Por lo tanto, una pequeña fracción de las SUVs puede perder su contenido soluble durante el período entre su preparación y su uso en los experimentos.
En más de 80% de los eventos (74 de 91) para el que tanto SRB y las señales hizo podrían ser seguidas, la fluorescencia SRB aumentó y se mantuvo en una meseta estable 27. En ~ 20% de tstos casos la fluorescencia SRB restante fue liberado repentinamente hasta unos pocos segundos más tarde. Esto podría ser debido a una reapertura del poro que permite una liberación rápida, completa el plazo de 1-2 fotogramas (18-36 ms) o una explosión de la camioneta debido a la foto-daño acumulado 16. Cualquiera que sea su origen, este segundo, liberación rápida de SRB descarta la posibilidad de que la señal de SRB residual después de la liberación inicial podría ser debido a la SRB restante atrapado en el espacio entre el SBL y el sustrato de vidrio después de la liberación completa. Tal mecanismo se sugirió en un estudio anterior en el que el SBL fue apoyada directamente sobre el vidrio, con poco espacio entre las dos 16. Por el contrario, los lípidos PEGilados utilizados en este ensayo deben proporcionar 4-5 nm de espacio entre el SBL y el vaso 25, permitiendo que el SRB se difunda lejos del sitio de fusión.

Figura 7. SIMULTAN eous lípidos y la liberación de contenidos. Las señales de fluorescencia de lípidos (azul) y el contenido (rojo) marcador para un evento de fusión SUV-SBL (para más detalles véase el texto). Las instantáneas de cada canal se muestran en la parte inferior (4,0 m de ancho). Dibujos animados (centro) ilustran las diferentes etapas. fluorescencia (1) muelles de SUV, DiD es baja. (2) La fusión permite la transferencia de DiD en el SBL y DiD fluorescencia aumenta. señal de SRB también aumenta a medida auto-extinción se alivia con el escape de SRB través del poro. (3) la señal de SRB permanece moléculas estables, mientras que hizo difundir dentro de la SBL que indican una posible resellado del poro. (4) Las señales de SRB disminuyen muy rápidamente, debido a la fusión completa rápida o rotura de los liposomas. La composición de las v-SUV era POPC: SAPE: DOPS: PEG2000-DOPE: DiD (67: 15: 12: 5: 1 mol%), LP 200, mientras que T-SBLS estaban compuestos de POPC: SAPE: DOPS: PI cerebro (4,5) P 2: PEG2000-DOPE: NBD-PE (64,5: 15: 12: 3: 5: 0,5% en moles) y LP = 20.000. = "_ Blank"> Haga clic aquí para ver una versión más grande de esta figura.
FRAP_instructions.pdf -. Las instrucciones para configurar una secuencia FRAP Haga clic aquí para descargar este archivo.
ReadMe_FRAP.txt -. Archivo de texto que describe el procedimiento para analizar los datos de recuperación del FRAP utilizando el código de MATLAB que se proporciona por favor haga clic aquí para descargar este archivo.
EK_FRAPbatch.m - archivo de MATLAB para analizar lotes de recuperación después de fluorescencia photobleaching. Este archivo llama a la MATLAB subrutinas EK_getFRAPvals.m y EK_SoumpassisFit.m."_blank"> Haga clic aquí para descargar este archivo.
EK_getFRAPvals.m -. Archivo de MATLAB, subrutina llamada por EK_FRAPbatch.m Haga clic aquí para descargar este archivo.
EK_SoumpassisFit.m -. Archivo de MATLAB, subrutina llamada por EK_FRAPbatch.m Haga clic aquí para descargar este archivo.
JN150923c1_FRAP_list.txt -. Un ejemplo de una lista de nombres de archivo a ser por el script principal MATLAB EK_FRAPbatch.m analizada por lotes favor, haga clic aquí para descargar este archivo.
losLos archivos siguientes pilas TIF y los archivos de texto correspondientes para cinco ejemplos de secuencias de FRAP que pueden ser analizados mediante la ejecución de EK_FRAPbatch.m y elegir el archivo JN150923c1_FRAP_list.txt cuando se le pida que elija una lista de archivos.
JN150923c1_F1_MMStack_Pos0.ome.tif -. FRAP imagen Pila Haga clic aquí para descargar este archivo.
JN150923c1_F1_MMStack_Pos0_metadata.txt -. Archivo de texto con metadatos correspondientes Haga clic aquí para descargar este archivo.
JN150923c1_F2_MMStack_Pos0.ome.tif -. FRAP imagen Pila Plaliviar clic aquí para descargar este archivo.
JN150923c1_F2_MMStack_Pos0_metadata.txt -. Archivo de texto con metadatos correspondientes Haga clic aquí para descargar este archivo.
JN150923c1_F3_MMStack_Pos0.ome.tif -. FRAP imagen Pila Haga clic aquí para descargar este archivo.
JN150923c1_F3_MMStack_Pos0_metadata.txt -. Archivo de texto con metadatos correspondientes Haga clic aquí para descargar este archivo.
JN150923c1_F4_MMStack_Pos0.ome.tif - FRAP pila de imágenes. Por favor, haga clic aquí para descargar este archivo.
JN150923c1_F4_MMStack_Pos0_metadata.txt -. Archivo de texto con metadatos correspondientes Haga clic aquí para descargar este archivo.
JN150923c1_F5_MMStack_Pos0.ome.tif -. FRAP imagen Pila Haga clic aquí para descargar este archivo.
JN150923c1_F5_MMStack_Pos0_metadata.txt -. Archivo de texto con metadatos correspondientes Haga clic aquí para descargar este archivo.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
La implementación exitosa del ensayo de fusión SUV-SBL descrito aquí depende críticamente de varios pasos clave, tales como la reconstitución funcional de las proteínas en liposomas, obteniendo SBLS de buena calidad, y la elección de los parámetros de imagen adecuados para detectar moléculas individuales. Aunque puede tomar algún tiempo y esfuerzo para tener éxito, una vez que el ensayo se lleva a cabo con éxito, proporciona una gran cantidad de información sobre el proceso de fusión no disponible en cualquier otro ensayo de fusión in vitro se discutió en Introducción. Las tasas a las que los SUV acoplarse al y se fusionan con el SBL, la distribución de los tiempos-docking-a la fusión, difusividad de lípidos, el tamaño de cada liposoma de fusión, y si la liberación de lípidos para un evento de fusión es dada por difusión lata o poro controlado ser determinado. Si la liberación es mucho más lento de lo esperado de difusión, un modelo que asume el parpadeo de poro subyace en el retraso de los rendimientos de liberación de los lípidos, P 0, la fracción de tiempo que el poro is abierta durante la fusión 27.
El trabajo previo de sondeo sola fusión SUV SUV 18-20,28 o fusión de liposomas y partículas virales para SBLS 15-17,21,22,33,44 dependían de FRET y la auto-extinción entre las moléculas de colorante. Por el contrario, se evita en gran medida desatenuación tinte para investigar la cinética de transferencia de etiquetas de lípidos en el SBL, por dos razones. En primer lugar, la eficiencia de FRET es un reportero altamente no lineal de la liberación de lípidos. Pequeños cambios en el resultado distancia d entre tinte en grandes cambios en la eficiencia de FRET, pero sólo para una gama limitada de D / R 0 valores, en la que R0 es la distancia Forster. Esto es, un tiempo de retraso entre la dilución de colorante real y un aumento observado en la señal de desatenuación puede ocurrir, debido al tiempo requerido para bajar a un valor cercano a 0 R. Del mismo modo, hay poco cambio en la señal de desatenuación una vez que la densidad de tinte disminuye suficientemente después de la liberación de lípidos tal que  1,4, incluso si una cantidad significativa de colorante todavía permanece en la vesícula. Estas propiedades hacen desatenuación una buena herramienta para obtener un aumento en la señal para detectar eventos de fusión, pero complicar el análisis cuantitativo de la cinética de liberación de lípidos mediante resolución temporal alta. Por el contrario, los cambios en la intensidad debido a la reorientación dipolo y los efectos del campo evanescente como un fluoróforo transferencias de un SUV en el SBL se relaciona linealmente con la fracción de colorante que queda en la camioneta. En segundo lugar, las altas concentraciones de etiquetas de lípidos requeridas para la auto-extinción pueden afectar el proceso de fusión en sí.
1,4, incluso si una cantidad significativa de colorante todavía permanece en la vesícula. Estas propiedades hacen desatenuación una buena herramienta para obtener un aumento en la señal para detectar eventos de fusión, pero complicar el análisis cuantitativo de la cinética de liberación de lípidos mediante resolución temporal alta. Por el contrario, los cambios en la intensidad debido a la reorientación dipolo y los efectos del campo evanescente como un fluoróforo transferencias de un SUV en el SBL se relaciona linealmente con la fracción de colorante que queda en la camioneta. En segundo lugar, las altas concentraciones de etiquetas de lípidos requeridas para la auto-extinción pueden afectar el proceso de fusión en sí.
Para excitación TIRF polarizada, una configuración construido en casa como en la figura 4 es rentable y proporciona un excelente control de propiedades de polarización. Sin embargo, una configuración de fabricación casera no es necesario, ya que al menos algunos microscopios TIRF comerciales utilizan la excitación polarizada y off-the-shelfmódulos de emisión de división de dos colores están disponibles. Un microscopio comercial que utiliza una fibra mantenedora de polarización (PM) para acoplar la luz de excitación polarizada que emerge del láser en el microscopio fue utilizado con éxito anteriormente 25-27. En tal instrumento, la polarización se puede girar simplemente girando la entrada de fibra PM a la unidad TIRF del microscopio. Por el contrario, algunas otras configuraciones comerciales pueden emplear un divisor de haz polarizante en la unidad TIRF, haciendo ajustes de polarización más difícil o imposible. Por desgracia, las propiedades de polarización a menudo no se especifican para los sistemas comerciales. Por lo tanto, es muy importante para los usuarios que no quieren construir un sistema de fabricación casera para analizar propiedades de polarización y de probar una unidad de demostración antes de invertir en una opción comercial particular.
control simultáneo de los lípidos y la liberación de la carga soluble proporciona más información sobre el proceso de fusión. Muchos poros parecen volver a cerrar después de releAsing sólo una fracción de la etiqueta soluble encapsulada en un SUV 27. Por lo general, todas las etiquetas de lípidos son liberados en el momento en que el poro se vuelve a sellar, por lo que volver a sellar los poros no se sospechaba previamente de seguimiento de la liberación de lípidos solo. De hecho la transferencia, el seguimiento de la liberación de lípidos solo, Kiessling et al. 29 habían sugerido de lípidos marcados en el SBL se produjo a través de muy rápido colapso de toda la membrana SUV en el SBL.
Resellado de la fusión de los poros después de la liberación parcial de los contenidos se observó previamente en un ensayo de fusión parche bicapa liposoma-atado. Rawle et al. 54 parches preparados bicapa independientes que fueron atados a un cubreobjetos por ~ 8 nm espaciadores largos de ADN. Se introducen entonces liposomas cuya fusión con el parche bicapa fue impulsado por la hibridación de ADN-conjugados de lípidos de cadena sencilla complementarios anclados a las membranas de los liposomas y de la corrección. 12% de todos los eventos fueron fusiones transitorios, lo que resulta en parteial liberación del contenido de liposomas. Eventos de ruptura de liposomas, visto en algunos ensayos de fusión SUV-SBL anteriores 44, eran muy raros. Los autores sugirieron que el ~ 8 nm espacio debajo del parche bicapa, evitando interacciones bicapa-sustrato, pueden haber permitido la transferencia no gotea del contenido de los liposomas al espacio por debajo de la bicapa. Además, la difusión de las etiquetas solubles liberados no se ve obstaculizada significativamente en el espacio entre la bicapa planar y el sustrato de vidrio. En el ensayo descrito aquí, la bicapa planar es igualmente mantuvo ~ 5 nm de distancia del cubreobjetos por la presencia de poli (etilenglicol) conjugados -lipid.
La información adicional proporcionada por la vigilancia de dos colores de la carga soluble en lípidos y viene con algunas ventajas y desventajas. En primer lugar, la preparación de los SUV con las dos etiquetas es algo más complicado que incluyendo etiquetas de lípidos solo. En segundo lugar, hay una cierta pérdida inevitable de señal-ruido en el seguimiento de las señales de las etiquetas de lípidos en tél mismo la velocidad de adquisición como en un solo color experimentos de liberación de lípidos (15-20 milisegundos por cuadro). Algunas de esta pérdida es debido al hecho de que no, que se utiliza como una etiqueta de lípidos junto con el marcador de SRB soluble, no es tan brillante como LR-PE usado en los experimentos de un solo color. Las pérdidas adicionales resultan de mayor fondo debido a la simultánea excitación de dos láser y componentes adicionales en la trayectoria óptica (Figura 4). Hasta el momento, la relación señal a ruido inferior nos impidió un seguimiento fiable de un solo DiD etiquetas en experimentos de dos colores. Esto a su vez ha impedido un análisis exhaustivo para extraer toda la información que puede ser con las mediciones LR-PE de un solo color. Este reto puede ser superado en el futuro mediante la optimización de las condiciones de adquisición y prueba de otros pares de etiquetas compatibles con propiedades mejoradas.
control simultáneo de liberación de carga soluble en lípidos y también puede detectar hemifusion, un estado en el que los folletos proximales han fusionado, pero el distalfolletos no tienen. De hecho, estos experimentos de dos colores pueden proporcionar la única evaluación concluyente de la presencia de hemifusion en otros en ensayos de fusión in vitro 18,20. Sin embargo, hemifusion fiable se puede detectar en un solo color SUV-SBL liberación monitoreo experimentos de lípidos solo 22,44. En estos experimentos, aproximadamente 1/2 de la intensidad etiqueta lipídica inicial de un SUV se mantiene en el punto de fusión. La intensidad restante puede desaparecer cuando las valvas se fusionan distales algún tiempo más tarde, si la mancha aún no ha blanqueado.
El uso de lípidos PEGilados para introducir un cojín suave entre el SBL y el soporte de vidrio ha sido clave en la reproducción de la exigencia de SNAP25 exocitosis. Los resultados de protocolo en un PEG cepillan que cubre el lado de la SBL que enfrenta el canal de flujo también. Esto tiene algunas ventajas, como la reducción de las interacciones no específicas entre terrenos y la LPS, como se explica en Introducción. Si las proteínas que interactúan con los fosfolípidos de laSBL se introducen en el ensayo, sin embargo, el cepillo PEG presentará una barrera estérica contra tales interacciones. Por lo tanto, para algunas aplicaciones, sería deseable incluir la capa de PEG asimétricamente, sólo entre el SBL y el cubreobjetos de vidrio. El lado de la SBL frente al canal de flujo sería entonces capaz de interactuar con proteínas de unión a fosfolípidos, tales como sinaptotagmina. Esto se puede lograr mediante el uso de cadenas de PEG bi-funcionales que pueden ser unidos covalentemente a la cubreobjetos en un extremo y que lleva un ancla lipídica en el otro. Métodos de Langmuir-Blodgett luego se pueden utilizar para depositar una monocapa de lípido sobre la capa de PEG 55. Los liposomas introducidos por encima de este fusible superficie hidrófoba con ella, formando una bicapa 56. Se espera que este enfoque se combinará con la microfluídica en el futuro.
Se espera que el ensayo de SUV-SBL que aquí se presenta será útil para explorar el papel de las proteínas adicionales que interactúan con trampas,como la familia Munc18 / Sec1, el sensor de calcio sinaptotagmina-1, y complexina.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Los autores declaran que no tienen intereses financieros en competencia.
Materials
| Name | Company | Catalog Number | Comments |
| Reagents | |||
| Milli-Q (MQ) water | Millipore | ||
| KOH | J.T. Baker | 3040-05 | |
| Ethanol 190 Proof | Decon | ||
| Isopropanol | Fisher Chemical | A416P4 | |
| HEPES | AmericanBio | AB00892 | |
| Sodium Cholride (KCl) | AmericanBio | AB01915 | |
| Dithiothreitol | AmericanBio | AB00490 | |
| N-[2-hydroxyethyl] piperazine-N'-[2-ethanesulfonic acid] (HEPES) | AmericanBio | AB00892 | |
| EGTA | Acros Organics | 409911000 | |
| Buffers | |||
| HEPES-KOH buffer (pH 7.4) | 25 mM HEPES-KOH, 140 mM KCl, 100 μM EGTA, 1 mM DTT | ||
| Solvents | |||
| Chloroform | J.T. Baker | 9180-01 | in glass bottle, CAUTION, wear PPE |
| Methanol | J.T. Baker | 9070-03 | in glass bottle, CAUTION, wear PPE |
| Liposome preparation | |||
| Gastight Hamilton syringe | Hamilton | var. sizes | only use glass sringe with solents (Chlorophorm/ Methanon, 2:1, v/v) |
| Glass tubes Pyrex Vista 11 ml, 16x100 mm screw cap culture tube | Pyrex | 70825-16 | clean thoroughly, rinse with chloroform |
| 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine, 16:0-18:1 PC (POPC) | Avanti Polar Lipids | 850457 | Lipids come dissolved in CHCl3 or as lyphilized powder in sealed vials. Aliquot upon opening. Store extra as dried lipid films under inert atmosphere at -20 °C. Keep stocks in CHCl3/MeOH (2:1, v/v) at -20 °C. let come to RT before opening |
| 1,2-dioleoyl-sn-glycero-3-phospho-L-serine (sodium salt), 18:1 PS (DOPS) | Avanti Polar Lipids | 840035 | Same as above. |
| 1-stearoyl-2-arachidonoyl-sn-glycero-3-phosphoethanolamine, 18:0-20:4 PE (SAPE) | Avanti Polar Lipids | 850804 | Same as above. |
| L-α-phosphatidylinositol-4,5-bisphosphate (Brain, Porcine) (ammonium salt), Brain PI(4,5)P2 | Avanti Polar Lipids | 840046 | Same as above. |
| 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(7-nitro-2-1,3-benzoxadiazol-4-yl) (ammonium salt), 18:1 NBD PE | Avanti Polar Lipids | 810145 | Same as above. |
| 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (ammonium salt), 18:1 PEG2000 PE | Avanti Polar Lipids | 880130 | Same as above. |
| cholesterol (ovine wool, >98%) | Avanti Polar Lipids | 700000 | Same as above. |
| DiD' oil; DiIC18(5) oil (1,1'-Dioctadecyl-3,3,3',3'-Tetramethylindodicarbocyanine Perchlorate) | Molecular Probes | D-307 | |
| Rotavapor R-210 | Buchi | R-210 | heat bath above Tm of lipids used |
| OG n-Octyl-β-D-Glucopyranoside | Affymetrix | 0311 | store at -20 °C, let come to RT before opening |
| Shaker - Eppendorf Thermomixer R | Eppendorf | ||
| Slide-A-Lyze Dialysis Cassettes, 20k MWCO, 3 ml | life technologies | 66003 | |
| Bio-Beads SM-2 Adsorbents | Bio-Rad | 1523920 | |
| OptiPrep Density Gradient Medium | Sigma-Aldrich | D1556 | |
| Ultracentrifugation tube, Thinwall, Ultra-Clear, 13.2 ml, 14 x 89 mm | Beckman Coulter | 41121703 | |
| Beckman SW41 Ti rotor | |||
| SuflorhodamineB | Molecular Probes | S-1307 | |
| Econo-Column Chromatography Columns, 2.5 × 10 cm | Bio-Rad | 7372512 | |
| Sepharose CL-4B | GE Healthcare | 17-0150-01 | |
| SYPRO Orange Protein Gel Stain | Molecular Probes | S-6650 | 5,000x Concentrate in DMSO |
| PDMS block | |||
| Sylgard 184 Silicone elastomer kit, PDMS | Dow Corning | 3097358-1004 | |
| Pyrex glass petri dish, 150 x 20 mm, complete with cover | Corning | 3160-152 | |
| Hole puncher - Reusable Biopsy Punch, 0.75 mm | World Precision Instruments | 504529 | |
| Manual Hole Punching Machine | SYNEO | MHPM-UNV | |
| Drill .035 x .026 x 1.5 304 SS TiN coated round punch | SYNEO | CR0350265N20R4 | drill diameter: 0.9 mm, punch bore size: 0.74 mm |
| Tygon Microbore tubing, 0.25 mm ID, 0.76 mm OD | Cole-Parmer | 06419-00 | 0.010" ID, 0.030" OD |
| Silicone Tubing (0.51 mm ID, 2.1 mm OD | Cole-Parmer | 95802-00 | 0.020" ID, 0.083" OD |
| Cover glass - cleanroom cleaned | |||
| Schott Nexterion cover slip glass D | Schott | 1472305 | |
| plasma cleaner | Harrick | PDC-32G | |
| pTIRF setup and accessories | |||
| IX81 microscope body | Olympus | IX81 | |
| EM CCD camera | Andor | ixon-ultra-897 | |
| Thermo Plate, heated microscope stage | Tokai Hit | MATS-U52RA26 | |
| 1 ml hamilton glass syringes (4x) | Hamilton | 81365 | |
| syringe pump | kd Scientific | KDS-230 | |
References
- Sudhof, T. C., Rothman, J. E. Membrane fusion: grappling with SNARE and SM proteins. Science. 323, 474-477 (2009).
- Wickner, W., Schekman, R. Membrane fusion. Nat Struct Mol Biol. 15, 658-664 (2008).
- Harrison, S. C. Viral membrane fusion. Nat Struct Mol Biol. 15, 690-698 (2008).
- Jahn, R., Scheller, R. H. SNAREs--engines for membrane fusion. Nat Rev Mol Cell Biol. 7, 631-643 (2006).
- Lindau, M., Alvarez de Toledo, G. The fusion pore. Biochim Biophys Acta. 1641, 167-173 (2003).
- Staal, R. G., Mosharov, E. V., Sulzer, D. Dopamine neurons release transmitter via a flickering fusion pore. Nat Neurosci. 7, 341-346 (2004).
- Wu, Z., et al. Nanodisc-cell fusion: Control of fusion pore nucleation and lifetimes by SNARE protein transmembrane domains. Sci. Rep. 6, 27287 (2016).
- Alabi, A. A., Tsien, R. W. Perspectives on kiss-and-run: role in exocytosis, endocytosis, and neurotransmission. Ann Rev Physiol. 75, 393-422 (2013).
- Rossetto, O., Pirazzini, M., Montecucco, C. Botulinum neurotoxins: genetic, structural and mechanistic insights. Nature Rev Microbiol. 12, 535-549 (2014).
- Weber, T., et al. SNAREpins: minimal machinery for membrane fusion. Cell. 92, 759-772 (1998).
- Nickel, W., et al. Content mixing and membrane integrity during membrane fusion driven by pairing of isolated v-SNAREs and t-SNAREs. Proc Natl Acad Sci U S A. 96, 12571-12576 (1999).
- McNew, J. A., et al. Compartmental specificity of cellular membrane fusion encoded in SNARE proteins. Nature. 407, 153-159 (2000).
- Melia, T. J., You, D. Q., Tareste, D. C., Rothman, J. E. Lipidic antagonists to SNARE-mediated fusion. J Biol Chem. 281, 29597-29605 (2006).
- Hernandez, J. M., et al. Membrane fusion intermediates via directional and full assembly of the SNARE complex. Science. 336, 1581-1584 (2012).
- Fix, M., et al. Imaging single membrane fusion events mediated by SNARE proteins. Proc Natl Acad Sci U S A. 101, 7311-7316 (2004).
- Bowen, M. E., Weninger, K., Brunger, A. T., Chu, S. Single molecule observation of liposome-bilayer fusion thermally induced by soluble N-ethyl maleimide sensitive-factor attachment protein receptors (SNAREs). Biophys J. 87, 3569-3584 (2004).
- Liu, T., Tucker, W. C., Bhalla, A., Chapman, E. R., Weisshaar, J. C. SNARE-driven, 25-millisecond vesicle fusion in vitro. Biophys J. 89, 2458-2472 (2005).
- Yoon, T. Y., Okumus, B., Zhang, F., Shin, Y. K., Ha, T. Multiple intermediates in SNARE-induced membrane fusion. Proc Natl Acad Sci U S A. 103, 19731-19736 (2006).
- Diao, J., et al. A single-vesicle content mixing assay for SNARE-mediated membrane fusion. Nat Commun. 1, 1-6 (2010).
- Kyoung, M., et al. In vitro system capable of differentiating fast Ca2+-triggered content mixing from lipid exchange for mechanistic studies of neurotransmitter release. Proc Natl Acad Sci U S A. 108, E304-E313 (2011).
- Domanska, M. K., Kiessling, V., Stein, A., Fasshauer, D., Tamm, L. K. Single vesicle millisecond fusion kinetics reveals number of SNARE complexes optimal for fast SNARE-mediated membrane fusion. J Biol Chem. 284, 32158-32166 (2009).
- Kreutzberger, A. J., Kiessling, V., Tamm, L. K. High Cholesterol Obviates a Prolonged Hemifusion Intermediate in Fast SNARE-Mediated Membrane Fusion. Biophys J. 109, 319-329 (2015).
- Schwenen, L. L., et al. Resolving single membrane fusion events on planar pore-spanning membranes. Sci Rep. 5, 12006 (2015).
- Karatekin, E., et al. A fast, single-vesicle fusion assay mimics physiological SNARE requirements. Proc Natl Acad Sci U S A. 107, 3517-3521 (2010).
- Karatekin, E., Rothman, J. E. Fusion of single proteoliposomes with planar, cushioned bilayers in microfluidic flow cells. Nat Protoc. 7, 903-920 (2012).
- Smith, M. B., et al. Interactive, computer-assisted tracking of speckle trajectories in fluorescence microscopy: application to actin polymerization and membrane fusion. Biophys J. 101, 1794-1804 (2011).
- Stratton, B. S., et al. Cholesterol Increases the Openness of SNARE-mediated Flickering Fusion Pores. Biophysical journal. 110, (2016).
- Diao, J., et al. Synaptic proteins promote calcium-triggered fast transition from point contact to full fusion. Elife. 1, e00109 (2012).
- Kiessling, V., Domanska, M. K., Tamm, L. K. Single SNARE-mediated vesicle fusion observed in vitro by polarized TIRFM. Biophys J. 99, 4047-4055 (2010).
- Blasi, J., et al. Botulinum neurotoxin A selectively cleaves the synaptic protein SNAP-25. Nature. 365, 160-163 (1993).
- Washbourne, P., et al. Genetic ablation of the t-SNARE SNAP-25 distinguishes mechanisms of neuroexocytosis. Nat Neurosci. 5, 19-26 (2002).
- Diaz, A. J., Albertorio, F., Daniel, S., Cremer, P. S. Double cushions preserve transmembrane protein mobility in supported bilayer systems. Langmuir. 24, 6820-6826 (2008).
- Floyd, D. L., Ragains, J. R., Skehel, J. J., Harrison, S. C., van Oijen, A. M. Single-particle kinetics of influenza virus membrane fusion. Proc Natl Acad Sci U S A. 105, 15382-15387 (2008).
- Albertorio, F., et al. Fluid and air-stable lipopolymer membranes for biosensor applications. Langmuir. 21, 7476-7482 (2005).
- Daniel, S., Albertorio, F., Cremer, P. S. Making lipid membranes rough, tough, and ready to hit the road. Mrs Bulletin. 31, 536-540 (2006).
- Gao, Y., et al. Single reconstituted neuronal SNARE complexes zipper in three distinct stages. Science. 337, 1340-1343 (2012).
- Kenworthy, A. K., Hristova, K., Needham, D., Mcintosh, T. J. Range and Magnitude of the Steric Pressure between Bilayers Containing Phospholipids with Covalently Attached Poly(Ethylene Glycol). Biophys J. 68, 1921-1936 (1995).
- Knoll, W., et al. Solid supported lipid membranes: New concepts for the biomimetic functionalization of solid surfaces. Biointerphases. 3, Fa125-Fa135 (2008).
- Quinn, P., Griffiths, G., Warren, G. Density of newly synthesized plasma membrane proteins in intracellular membranes II. Biochemical studies. J Cell Biol. 98, 2142-2147 (1984).
- Sund, S. E., Swanson, J. A., Axelrod, D. Cell membrane orientation visualized by polarized total internal reflection fluorescence. Biophys J. 77, 2266-2283 (1999).
- Johnson, D. S., Toledo-Crow, R., Mattheyses, A. L., Simon, S. M. Polarization-controlled TIRFM with focal drift and spatial field intensity correction. Biophys J. 106, 1008-1019 (2014).
- Anantharam, A., Onoa, B., Edwards, R. H., Holz, R. W., Axelrod, D. Localized topological changes of the plasma membrane upon exocytosis visualized by polarized TIRFM. J Cell Biol. 188, 415-428 (2010).
- Axelrod, D. Carbocyanine dye orientation in red cell membrane studied by microscopic fluorescence polarization. Biophys J. 26, 557-573 (1979).
- Wang, T., Smith, E. A., Chapman, E. R., Weisshaar, J. C. Lipid mixing and content release in single-vesicle, SNARE-driven fusion assay with 1-5 msec resolution. Biophys J. 96, 4122-4131 (2009).
- Chernomordik, L. V., Frolov, V. A., Leikina, E., Bronk, P., Zimmerberg, J. The pathway of membrane fusion catalyzed by influenza hemagglutinin: restriction of lipids, hemifusion, and lipidic fusion pore formation. J Cell Biol. 140, 1369-1382 (1998).
- Takamori, S., et al. Molecular anatomy of a trafficking organelle. Cell. 127, 831-846 (2006).
- Wilhelm, B. G., et al. Composition of isolated synaptic boutons reveals the amounts of vesicle trafficking proteins. Science. 344, 1023-1028 (2014).
- Scott, B. L., et al. Liposome fusion assay to monitor intracellular membrane fusion machines. Methods Enzymol. 372, 274-300 (2003).
- Linkert, M., et al. Metadata matters: access to image data in the real world. J Cell Biol. 189, 777-782 (2010).
- Soumpasis, D. M. Theoretical analysis of fluorescence photobleaching recovery experiments. Biophys J. 41, 95-97 (1983).
- Ohki, S. A mechanism of divalent ion-induced phosphatidylserine membrane fusion. Biochim Biophys Acta. 689, 1-11 (1982).
- Berquand, A., et al. Two-step formation of streptavidin-supported lipid bilayers by PEG-triggered vesicle fusion. Fluorescence and atomic force microscopy characterization. Langmuir. 19, 1700-1707 (2003).
- Tamm, L. K., McConnell, H. M. Supported phospholipid bilayers. Biophys J. 47, 105-113 (1985).
- Rawle, R. J., van Lengerich, B., Chung, M., Bendix, P. M., Boxer, S. G. Vesicle fusion observed by content transfer across a tethered lipid bilayer. Biophys J. 101, L37-L39 (2011).
- Wagner, M. L., Tamm, L. K. Reconstituted syntaxin1a/SNAP25 interacts with negatively charged lipids as measured by lateral diffusion in planar supported bilayers. Biophys J. 81, 266-275 (2001).
- Kalb, E., Frey, S., Tamm, L. K. Formation of Supported Planar Bilayers by Fusion of Vesicles to Supported Phospholipid Monolayers. Biochimica Et Biophysica Acta. 1103, 307-316 (1992).
