

Summary
We provide a method to simultaneously screen a library of antibody fragments for binding affinity and cytoplasmic solubility by using the Escherichia coli twin-arginine translocation pathway, which has an inherent quality control mechanism for intracellular protein folding, to display the antibody fragments on the inner membrane.
Abstract
Antibodies engineered for intracellular function must not only have affinity for their target antigen, but must also be soluble and correctly folded in the cytoplasm. Commonly used methods for the display and screening of recombinant antibody libraries do not incorporate intracellular protein folding quality control, and, thus, the antigen-binding capability and cytoplasmic folding and solubility of antibodies engineered using these methods often must be engineered separately. Here, we describe a protocol to screen a recombinant library of single-chain variable fragment (scFv) antibodies for antigen-binding and proper cytoplasmic folding simultaneously. The method harnesses the intrinsic intracellular folding quality control mechanism of the Escherichia coli twin-arginine translocation (Tat) pathway to display an scFv library on the E. coli inner membrane. The Tat pathway ensures that only soluble, well-folded proteins are transported out of the cytoplasm and displayed on the inner membrane, thereby eliminating poorly folded scFvs prior to interrogation for antigen-binding. Following removal of the outer membrane, the scFvs displayed on the inner membrane are panned against a target antigen immobilized on magnetic beads to isolate scFvs that bind to the target antigen. An enzyme-linked immunosorbent assay (ELISA)-based secondary screen is used to identify the most promising scFvs for additional characterization. Antigen-binding and cytoplasmic solubility can be improved with subsequent rounds of mutagenesis and screening to engineer antibodies with high affinity and high cytoplasmic solubility for intracellular applications.
Introduction
Antibodies capable of folding and functioning in the intracellular environment are promising tools for both research and therapeutic applications. They have the ability to modulate protein activity by binding to a target protein inside cells to prevent protein-protein interactions, disrupt protein-nucleic acid interactions, or prevent substrate access to enzymes1-5.
Although antibodies have much potential for intracellular applications, engineering them for proper folding and solubility in the intracellular environment while maintaining the ability to bind to a target antigen is challenging. The reducing cytoplasmic environment prevents the formation of the disulfide bonds normally required for the stable folding of full-length antibodies and antibody fragments, including single-chain variable fragment (scFv) antibodies6,7. A number of directed evolution approaches have been employed to engineer antibodies with high affinities for target antigens8-10. These approaches commonly use phage display, yeast surface display, or bacterial surface display to screen large libraries of antibodies11-13. These methods are powerful and effective for identifying antibodies that bind to targets, yet they depend on the secretory pathway to transport proteins that will be displayed14-16. The secretory pathway translocates unfolded proteins from the reducing cytoplasm into the endoplasmic reticulum lumen in yeast or into the periplasm in bacteria. The proteins then fold under oxidizing conditions and are displayed on the cell surface or packaged into phage particles to screen for binding affinity17,18. As a result, antibodies isolated using these techniques will not necessarily fold well in the cytoplasm, and intracellular solubility must often be engineered separately if the antibodies will be used in intracellular applications.
To improve the efficiency of engineering antibodies that are well folded in the cytoplasm, we previously reported the success of MAD-TRAP (membrane-anchored display for Tat-based recognition of associating proteins), a method for screening an scFv antibody library using Escherichia coli inner-membrane display19. Bacterial inner-membrane display relies on the twin-arginine translocation (Tat) pathway for transporting displayed antibodies, in contrast to other common display methods that use the secretory pathway. The Tat pathway contains a quality control mechanism that only allows soluble, correctly folded proteins to be transported from the E. coli cytoplasm, across the inner membrane, and into the periplasm20,21. Overexpressed Tat substrates (i.e., proteins targeted to the Tat pathway with an N-terminal fusion to the Tat signal peptide ssTorA) that are well folded in the cytoplasm form a long-lived translocation intermediate with the N-terminus in the cytoplasm and the C-terminus in the periplasm19. This allows display of correctly folded Tat substrates, including antibody fragments, on the periplasmic face of the E. coli inner membrane. After removing the outer membrane by enzymatic digestion to generate spheroplasts, antibodies are exposed to the extracellular space (Figure 1). This allows Tat substrates displayed on the inner membrane to be screened for binding to a specific target. Importantly, harnessing the Tat pathway for cell-surface display ensures that only the antibodies in the library that are well folded in the cytoplasm will be interrogated for binding, allowing simultaneous engineering of binding affinity and intracellular folding. In this protocol, we describe how to display an scFv library on the E. coli inner membrane, pan the library against a target antigen, and perform a secondary screen to identify the most promising constituents of the library. While we focus the protocol on scFvs, the method could be applied to engineering any protein whose application requires binding and intracellular folding.
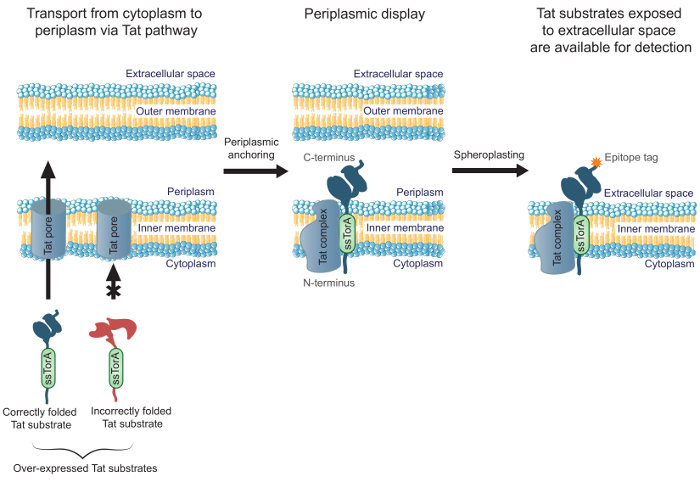
Figure 1. Tat inner-membrane display. In E. coli, scFv antibodies that are expressed as a fusion to the ssTorA signal sequence and correctly folded in the cytoplasm are transported across the inner membrane. A translocation intermediate forms, where the scFvs are anchored in the inner membrane with the N-terminus in the cytoplasm and the C-terminus in the periplasm. The E. coli outer membrane is enzymatically digested to form spheroplasts, thereby exposing the anchored antibodies to the extracellular space and making them available for detection by using an antibody that binds to the C-terminally fused epitope tag on the displayed antibody. Please click here to view a larger version of this figure.
Protocol
1. Prepare the scFv Library as a Fusion to the ssTorA Signal Sequence
- Obtain a deoxyribonucleic acid (DNA) library containing variants of an scFv gene.
NOTE: The library may also be constructed using any appropriate mode to generate diversity over the entire scFv gene or targeted domains (e.g., the third complementarity determining regions, CDR3s)22. - Insert the DNA library into the pIMD plasmid (Figure 2) using standard molecular cloning methods23.
NOTE: This plasmid expresses scFvs as a genetic fusion to the ssTorA signal sequence (N-terminal to scFv) and the FLAG epitope tag (C-terminal to the scFv). The design of the plasmid for inner membrane display has been described previously19. The pIMD plasmid is available from the authors.

Figure 2. Inner-membrane display plasmid (pIMD) map (Steps 1.2 through 1.3). This plasmid contains a lac promotor, ColE1 origin of replication, and a chloramphenicol resistance gene. The inserted scFv gene is fused to the ssTorA signal sequence to target the scFv to the Tat pathway and to a FLAG epitope tag, with all three in the same reading frame. Restriction enzyme sites are indicated. For a library inserted between the XbaI and NotI restriction enzyme sites, the size of the plasmid is 2219 bp plus the size of the scFv. Please click here to view a larger version of this figure.
- Transform the plasmid DNA containing the library into MC4100 E. coli cells23. Recover and grow this bacterial form of the library. Centrifuge at 4,000× g for 15 min at RT to collect the cells. Remove the supernatant, and resuspend the collected cells in 25% glycerol in Luria-Bertani (LB) media. Store aliquots at -80 °C until needed, or proceed to Step 2.
NOTE: The protocol has been verified with MC4100 cells, though other E. coli strains are also expected to be compatible with the protocol. Electroporation is the preferred method for transformation, due to its high transformation efficiency. The library should typically consist of at least 109 scFv variants at this stage, and each aliquot should contain enough cells such that the library is covered 100-fold.
2. Express the Library and Prepare Spheroplasts
- Thaw one aliquot of the bacterial library (from Step 1.3) at RT, and add the aliquot to a flask containing 100 ml LB media with 20 µg/ml chloramphenicol (Cm). Grow for 3 hr at 37 °C and 225 rpm in an incubated shaker.
- After 3 hr, remove the flask from the 37 °C incubated shaker. Allow expression of the scFv library to proceed O/N for 15 to 22 hr at 20 °C and 225 rpm in an incubated shaker.
NOTE: No inducer is needed when using the pIMD plasmid, as the promoter is leaky. Note that MC4100 cells do not overexpress the Lac repressor (and LacI is not found on the plasmid).

Figure 3. E.coli cells and spheroplasts. (A) E. coli cells are cylindrical in shape. (B) After spheroplasting using EDTA and lysozyme, the outer membrane of the E. coli cells is ruptured, and the resulting spheroplasts are spherical in shape. Differential interference contrast (DIC) microscopy images were obtained using a 100X objective on an inverted microscope. Please click here to view a larger version of this figure.
- Prepare the library spheroplasts.
NOTE: Spheroplasts are formed by rupturing the outer membrane of E. coli and are spherical in shape (Figure 3).- Prepare the necessary buffers.
NOTE: All buffers should be sterile.- Prepare 1× phosphate-buffered saline (PBS; pH 7.4) by dissolving 8 g NaCl, 0.2 g KCl, 1.44 g Na2HPO4, and 0.24 g KH2PO4 in distilled H2O to a final volume of 1,000 ml. Keep on ice.
- Prepare PBS with 0.1% (w/v) bovine albumin serum (BSA) by dissolving 0.2 g BSA into 200 ml 1× PBS. Keep on ice.
- Prepare the fractionation buffer (FB) by mixing 7.5 ml of sterile-filtered 1 M sucrose, 1 ml of 1 M Tris buffer (pH 8.0), and 1.5 ml distilled H2O. Keep on ice.
- Prepare 1 mM ethylenediaminetetraacetic acid (EDTA) by adding 30 µl of 0.5 M EDTA to 14.97 ml distilled H2O.
- Prepare 0.5 M MgCl2 by dissolving 4.76 g MgCl2 in 100 ml distilled H2O. Keep on ice.
- Remove the flask from the shaker, and measure the optical density (OD) at 600 nm using a spectrophotometer to determine the cell density. Calculate the volume of induced culture needed such that each sample for spheroplasting has 1×1010 cells.
NOTE: The approximation of an OD600 of 1 indicating a concentration of 109 cells/ml for E. coli can be used24. - Centrifuge the calculated volume of induced culture in a 1.5 ml microcentrifuge tube at 12,000 × g at RT for 5 min. Prepare at least two samples in the event that an issue arises in sample preparation.
- Remove the supernatant from the centrifuged cultures and resuspend each cell pellet in 100 µl of ice-cold FB. Centrifuge at 12,000 × g at RT for 1 min, and then remove the supernatant by pipetting. Resuspend each pellet in 350 µl of ice-cold FB supplemented with 3.5 µl of 10 mg/ml lysozyme.
- Slowly vortex each tube while adding, dropwise, 700 µl of 1 mM EDTA, and then incubate the tubes at RT for 20 min while slowly rotating on a tube rotator to mix the samples. Remove the tubes from the rotator, add 50 µl of ice-cold 0.5 M MgCl2 to each tube, and incubate them on ice for 10 min. Centrifuge the tubes at 11,000 × g at 4 °C for 10 min.
- Isolate the spheroplast pellet.
- Use a micropipette with a 1 ml tip to slowly pull up part of pellet. While holding the tube at an angle with the opening directly above a new 1.5 ml tube, slowly lift the pipette tip out of the supernatant and slide the pellet into the new tube.
- If a significant volume of supernatant is transferred to the new tube, remove it by pipetting. If the pellet is not firm enough to transfer, re-centrifuge at 11,000 × g for 2 min and attempt pellet isolation again.
- Resuspend the spheroplast pellet in each tube in 1 ml of ice-cold 1× PBS. Alternate between pipetting and slowly vortexing on a vortex holder until the pellet is fully resuspended. Do not keep samples off of the ice for more than 2 min at a time, and return to the ice for at least 5 min before removing from the ice again. Keep the spheroplasts at 4 °C (for up to 2 days) until used for panning in Step 4.
- Prepare the necessary buffers.
3. Immobilize the Target Antigen onto Magnetic Beads
- Biotinylate the target antigen in vivo during recombinant production in E. coli cells. Alternatively, use chemical conjugation25 or purchase target antigen that has already been biotinylated, and proceed to Step 3.2.
- Add 816 g bicine to 50 ml water to make 10× bicine buffer. Dilute the buffer to 1× in distilled H2O and heat to 50 °C. Add 14.7 mg biotin to 12 ml of the heated 1× bicine buffer to make a biotin solution that is 5 mM biotin in 10 mM bicine buffer. Store at -20 °C until needed.
- Express and biotinylate the target protein using the pAK400cb-BCCP plasmid26, which allows production of the target antigen as a fusion to the biotin carboxyl carrier protein (BCCP).
NOTE: E. coli cells natively biotinylate BCCP, eliminating the need to purify and chemically biotinylate the target protein prior to immobilization on streptavidin-coated beads. The native E. coli biotin ligase BirA is sufficient for biotinylating the fusion protein.- Grow E. coli containing the biotinylation plasmid (with the target antigen inserted as a fusion to the N-terminus of BCCP) O/N for 15 to 18 hr in 5 ml of LB media supplemented with 20 µg/ml Cm at 37 °C while shaking at 225 rpm.
- Measure the OD at 600 nm using a spectrophotometer and calculate the volume of culture needed (Vadd) to subculture at a starting OD of 0.05 in 25 ml fresh LB media with 20 µg/ml Cm using the equation: Vadd = (0.05 × 25 ml)/(OD600 - 0.05), where OD600 is the optical density of the O/N culture and Vadd is the volume of the O/N culture to add to the fresh LB. Subculture and grow to an OD of 0.5 to 0.8 in an incubated shaker at 37 °C and 225 rpm.
- Add isopropyl β-D-1-thiogalactopyranoside to a final concentration of 100 µM and biotin to a final concentration of 5 µM. Induce expression in an incubated shaker for 15 to 22 hr at 20 °C and 225 rpm.
- Harvest bacteria by centrifugation at 4,000 × g at 4 °C for 10 min. Remove the supernatant. Store the pellet at -20 °C until ready for use.
- Add 1 ml of a cell lysis detergent per 0.2 g of cell pellet. Resuspend by pipetting and gently rotate for 20 min to lyse the cells. After lysis, centrifuge at 16,000 × g and 4 °C for 20 min. Pipette the soluble lysate (supernatant) into a new 1.5 ml tube.
- Use a 3 kDa molecular weight cutoff column to remove the unbound biotin. Pipette the lysate into the column, and centrifuge at 20 °C according to the manufacturer's instructions. Wash with 1× PBS until the biotin in the lysate has been diluted 100-fold and the volume of the washed lysate is equal to the original volume of the lysate. Transfer the lysate to a new tube.
- Immobilize the biotinylated target antigen onto streptavidin-coated magnetic beads.
- Prepare 1× PBS and 1× PBS with 0.1% (w/v) BSA as described in Step 2.3.1.
- Prepare the magnetic beads.
NOTE: This requires the use of a magnetic separation rack.- Resuspend streptavidin-coated magnetic beads in their original vial. Either vortex for at least 30 sec or rotate for 5 min.
- Transfer 7-10 × 109 beads to a 1.5 ml tube.
NOTE: The volume required will be dependent on the bead concentration supplied by the manufacturer. - Place the tube containing the beads on the magnet rack for 2 min to collect the beads on the side of the tube. With the tube still on the magnet, carefully remove the supernatant by pipetting without disrupting the beads.
- To wash, remove the tube from the magnet, and resuspend the beads in 1 ml of 1× PBS by pipetting without generating bubbles. Return the tube to the magnet for 2 min to collect the beads, and carefully remove the supernatant by pipetting. Repeat the process two more times for a total of three washes. Ensure that no liquid is left in the tube after the final wash.
- Add the lysate containing the biotinylated antigen to the magnetic beads.
- Remove the tube from the magnet and resuspend the beads in 1 ml of lysate (from Step 3.1.5). Incubate at RT for 30 min while gently rotating.
- Place the tube on the magnet for 3 min to collect the antigen-coated beads. Wash the coated beads five times with 1× PBS with 0.1% BSA in the same manner as described in Steps 3.2.2.3 to 3.2.2.4. After the final wash, resuspend the beads in 1× PBS with 0.1% BSA up to the same volume used in Step 3.2.2.2.
- If the immobilized target antigen is stable at 4 °C, store the coated beads at 4 °C until needed for panning. Otherwise, proceed to Step 4.
4. Screen the scFv Library by Panning against the Target Antigen (Figure 4)

Figure 4. Panning (Step 4). Antigen-coated magnetic beads are incubated with spheroplasts expressing antibody library variants. Plasmid DNA from bead-bound spheroplasts is recovered and used to generate a sublibrary, which is screened using the ELISA-based secondary screen. Corresponding protocol steps are noted. Please click here to view a larger version of this figure.
- Incubate the coated beads with spheroplasts.
- Use a spheroplast to bead ratio of approximately 5:1. Add 4 × 109 spheroplasts and 8 × 108 beads to a sterile 15 ml tube.
NOTE: Assume that no cells were lost during the spheroplasting process, so the concentration is still 1 × 1010 spheroplasts/ml. - Add 1× PBS with 0.1% BSA to bring the total volume to 4 ml. Aliquot into four 1.5 ml tubes with 1 ml each. Incubate the reactions at 4 °C for 5 hr while gently rotating.
- Use a spheroplast to bead ratio of approximately 5:1. Add 4 × 109 spheroplasts and 8 × 108 beads to a sterile 15 ml tube.
- Prepare the bead-bound spheroplasts for polymerase chain reaction (PCR).
- Place the panning reaction tubes on the magnet for 3 min. Remove the supernatant by pipetting, and wash the bead-bound spheroplasts four times with ice cold 1× PBS with 0.1% BSA in the same manner as described in Steps 3.2.2.3 to 3.2.2.4. Resuspend the bead-bound spheroplasts in each tube in 25 µl of distilled H2O. Store the beads at -20 °C or proceed to Step 4.3.
- Perform whole-plasmid PCR on bead-bound spheroplasts to amplify the plasmids containing the genes for bead-bound scFvs.
- Obtain primers with the following sequences: 5'CCAACTCTTTTTCCGAAGGTAACTG3' (forward primer) and 5'TAGCTCTTGATCCGGCAAACAAA3' (reverse primer).
NOTE: These will bind end-to-end on opposite strands of the pIMD plasmid (Figure 2) and are designed to anneal to a common feature of pIMD, so amplification will occur regardless of the scFv variant sequence. - Phosphorylate the primers.
NOTE: Without phosphorylation, re-ligation will not occur. Primers may also be ordered with 5'-phosphorylation, rather than using this phosphorylation method in this protocol.- In a 0.5 ml tube, set up a phosphorylation reaction for the forward PCR primer as described in Table 1. Repeat this process for the reverse primer.
- Incubate the reactions at 37 °C for 1 hr. Then incubate them at 65 °C for 20 min to deactivate the T4 polynucleotide kinase (PNK). Store the phosphorylated primers at -20 °C.
- Perform PCR.
- In a PCR tube, prepare the PCR reaction as described in Table 2.
NOTE: Multiple reactions can be prepared for higher yield. The unused bead-bound spheroplasts can be stored at -20 °C. - Heat the PCR reactions at 98 °C for 15 min in a thermal cycler to ensure full lysis of the spheroplasts. Remove the tubes from the thermal cycler, and add 0.5 µl of a high fidelity polymerase to each. Return tubes to the thermal cycler and run using the program detailed in Table 3.
- Pool the PCR products as appropriate. Store at -20 °C or proceed to Step 4.4.
- In a PCR tube, prepare the PCR reaction as described in Table 2.
- Obtain primers with the following sequences: 5'CCAACTCTTTTTCCGAAGGTAACTG3' (forward primer) and 5'TAGCTCTTGATCCGGCAAACAAA3' (reverse primer).
Table 1. PNK phosphorylation reaction (Step 4.3.2.1).
| Reagent | Volume (μl) |
| Distilled H2O | 15 |
| 10x T4 DNA ligase reaction buffer | 2 |
| 100 μM primer | 2 |
| T4 Polynucleotide kinase (PNK) | 1 |
Table 2. Whole-plasmid PCR reaction (Step 4.3.3.1).
| Reagent | Volume (µl) |
| Distilled H2O | 28.5 |
| 5x High-fidelity polymerase buffer | 10 |
| 10 μM Phosphorylated forward primer | 2.5 |
| 10 μM Phosphorylated reverse primer | 2.5 |
| 40 mM dNTP mix (10 mM each dNTP) | 1 |
| Bead-bound spheroplasts | 5 |
Table 3. PCR program (Step 4.3.3.2).
| Step | Temperature (°C) | Time (min:sec) | Number of cycles |
| Initial denature | 98 | 0:30 | 1 |
| Denature | 98 | 0:10 | 35 |
| Annealing | 69 | 0:30 | |
| Extension | 72 | 0:30 per kb | |
| Final extension | 72 | 6:00 | 1 |
| Hold | 12 | Infinite | 1 |
- Re-circularize the whole-plasmid PCR products, and use the ligated product to transform MC4100 E. coli cells.
- Purify the PCR product by running the PCR reaction on an agarose gel23, staining the DNA in the gel23, and using a gel cleanup kit to purify the linearized plasmid by following the instructions supplied by the manufacturer. Measure the concentration using a spectrophotometer at 260 nm. Store the purified fragment at -20 °C until needed, or continue to Step 4.4.2.
- Re-circularize plasmid from the PCR product.
- To prevent intermolecular ligation of the PCR product, carry out the ligation reaction with a low concentration27 of 1 ng/µl of PCR product. Calculate the volume needed for preparing an 800 µl ligation reaction at this concentration.
- Prepare the ligation reaction on ice. In a tube, add the volume calculated in Step 4.4.2.1 of the PCR product, 80 µl of 10× DNA ligase buffer, and distilled H2O up to 800 µl. Add 4 µl of T4 DNA ligase, and immediately place tubes at 16 °C in a water bath or thermal cycler. Incubate at 16 °C O/N for 14 to 18 hr. Store the completed ligation reactions at -20 °C until needed, or proceed to Step 4.4.3.
- Place the ligation reaction on a heat block at 65 °C for 15 min to heat-inactivate the DNA ligase. Then use a microdialysis membrane or DNA cleanup kit to de-salt the ligated DNA. Store at 20 °C or proceed to Step 4.4.4.
- Use the entire heat-inactivated, de-salted ligation product to transform MC4100 E. coli cells23. Prepare glycerol stocks, as described in Step 1.3, of the cells containing the resulting panned sublibrary, and store aliquots at -80 °C.
- Repeat Step 4 in its entirety using an aliquot from Step 4.4.4 to do a second panning on the sublibrary.
NOTE: A second panning helps enrich for library constituents that bind well to the target antigen19.
5. Perform a Secondary Screen Using an Enzyme-linked Immunosorbent Assay Method to Identify Promising Clones for Further Characterization (Figure 5)

Figure 5. ELISA-based secondary screening (Step 5). (A) Library variants from the sublibrary enriched during panning are inoculated in individual wells of a culture plate for growth and expression. (B) An ELISA plate is coated with target antigen. (C) The library variants are screened using the ELISA-based secondary screen described in the protocol. Upon analysis of data obtained from the secondary screen, variants of interest are selected and characterized further. Corresponding protocol steps are noted. Please click here to view a larger version of this figure.
- Thaw one tube of the panned sublibrary (from Step 4.4.4) and plate onto LB agar plates. Plate several dilutions at concentrations low enough to ensure individual colonies (e.g., 102- to 106-fold dilutions). Incubate the plates for 15 to 18 hr at 37 °C. Store the plates at 4 °C or proceed to Step 5.2.
- Culture and induce colonies from the panned sublibrary. Perform all the steps under sterile conditions. Use a multichannel pipettor for steps involving 96-well plates.
- Add 200 µl of LB with 20 µg/ml Cm into each well of a round-bottom 96-well culture plate.
- Pick an individual colony from the agar plate with a pipette tip, place the tip in the first well of the 96-well plate, and gently stir to inoculate. Use a new tip for each well. Inoculate one colony into each well. As a control, include at least one sterility control well with no colony inoculated.
- Repeat Steps 5.2.1 and 5.2.2 to inoculate several 96-well plates.
- Place the 96-well plates on a microplate shaker at 310 rpm. Incubate at 37 °C for 20 to 24 hr to express the scFvs.
- For each culture plate prepared in Step 5.2, coat one 96-well ELISA plate with the target antigen.
- Dilute purified target antigen to an appropriate concentration (e.g., 1 µg/ml to 4 µg/ml) in 1× PBS to make the coating solution. Make 5 ml of coating solution for each 96-well plate.
NOTE: The appropriate concentration depends on the specific antigen being used and may need to be adjusted. - Add 50 µl of the coating solution to each well of a 96-well high-binding clear polystyrene ELISA plate. Gently tap the plate on the benchtop surface to ensure that the entire surface of each well is coated. Repeat for each plate. Incubate the plates at 4 °C O/N.
- Dilute purified target antigen to an appropriate concentration (e.g., 1 µg/ml to 4 µg/ml) in 1× PBS to make the coating solution. Make 5 ml of coating solution for each 96-well plate.
- Replicate the colonies from the 96-well culture plates onto agar plates.
- Place a sterile polystyrene replicator into the wells of a culture plate to collect a small amount of liquid. Carefully raise the replicator and transfer to a 15 cm LB agar plate such that all tips are touching the plate. Once the liquid has transferred, lift the replicator straight up. Repeat for each culture plate.
- Label the agar plate with the correct orientation so that results from the secondary screen in the 96-well plate can be matched with the correct replicated colony on the plate, if further characterization is desired. Grow at 37 °C for 15 to 18 hr, and then store at 4 °C until needed.
- Perform the ELISA secondary screen.
- Prepare the blocking solution by making 2% (w/v) dry milk in 1× PBS. Empty the coating solution from the ELISA plates. Add 100 µl of the blocking solution to each well. Incubate at RT for at least 2 hr, or block O/N at 4 °C.
- Prepare the wash buffer by adding polysorbate 20 to a final concentration of 0.05% in 1× PBS. Make 250 ml per ELISA plate.
- Add 20 µl of a concentrated cell lysis detergent to each well of the round-bottom culture plate, and incubate the culture plate on a microplate shaker at RT for 15 to 20 min. Begin the lysis at the same time that the blocking of the ELISA plates is complete so that the lysis and the wash Step 5.5.4 can be performed concurrently.
- Empty the blocking solution from the ELISA plates. Wash the blocked ELISA plates four times with 200 µl of wash buffer per well per wash. Empty the wash buffer from the wells.
- Transfer 50 µl from each well of the cell lysis plate to the corresponding well of the ELISA plate, using a new tip for each well. Incubate the ELISA plate at RT for 1 to 2 hr.
- Prepare the antibody solution to detect bound scFvs.
- Use a horseradish peroxidase (HRP)-conjugated primary antibody that binds to the FLAG epitope tag fused to the library scFvs.
- Dilute the antibody to the appropriate dilution to use in an ELISA (see supplier's recommendations) in 2% (w/v) dry milk in 0.05% polysorbate 20 in 1× PBS. Prepare 5 ml for each plate.
- Wash the ELISA plates four times with wash buffer as described in Step 5.5.4.
- Add 50 µl of the antibody solution to each well of the ELISA plate. Incubate for 1 to 2 hr at RT.
- Prepare the HRP substrate by dissolving o-phenylenediamine dihydrochloride (OPD) tablets in distilled H2O per the manufacturer's protocol while avoiding light. Prepare 20 ml per ELISA plate.
- Prepare 3 M H2SO4 by diluting concentrated H2SO4 with distilled H2O as necessary. Prepare 5 ml per ELISA plate.
Caution: H2SO4 is a strong acid. Be sure to wear appropriate personal protective equipment. - Wash the ELISA plates four times with wash buffer, as described in Step 5.5.4.
- Incubate ELISA plates with the HRP substrate.
- Add 200 µl of the HRP substrate to each well. To minimize light exposure, add the substrate to one ELISA plate at a time, and wrap with aluminum foil before proceeding to the next plate. Incubate the plates for 30 to 60 min at RT in the dark.
- After the first 30 min, check the plates for darkening of the substrate, and incubate longer if needed to visualize developing color.
- Add 50 µl of 3 M H2SO4 to each well to quench the reaction. Using a different tip for each well, mix the solution in the wells by gently pipetting up and down without frothing. For consistency and to prevent saturation, add the H2SO4 quickly and carefully to all of the ELISA plates prior to mixing the solution for each plate.
- Measure the absorbance of the solution in the wells of each plate at 492 nm using a plate reader.
- Analyze the absorbance data to identify scFv variants that exhibit promising binding signals and characterize these promising scFvs. Select scFvs that exhibit absorbance signals higher than the background signal and higher than the average signal on each plate.
NOTE: The absorbance level will be dependent on the properties of the antigen and anti-FLAG antibody used, along with the strength of the scFv variants that were isolated in the screening.
Representative Results
The intracellular protein folding quality control mechanism of the Tat pathway in E. coli limits transport across the inner cell membrane to proteins that are well folded in the reducing cytoplasmic environment. By overexpressing a fusion of an scFv to the ssTorA signal sequence (the signal sequence from the TorA protein, which is naturally transported by the Tat pathway20), translocation is stalled, resulting in display of scFvs on the inner membrane19. After enzymatic disruption of the outer membrane, the displayed antibodies are made available for screening for antigen-binding activity. The ability to take advantage of the Tat pathway for scFv display was shown by Karlsson et al.19 (Figure 6). The scFv antibodies scFv13 and scFv13.R4 were fused to either the native ssTorA sequence or a modified ssTorA that lacks the arginine-arginine residue pair recognized by the Tat pathway. scFv13.R4 was engineered by Martineau et al. from scFv13 through four rounds of directed evolution and is known to fold well in the cytoplasm9. This scFv was displayed on the inner membrane, but only when expressed as a fusion to the native ssTorA signal sequence (Figure 6). Contrarily, scFv13 is not well folded cytoplasmically9, so it is not displayed well on the inner membrane, regardless of the signal sequence to which it is fused. Additionally, if the scFvs were expressed in cells that lacked the TatC protein, a vital component of the Tat machinery20,28, display was not observed, showing the important link between inner-membrane display and the Tat pathway. These results demonstrate that only proteins that contain the Tat signal peptide and that are correctly folded in the cytoplasm are displayed on the inner membrane, allowing transport through the Tat pathway to function as a screen for intracellular folding.

Figure 6. Detection of displayed scFvs on the inner membrane. Flow cytometry analysis was performed to detect the display of poorly folded scFv13 and well-folded scFv13.R4 on the inner membrane. scFvs were fused to native ssTorA or ssTorA(KK), where the Arg-Arg pair in the ssTorA sequence was modified to Lys-Lys. The C-terminal FLAG epitope tags on the scFvs were detected with a fluorescein isothiocyanate (FITC)-conjugated anti-FLAG antibody. Cells without the TatC protein (ΔtatC) and ssTorA-scFv13 without the FLAG tag were tested as controls. M indicates the median fluorescence value. Reprinted from reference 19 with permission. Please click here to view a larger version of this figure.
Inner-membrane display can successfully isolate scFv antibodies with high levels of affinity for a target protein and high levels of cytoplasmic solubility. Additionally, subsequent rounds of directed evolution using inner-membrane display improve antibody characteristics19. To demonstrate this, an error-prone PCR library based on scFv13, which has a low level of binding affinity for β-galactosidase, was panned against the target antigen β-galactosidase using the display and panning method described in the protocol. scFv 1-4 was isolated after one round of mutagenesis and panning, and exhibited higher binding affinity to β-galactosidase than scFv13 (Figure 7A) and a higher level of cytoplasmic solubility (Figure 7B).
A new library, based on scFv 1-4, was made using error-prone PCR, and panning of this second-generation library against β-galactosidase was done using a modification of the described protocol. The panning against β-galactosidase for the second round of evolution was done in the presence of purified, soluble scFv 14 as a competitor to improve the likelihood of isolating clones with higher affinity than scFv 1-4. After this second round of mutagenesis and panning, scFv 2-1 and scFv 2-3 were isolated using the ELISA-based secondary screening. These scFvs not only exhibited higher binding affinity for β-galactosidase than scFv13, but also exhibited better binding than the first-round clone scFv 1-4. scFv 2-1 exhibited β-galactosidase binding comparable to that of scFv13.R4 (Figure 7A). scFv 2-3 also shows a further increase in cytoplasmic solubility compared to scFv 14, highlighting the simultaneous engineering of solubility and antigen-binding. Since affinity and soluble expression of the scFvs are screened for simultaneously, it is possible that a selected scFv has moderate solubility but high binding or vice versa. For example, scFv 2-1 has lower soluble expression than scFv 2-3, but it exhibits higher binding affinity to β-galactosidase.

Figure 7. Target-binding and cytoplasmic expression of scFv variants isolated using inner-membrane display. (A) scFvs were expressed in the cytoplasm of E. coli cells (e.g., without the ssTorA signal sequence) with a hexahistidine (6×-His) tag and purified using nickel-nitrilotriacetic acid spin-columns. The binding of the purified scFvs to β-galactosidase was measured with an ELISA. Purified scFvs were loaded onto β-galactosidase-coated ELISA plates, and the bound scFvs were detected with an anti-6×-His antibody. The data are an average of six replicates, and the error bar shows standard error of the mean. (B) The soluble and insoluble fractions of the cell lysates from cells expressing the scFvs cytoplasmically were analyzed by a Western blot probed with an anti-6×-His antibody. Total protein concentration was used to normalize the loading of the samples. Reprinted (A) and adapted (B) from reference 19 with permission. Please click here to view a larger version of this figure.
Discussion
Engineering antibodies for cytoplasmic activity is a difficult task due to the reducing milieu of the cytoplasm, which impedes the formation of stabilizing disulfide bonds6,7. This causes most antibodies to be cytoplasmically inactive unless they are engineered for stability and solubility in the cytoplasm, in addition to being engineered for binding affinity. The existing methods of phage display, bacterial surface display, and yeast surface display methods all use the secretory pathway14-16 for the display of engineered antibodies, but these methods have no means to engineer intracellular folding. Antibodies engineered using inner-membrane display have improved cytoplasmic stability and solubility because the folding quality control of the Tat pathway prevents translocation of antibodies that are poorly folded and unstable in the cytoplasm. This method simplifies the iterative process of engineering intracellular antibodies for affinity and solubility, as the two properties are engineered in one step. Although this method was designed for engineering antibodies with solubility in the reducing intracellular environment, it could also be applied to engineering antibodies to function in non-reducing conditions, since the proteins engineered using this method maintain their folding in the oxidizing environment of the periplasm.
Although this technique simplifies the process of engineering antibodies with high affinity and high cytoplasmic solubility, several limitations are important to consider when using this protocol. When analyzing the secondary screen ELISA signals to identify promising scFv variants, the threshold for discerning between potentially interesting variants and those that may not exhibit adequate antigen-binding is not likely to be apparent until after several clones have been further characterized. It is important to look for improved binding over the parent antibody; however, an abnormally high signal could be indicative of avidity29 or aggregation effects30, a challenge which is not unique to the inner-membrane display screening approach. A key limitation to remember when using this protocol is the inability to recover spheroplasts after panning, as they are non-viable (unpublished data). This necessitates the DNA amplification and transformation steps to recover the antibody-encoding plasmids.
Several critical steps of the protocol enable the simultaneous engineering of folding and binding of antibodies. For screening to be successful, the scFv library being screened must be expressed as a fusion to the ssTorA signal peptide. Without this sequence, antibodies will not be directed to the Tat pathway and thus will not be translocated to the periplasm19. Additionally, it is imperative that a C-terminal epitope tag is fused to the antibodies to allow detection of the displayed antibodies in the binding assays. Clearly, the E. coli strain used to express the scFvs must also have the necessary Tat pathway machinery, but this is true of the commonly used E. coli strains.
Modifications to this protocol are possible to improve its potential to isolate antibodies with the desired characteristics. A subtractive panning step may be completed prior to panning against the target antigen to deplete the scFv library of non-desired constituents. The library spheroplasts can be incubated with magnetic beads coated with BCCP alone or coated with a non-desired protein, and the spheroplasts that bind to those beads can be discarded before screening the remaining unbound spheroplasts for binding to the desired target. As mentioned in the Representative Results, a method to improve the affinity of an isolated scFv is to include a soluble competitor in the panning reaction to compete with the scFvs displayed on the spheroplasts. Because the soluble competitor is a purified protein, no DNA is amplified from it, so only sequences of the scFvs displayed on the spheroplasts will be recovered in the PCR reaction. Additionally, this method could be extended to engineering other types of antibodies or to non-antibody binding proteins.
E. coli inner-membrane display is a powerful platform for engineering antibodies with high affinity and high levels of intracellular solubility. This method is particularly suited for efficient engineering of antibodies designed to function in the intracellular environment. These intracellular antibodies are already being explored as potential therapeutics in a number of fields, including neurodegenerative diseases, cancer, and viral infections31. This technique will enable more widespread use of intracellular antibodies as tools for research and medicine in these fields and any other field where studying a protein target in situ is desired.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
We thank Tomer Zohar for work on scFv screening and characterization assays. A portion of this work was supported by award number F32CA150622 from the National Cancer Institute (to AJK). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health.
Materials
| Name | Company | Catalog Number | Comments |
| scFv library | Varies | A suitable scFv library should be obtained from a commercial or academic source. | |
| MC4100 E. coli cells | Coli Genetic Stock Center | 6152 | Cells need to be chemically competent or electrocompetent, depending on the selected transformation method. |
| Glycerol | Fisher Scientific | BP229-4 | |
| Difco dehydrated culture media LB Broth, Miller (Luria-Bertani) | BD | 244610 | |
| Chloramphenicol (Cm) | Fisher Scientific | BP904-100 | |
| Sodium chloride (NaCl) | Fisher Scientific | BP358-1 | |
| Potassium chloride (KCl) | Fisher Scientific | BP366-500 | |
| Sodium phosphate, dibasic (Na2HPO4) | Fisher Scientific | BP332-500 | |
| Potassium phosphate, monobasic (KH2PO4) | Fisher Scientific | BP362-500 | |
| Bovine serum albumin (BSA) | Fisher Scientific | BP9706-100 | |
| Sucrose | Fisher Scientific | BP220-1 | |
| Tris base | Fisher Scientific | BP1521 | |
| Ethylenediaminetetraacetic acid (EDTA), 0.5 M | Fisher Scientific | BP2482-500 | |
| Magnesium chloride (MgCl2) | Fisher Scientific | BP214-500 | |
| Lysozyme | Sigma Aldrich | L3790-10X1ML | |
| Vortex mixer | VWR | 97043-564 | |
| Bicine | Fisher Scientific | BP2646100 | |
| D-Biotin | Fisher Scientific | BP232-1 | |
| Isopropyl β-D-1-thiogalactopyranoside | Fisher Scientific | BP1755-1 | |
| BugBuster Master Mix (cell lysis detergent) | EMD Millipore | 71456 | |
| Vivaspin 2 MWCO, 3,000 daltons | GE Healthcare Sciences | 28932240 | |
| Target antigen | Varies | N/A | Purified target antigen may be purchased or produced/purified. |
| Dynabeads MyOne Streptavidin T1 | Invitrogen | 65601 | |
| Dynamag-2 magnet | Invitrogen | 12321D | |
| Tube rotator | VWR | 13916-822 | |
| PCR primers | IDT | N/A | Primer sequences are as described in the protocol. |
| 10x T4 DNA ligase reaction buffer | New England BioLabs | B0202S | |
| T4 Polynucelotide kinase (PNK) | New England BioLabs | M0201S | Make sure the T4 ligase buffer used in the primer phosphorylation reaction contains 1 mM ATP. |
| 5x Phusion HF buffer pack | New England BioLabs | B0518S | |
| Deoxynucleotide (dNTP) solution mix, 10 mM each dNTP | New England BioLabs | N0447L | |
| Phusion DNA polymerase | New England BioLabs | M0530S | Other high-fidelity polymerases may be used as an alternative, but the annealing temperature in Table 3 must be adjusted. |
| C1000 Touch thermal cycler with dual 48/48 fast reaction module | Bio-Rad | 185-1148 | |
| Agarose | Promega | V3121 | |
| SYBR Safe DNA gel stain | Invitrogen | S33102 | |
| Wizard SV gel and PCR clean-up system | Promega | A9281 | |
| T4 DNA ligase | New England BioLabs | M0202S | |
| Microdialysis membrane filter | EMD Millipore | VSWP04700 | |
| Agar | BD | 214030 | |
| 96-well polystyrene round-bottom cell culture plates | VWR | 10062-902 | |
| Costar general polystyrene assay plate lids | Corning | 3931 | |
| Microtitre plate shaker | VWR | 12620-926 | |
| Costar 96 well EIA/RIA Easy Wash clear flat bottom polystyrene high bind microplate | Corning | 3369 | |
| Bel-blotter polycarbonate 96-well replicating tool | Bel-Art Products | 378760002 | |
| Instant nonfat dry milk | Quality Biological | A614-1000 | |
| Tween 20 (polysorbate 20) | Fisher Scientific | BP337-500 | |
| PopCulture reagent (concentrated cell lysis detergent) | EMD Millipore | 71092-3 | |
| Monoclonal ANTI-FLAG M2-Peroxidase(HRP) antibody produced in mouse | Sigma Aldrich | A8592 | |
| SigmaFast OPD | Sigma Aldrich | P9187-50SET | |
| Sulfuric acid (H2SO4), 10 N solution | Fisher Scientific | SA200-1 | |
| Reynolds Wrap aluminum foil | VWR | 89079-075 | |
| BioTek Epoch microplate spectrophotometer | Fisher Scientific | 11120570 |
References
- Biocca, S., Pierandrei-Amaldi, P., Campioni, N., Cattaneo, A. Intracellular immunization with cytosolic recombinant antibodies. Biotechnology (NY). 12 (4), 396-399 (1994).
- Chen, S. Y., Bagley, J., Marasco, W. A. Intracellular antibodies as a new class of therapeutic molecules for gene-therapy. Hum. Gene Ther. 5 (5), 595-601 (2008).
- Gargano, N., Biocca, S., Bradbury, A., Cattaneo, A. Human recombinant antibody fragments neutralizing human immunodeficiency virus type 1 reverse transcriptase provide an experimental basis for the structural classification of the DNA polymerase family. J Virol. 70 (11), 7706-7712 (1996).
- Mhashilkar, A. M., et al. Inhibition of HIV-1 Tat-mediated LTR transactivation and HIV-1 infection by anti-Tat single chain intrabodies. Embo J. 14 (7), 1542-1551 (1995).
- Strube, R. W., Chen, S. Y. Characterization of anti-cyclin E single-chain Fv antibodies and intrabodies in breast cancer cells: enhanced intracellular stability of novel sFv-F-c intrabodies. J. Immunol. Meth. 263 (1-2), 149-197 (2002).
- Mössner, E., Koch, H., Plückthun, A. Fast selection of antibodies without antigen purification: adaptation of the protein fragment complementation assay to select antigen-antibody pairs. J. Mol. Biol. 308 (2), 115-122 (2001).
- Wörn, A., et al. Correlation between in vitro stability and in vivo performance of anti-GCN4 intrabodies as cytoplasmic inhibitors. J Biol Chem. 275 (4), 2795-2803 (2000).
- Knappik, A., Plückthun, A. Engineered turns of a recombinant antibody improve its in vivo folding. Protein Eng. 8 (1), 81-89 (1995).
- Martineau, P., Jones, P., Winter, G. Expression of an antibody fragment at high levels in the bacterial cytoplasm. J Mol Biol. 280 (1), 117-127 (1998).
- Steipe, B., Schiller, B., Plückthun, A., Steinbacher, S. Sequence statistics reliably predict stabilizing mutations in a protein domain. J. Mol. Biol. 240 (3), 188-192 (1994).
- Daugherty, P. S. Protein engineering with bacterial display. Curr Opin Struct Biol. 17 (4), 474-480 (2007).
- Lener, M., et al. Diverting a protein from its cellular location by intracellular antibodies. Eur. J. Biochem. 267 (4), 1196-1205 (2000).
- Lynch, S. M., Zhou, C., Messer, A. An scFv intrabody against the nonamyloid component of α-synuclein reduces intracellular aggregation and toxicity. J. Mol. Biol. 377 (1), 136-147 (2008).
- Gai, S. A., Wittrup, K. D. Yeast surface display for protein engineering and characterization. Curr. Opin. Struc. Biol. 17 (4), 467-473 (2007).
- Kieke, M. C., et al. Selection of functional T cell receptor mutants from a yeast surface-display library. Proc. Natl. Acad. Sci. USA. 96 (10), 5651-5656 (1999).
- Steiner, D., Forrer, P., Stumpp, M. T., Pluckthun, A. Signal sequences directing cotranslational translocation expand the range of proteins amenable to phage display. Nat. Biotechnol. 24, 823-831 (2006).
- Pugsley, A. P. The complete general secretory pathway in gram-negative bacteria. Microbiol. Rev. 57 (1), 50-108 (1993).
- Rapoza, M. P., Webster, R. E. The filamentous bacteriophage assembly proteins require the bacterial SecA protein for correct localization to the membrane. J. Bacteriol. 175 (6), 1856-1859 (1993).
- Karlsson, A. J., et al. Engineering antibody fitness and function using membrane-anchored display of correctly folded proteins. J. Molec. Biol. 416 (1), 94-107 (2012).
- DeLisa, M. P., Tullman, D., Georgiou, G. Folding quality control in the export of proteins by the bacterial twin-arginine translocation pathway. Proc Natl Acad Sci U S A. 100 (10), 6115-6120 (2003).
- Fisher, A. C., Kim, W., DeLisa, M. P. Genetic selection for protein solubility enabled by the folding quality control feature of the twin-arginine translocation pathway. Protein Sci. 15 (3), 449-458 (2006).
- Maynard, J., Georgiou, G. Antibody engineering. Annu Rev Biomed Eng. 2, 339-376 (2000).
- Green, M. R., Sambrook, J. Molecular Cloning: A Laboratory Manual. 1, Fourth, Cold Spring Harbor Laboratory Press. (2012).
- Milo, R., Jorgensen, P., Moran, U., Weber, G., Springer, M. BioNumbers--the database of key numbers in molecular and cell biology. Nucleic Acids Res. 38, D750-D753 (2010).
- Hermanson, G. T. Bioconjugate Techniques. , Third, Elsevier/Academic Press. (2013).
- Tayapiwatana, C., Chotpadiwetkul, R., Kasinrerk, W. A novel approach using streptavidin magnetic bead-sorted in vivo biotinylated survivin for monoclonal antibody production. J Immunol Methods. 317 (1-2), 1-11 (2006).
- Zhu, G., Song, L., Lippard, S. J. Visualizing inhibition of nucleosome mobility and transcription by cisplatin-DNA interstrand crosslinks in live mammalian cells. Cancer Res. 73 (14), 4451-4460 (2013).
- Bogsch, E. G., et al. An essential component of a novel bacterial protein export system with homologues in plastids and mitochondria. J. Biol. Chem. 273, 18003-18006 (1998).
- Julian, M. C., et al. Co-evolution of affinity and stability of grafted amyloid-motif domain antibodies. Protein Eng. Des. Sel. 28 (10), 339-350 (2015).
- Garber, K. Bispecific antibodies rise again. Nat. Rev. Drug. Discov. 13 (11), 799-801 (2014).
- Marschall, A. L., Dübel, S., Böldicke, T. Specific in vivo knockdown of protein function by intrabodies. MAbs. 7 (6), 1010-1035 (2015).
