

Introduction
נוגדנים המסוגלים קיפול מתפקדים בסביבה התאית הם כלים מבטיחים הוא במחקר והן יישומים רפואיים. יש להם את היכולת לווסת את פעילות חלבונים באמצעות קשירה לחלבון יעד בתוך תאים על מנת למנוע אינטראקציות בין חלבונים, לשבש אינטראקציות חומצת חלבון גרעין, או למנוע גישת מצע אנזימי 1-5.
אמנם יש נוגדני פוטנציאל רב עבור יישומים תאיים, ההנדסה להם לקיפול נכון מסיס בסביבת תאיים תוך שמירה על היכולת להיקשר אנטיגן יעד היא מאתגר. סביבת ציטופלסמית הצמצום מונעת ההיווצרות של אג"ח דיסולפיד בדרך כלל נדרש עבור הקיפול היציב של נוגדנים באורך מלא ושברי נוגדן, כוללים שבר משתנה יחיד שרשרת (scFv) נוגדנים 6,7. מספר גישות אבולוציה מכוונת להיות מועסק להנדס נוגדנים עם hזיקות igh עבור היעד אנטיגנים 8-10. גישות אלה להשתמש תצוגה הפאג נפוצות, תצוגה משטחת שמרים, או תצוגת משטח חיידקי למסך ספריות גדולות של נוגדנים 11-13. שיטות אלה הם חזקים ויעילים לזיהוי נוגדנים הנקשרים מטרות, ובכל זאת הם תלויים מסלול הפרשה להובלת חלבונים אשר יוצג 14-16. מסלול הפרשה translocates חלבונים פרש מן הציטופלסמה צמצום לתוך לומן reticulum endoplasmic בשמרים או לתוך periplasm בחיידקים. החלבונים ואז לקפל בתנאי חמצון והם מוצגים על פני התא או ארוזים לחלקיקים הפאג למסך מחייב זיקת 17,18. כתוצאה מכך, נוגדנים המבודדים באמצעות טכניקות אלה לא בהכרח לקפל גם בציטופלסמה, מסיס תאי לעתים קרובות חייב להיות מהונדס בנפרד אם הנוגדנים לשמש ביישומים תאיים.
לשפראת היעילות של נוגדנים הנדסה כי מקופלות גם בציטופלסמה, אנו שדווחנו בעבר להצלחה-TRAP MAD (תצוגת מעוגני קרום להכרה מבוססת טאט של חלבוני שיוך), שיטה לסינון ספריית נוגדן scFv באמצעות Escherichia coli inner- תצוגת קרום 19. תצוגה פנימית-קרום חיידקים מסתמכת על טרנסלוקציה התאום ארגינין (Tat) המסלול להובלת נוגדנים מוצגים, בניגוד לשיטות להציג משותף אחרים המשתמשות מסלול ההפרשה. מסלול טאט מכיל מנגנון בקרת איכות שרק מאפשר מסיסים, חלבונים מקופלים כראוי להיות מועברים מן E. הציטופלסמה coli, דרך הממברנה הפנימית, ולתוך 20,21 periplasm. מצעי overexpressed טאט (כלומר., חלבונים ממוקדים מסלול טאט עם פיוז'ן N-terminal כדי הפפטיד אות טאט ssTorA) כי מקופלים גם בציטופלסמה יוצרים טרנסלוקציה חיים ארוך ביניים עם הסופית- N in בציטופלסמה ואת C- הסופית של 19 periplasm. זה מאפשר תצוגה של מצעים טאט מקופלים כהלכה, כוללים שברי נוגדנים, על פן periplasmic של E. הקרום הפנימי coli. לאחר הסרת הקרום החיצוני על ידי עיכול אנזימטי ליצור spheroplasts, נוגדנים נחשפים למרחב התאי (איור 1). זה מאפשר מצעי טאט מוצגים על הקרום הפנימי שיוקרו עבור מחייב יעד ספציפי. חשוב לציין, רתימת מסלול טאט לתצוגה על פני קרום תא מבטיחה כי רק את הנוגדנים בספרייה כי מקופלות גם בציטופלסמה ייחקרו לכריכה, המאפשר הנדסה סימולטני של מחייב זיקה וקיפול תאי. בפרוטוקול זה, אנו מתארים כיצד להציג ספריית scFv על E. הקרום הפנימי coli, במחבת את הספרייה נגד אנטיגן היעד, ולבצע מסך משני כדי לזהות את המרכיבים המבטיחים ביותר של הספרייה. בעוד אנו מתמקדים הפרוטוקול על scFvs, השיטה יכולה להיות מיושמת על הנדסת חלבון כלשהו שבקשתו דורשת קיפול מחייב תאי.
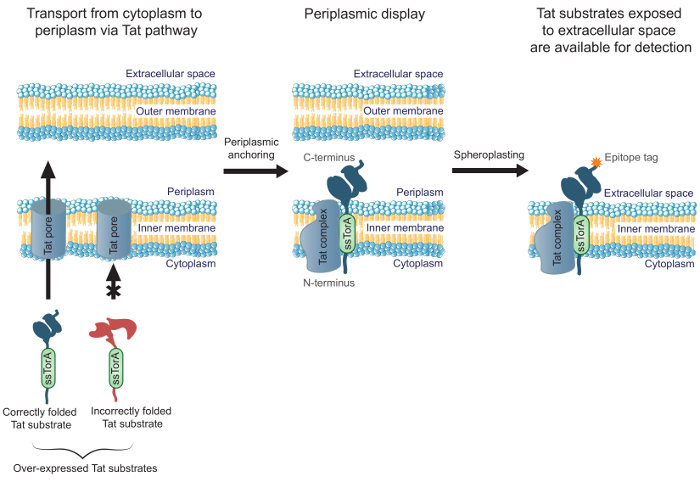
באיור 1. תצוגה פנימי-קרום טאט. בתוך א ' coli, נוגדנים scFv המתבטאים כמיזוג לרצף אות ssTorA ושילב כראוי בציטופלסמה מועברים דרך הממברנה הפנימית. טרנסלוקציה צורות ביניים, שבו scFvs מעוגנים בקרום הפנימי עם הסופית- N בציטופלסמה ואת C- הסופית של periplasm. .ה הממברנה חיצונית coli מתעכלת enzymatically לגבש spheroplasts, ובכך לחשוף את הנוגדנים המעוגנים במרחב התאי ולהפוך אותם לזמינים עבור זיהוי באמצעות נוגדן אשר נקשר תג epitope C- סופנים התמזג על הנוגדן המוצג.עומס / 54,583 / 54583fig1large.jpg "target =" _ blank "> לחץ כאן כדי לצפות בגרסה גדולה יותר של דמות זו.
Protocol
1. מכין את ספריית scFv כמיזוג אל רצף איתותי ssTorA
- השג חומצה deoxyribonucleic (DNA) הספרייה המכיל גרסאות של גן scFv.
הערה: הספרייה עשויה גם להיבנות באמצעות כל מצב מתאים כדי ליצור גיוון מעל גן scFv כולו או התחומים ממוקדים 22 (למשל, המשלים השלישי קביעת אזורים, CDR3s.). - הכנס את ספריית DNA לתוך פלסמיד pIMD (איור 2) שימוש בשיטות שיבוט מולקולרי תקן 23.
הערה: פלסמיד זה מבטא scFvs כמיזוג גנטי לרצף אות ssTorA (N-terminal כדי scFv) ואת תג epitope FLAG (C-terminal אל scFv). העיצוב של פלסמיד לתצוגת קרום פנימי תואר בעבר 19. הפלסמיד pIMD זמין אצל הסופרים.

איור 2. פלסמיד התצוגה Inner-קרום (pIMD) המפה (צעדים 1.2 דרך 1.3). פלסמיד זה מכיל promotor לאק, מוצא ColE1 של שכפול, ויש גן התנגדות chloramphenicol. גן scFv המוכנס הוא התמזג ברצף אות ssTorA למקד את scFv להתפתח בהתאם למסלול טאט וכדי תג epitope FLAG, עם כל השלוש באותה מסגרת קריאה. אתרי אנזים הגבלה מסומנים. עבור ספרייה המוכנסת בין אתרי אנזים הגבלת XbaI ו NotI, בגודל של הפלסמיד הוא 2219 נ"ב בתוספת גודל scFv. אנא לחץ כאן כדי לצפות בגרסה גדולה יותר של דמות זו.
- להפוך את ה- DNA פלסמיד המכיל את הספרייה לתוך MC4100 E. coli תאים 23. שחזור ולגדול בצורת החיידק הזה של הספרייה. צנטריפוגה ב 4000 גרם × במשך 15 דקות ב RT כדי לאסוף את התאים. הסר את supernatant ו resuspend התאים שנאספוגליצרול 25% ב לוריא- Bertani התקשורת (LB). חנות aliquots ב -80 ° C עד הצורך, או המשך לשלב 2.
הערה: הפרוטוקול אומת עם תאי MC4100, למרות E. האחר קולי זנים צפויים גם להיות תואם הפרוטוקול. Electroporation היא השיטה המועדפת לטרנספורמציה, בשל יעילות השינוי הגבוהה שלה. הספרייה צריכה בדרך כלל להתבסס על לפחות 10 9 scFv וריאנטים בשלב זה, וכל אחד aliquot צריך להכיל מספיק תאים כאלה שהספרייה מכוסית פי 100.
2. אין מהיר הספרייה ולהכין Spheroplasts
- להפשיר aliquot אחד של הספרייה בקטריאלי (מ שלב 1.3) ב RT, ומוסיפים את aliquot בבקבוק המכיל 100 מ"ל מדיה LB עם 20 מיקרוגרם / מ"ל chloramphenicol (ס"מ). לגדול במשך 3 שעות ב 37 מעלות צלזיוס, 225 סל"ד שייקר מודגרות.
- אחרי 3 שעות, להסיר את הבקבוק מן 37 ° C מודגרות שייקר. אפשר ביטוי של ספריית scFv כדילהמשיך O / N במשך 15 עד 22 שעות ב 20 מעלות צלזיוס, 225 סל"ד שייקר מודגרות.
הערה: אין inducer נדרשת בעת שימוש פלסמיד pIMD, כמו האמרגן הוא דולף. שים לב תאים MC4100 לא overexpress מדכא Lac (ו לאצי לא נמצא על פלסמיד).

איור 3. תאים E.coli ו spheroplasts. (א) א קולי תאים הם גליליים בכושר. (ב) לאחר spheroplasting באמצעות EDTA ו ליזוזים, הקרום החיצוני של E. קולי תאים מקרע, ואת spheroplasts וכתוצאה מכך הם בעלי צורה כדורית. תמונות מיקרוסקופיה התערבות ההפרש לעומת זאת (DIC) התקבלו באמצעות מטרה 100x על מיקרוסקופ הפוכה. אנא גללקק כאן כדי לצפות בגרסה גדולה יותר של דמות זו.
- הכן את spheroplasts הספרייה.
הערה: Spheroplasts נוצרת על ידי פקיעת הקרום החיצוני של E. coli והם בעלי צורה כדורית (איור 3).- כן המאגרים הצורכים.
הערה: כל המאגרים צריכים להיות סטרילי.- הכן 1 × פוספט שנאגרו מלוחים (PBS; pH 7.4) על ידי המסת 8 גרם NaCl, 0.2 KCl גרם, 1.44 גרם Na 2 HPO 4, ו 0.24 גרם KH 2 PO 4 ב מזוקקים H 2 O לנפח סופי של 1,000 מ"ל. שמור על הקרח.
- הכן PBS עם 0.1% (w / v) בסרום אלבומין שור (BSA) על ידי המסת BSA 0.2 גרם לתוך 200 מ"ל 1 × PBS. שמור על הקרח.
- הכן את החיץ חלוקה (FB) על ידי ערבוב 7.5 מ"ל של סטרילי, מסוננים 1 M סוכרוז, 1 מ"ל של 1 M טריס חיץ (pH 8.0), ו -1.5 מ"ל מזוקקים H 2 O. שמור על הקרח.
- כן 1 מ"מ חומצת ethylenediaminetetraacetic (EDTA) על ידי הוספת 30 μl of 0.5 M EDTA כדי 14.97 מ"ל מזוקקים H 2 O.
- הכן 0.5 M MgCl 2 על ידי המסת 4.76 גרם MgCl 2 ב 100 מ"ל מזוקקים H 2 O. שמור על הקרח.
- הסר את הבקבוק מן שייקר, ולמדוד את הצפיפות האופטית (OD) ב 600 ננומטר באמצעות ספקטרופוטומטר כדי לקבוע את צפיפות התאים. חשבתי את הנפח של תרבות המושרית צורך כזה כי כל דגימה עבור spheroplasting יש 1 × 10 10 תאים.
הערה: קירוב OD של 600 1 המעיד על ריכוז של 10 9 תאים / מ"ל עבור E. coli יכול לשמש 24. - צנטריפוגה הנפח המחושב של תרבות מושרה צינור 1.5 מיליליטר microcentrifuge בבית גרם 12,000 × ב RT במשך 5 דקות. הכן לפחות שתי דגימות במקרה בעיה מתעוררת הכנת מדגם.
- הסר את supernatant מן התרבויות centrifuged ו resuspend כל גלולה תא 100 μl של FB קר כקרח. צנטריפוגה ב 12,000 ×g ב RT עבור 1 דקות, ולאחר מכן להסיר את supernatant ידי pipetting. Resuspend כל גלולה ב 350 μl של FB קר כקרח בתוספת 3.5 μl של 10 מ"ג / מ"ל ליזוזים.
- לאט מערבולת כל צינור תוך הוספת, dropwise, 700 μl של 1 mM EDTA, ולאחר מכן דגירה צינורות ב RT במשך 20 דקות תוך כדי סיבוב לאט על הכתף צינור לערבב דגימות. הסר את הצינורות מן הכתף, להוסיף 50 μl של 0.5 קר כקרח M MgCl 2 על צינור אחד, דגירה אותם על קרח למשך 10 דקות. צנטריפוגה צינורות ב g 11,000 × ב 4 מעלות צלזיוס במשך 10 דקות.
- לבודד את גלולת spheroplast.
- השתמש micropipette עם טיפ 1 מ"ל להרים חלק גלולה לאט. תוך כדי לחיצה על הצינור בזווית עם הפתיחה ישירות מעל צינור 1.5 מיליליטר חדש, לאט להרים את קצה פיפטה מתוך supernatant וחלק גלול לתוך הצינור החדש.
- אם אמצעי אחסון של supernatant משמעותי מועבר הצינור החדש, להסיר אותו על ידי pipetting. אם הגלולה אינה חברת Enoאיכס להעביר, מחדש צנטריפוגות ב 11,000 × גרם במשך 2 דקות ובידוד גלולה ניסיון שוב.
- Resuspend גלולה spheroplast בצינור אחד ב 1 מ"ל של 1 קר כקרח × PBS. חלופי בין pipetting ולאט לאט vortexing על בעל מערבולת עד הגלולה היא resuspended לחלוטין. אין להשאיר דגימות הנחה של קרח למשך יותר מ -2 דקות בכל פעם, ולחזור קרח דק 5 לפחות לפני הסרת מן הקרח שוב. שמור את spheroplasts ב 4 ° C (עד 2 ימים) עד בשימוש עבור panning בשלב 4.
- כן המאגרים הצורכים.
3. לשתק את יעד Antigen על חרוזים מגנטיים
- Biotinylate האנטיגן היעד in vivo במהלך הייצור רקומביננטי ב E. קולי תאים. לחלופין, להשתמש נטייה כימית 25 או רכישת אנטיגן יעד שכבר biotinylated, והמשך לשלב 3.2.
- להוסיף 816 גרם bicine 50 מ"ל מים כדי להפוך 10 × bicine חיץ. לדלל את buffer עד 1 × ב מזוקקים H 2 O וחום עד 50 ° C. הוסף 14.7 מ"ג ביוטין עד 12 מ"ל של חיץ 1 × bicine מחוממת לבצע פתרון ביוטין כי הוא ביוטין 5 מ"מ ב -10 מ"מ חיץ bicine. חנות ב -20 ° C עד הצורך.
- לבטא biotinylate חלבון המטרה באמצעות פלסמיד pAK400cb-BCCP 26, אשר מאפשר ייצור של אנטיגן היעד כמיזוג לחלבון המוביל carboxyl ביוטין (BCCP).
הערה: E. קולי תאים מקורי biotinylate BCCP, ומבטל את הצורך לטהר וכימית biotinylate חלבון המטרה לפני קיבוע על חרוזים מצופים streptavidin. .ה הילידים האנזים ביוטין coli הבירה מספיקה biotinylating חלבון ההיתוך.- לגדול E. coli המכיל את הפלסמיד biotinylation (עם האנטיגן היעד מוכנס כמיזוג אל N הסופית- של BCCP) O / N עבור 15 עד 18 שעות ב 5 מ"ל של התקשורת LB בתוספת 20 מיקרוגרם / מ"ל ס"מ על 37 מעלות צלזיוס תוך רועד ב 225 סל"ד.
- מדוד את OD ב 600 ננומטר באמצעות ספקטרופוטומטר ולחשב את היקף נזקקה לתרבות (add V) כדי תת בכל OD התחלתי של התקשורת LB 0.05 ב 25 מ"ל טרי עם 20 מיקרוגרם / מ"ל ס"מ באמצעות המשוואה: V להוסיף = (0.05 × 25 מיליליטר) / (OD 600 - 0.05), שם OD 600 הוא הצפיפות האופטית של O / N הרחבת תרבות V הוא הנפח של תרבות O / N להוסיף LB. הטרי תת ולגדול כדי OD של 0.5 עד 0.8 שייקר וטופחו על 37 מעלות צלזיוס, 225 סל"ד.
- להוסיף איזופרופיל β-D-1-thiogalactopyranoside לריכוז סופי של 100 מיקרומטר ו ביוטין לריכוז סופי של 5 מיקרומטר. להשרות ביטוי שייקר וטופח במשך 15 עד 22 שעות ב 20 מעלות צלזיוס, 225 סל"ד.
- חיידקי קציר על ידי צנטריפוגה ב 4000 גרם × ב 4 מעלות צלזיוס במשך 10 דקות. הסר את supernatant. אחסן את הגלולה ב -20 ° C עד מוכן לשימוש.
- לְהוֹסִיף1 מ"ל של סבון תמוגה תא לכל 0.2 גרם של התא גלולה. Resuspend ידי pipetting ולסובב בעדינות במשך 20 דקות כדי lyse התאים. לאחר התפרקות צנטריפוגות ב g 16,000 × ו -4 מעלות צלזיוס למשך 20 דקות. פיפטה lysate המסיס (supernatant) לתוך צינור 1.5 מיליליטר חדש.
- השתמש בעמודת הפסקת משקל מולקולרי 3 kDa להסיר את ביוטין המאוגד. פיפטה lysate לתוך הטור, ו צנטריפוגות ב 20 ° C, בהתאם להוראות היצרן. לשטוף עם 1 × PBS עד ביוטין ב lysate דולל פי 100 ואת הנפח של lysate השטף הוא שווה לנפח המקורי של lysate. מעבירים את lysate לצינור חדש.
- לשתק את אנטיגן יעד biotinylated על חרוזים מגנטיים streptavidin מצופה.
- כן 1 × PBS ו 1 × PBS עם 0.1% (w / v) BSA כמתואר שלב 2.3.1.
- הכן את החרוזים המגנטיים.
הערה: זה מחייב השימוש מתל הפרדה מגנטית.- resuspend streptavIdin מצופה חרוזים מגנטיים ב הבקבוקון המקורי שלהם. מערבולת או למשך 30 שניות לפחות או לסובב במשך 5 דקות.
- העבר 7-10 × 10 9 חרוזים לצינור 1.5 מ"ל.
הערה: הנפח נדרש יהיה תלוי בריכוז החרוז המסופק על ידי היצרן. - מניח את הצינור המכיל את החרוזים על המדף השואב 2 דקות כדי לאסוף את החרוזים על הצד של הצינור. עם צינור עדיין על המגנט, להסיר את supernatant בקפידה על ידי pipetting מבלי לשבש את החרוזים.
- כדי לשטוף, להסיר את הצינור מתוך המגנט, ו resuspend את החרוזים ב 1 מ"ל של 1 × PBS על ידי pipetting מבלי ליצור בועות. החזר את הצינור כדי המגנט במשך 2 דקות כדי לאסוף את החרוזים, ולהסיר את supernatant בקפידה על ידי pipetting. חזור על התהליך פעמיים נוספות עבור סכום כולל של שלושה שוטף. ודא שאף נוזל נשאר בצינור לאחר השטיפה של הדבר.
- מוסיפים את lysate המכיל את האנטיגן biotinylated אל ביה מגנטיds.
- הסר את הצינור מתוך המגנט resuspend את החרוזים ב 1 מ"ל של lysate (משלב 3.1.5). לדגור על RT במשך 30 דקות תוך כדי סיבוב בעדינות.
- מניחים את הצינור על המגנט במשך 3 דקות כדי לאסוף את החרוזים מצופים אנטיגן. שטפו את החרוזים מצופה חמש פעמים עם 1 × PBS עם BSA 0.1% באותו אופן כפי שמתואר צעדים 3.2.2.3 כדי 3.2.2.4. לאחר שטיפה של דבר, resuspend את החרוזים 1 × PBS עם 0.1% BSA עד נפח זהה המשמשים שלב 3.2.2.2.
- אם אנטיגן היעד המשותק יציב ב 4 ° C, לאחסן את החרוזים צופה ב 4 ° C עד צורך עבור צילום פנורמי. אחרת, המשך לשלב 4.
מסך 4. הספרייה scFv ידי צילום פנורמי נגד יעד Antigen (איור 4)

איור 4. פאן (שלב 4). חרוזים מגנטיים מצופה Antigen arדואר מודגרות עם spheroplasts להביע גרסאות הספרייה נוגדן. פלסמיד דנ"א spheroplasts חרוז נכנס הוא התאושש המשמש לייצור sublibrary, אשר הוקרן באמצעות המסך משני המבוסס ELISA. מקבילים צעדי פרוטוקול מצוינים. אנא לחץ כאן כדי לצפות בגרסה גדולה יותר של דמות זו.
- דגירת חרוזי מצופי spheroplasts.
- השתמש spheroplast חרוז יחס של כ -5: 1. הוסף 4 × 10 9 spheroplasts ו -8 × 10 8 חרוזים לצינור 15 מ"ל סטרילי.
הערה: נניח כי אין תאים שאבדו במהלך תהליך spheroplasting, כך הריכוז הוא עדיין 1 × 10 10 spheroplasts / מ"ל. - הוסף 1 × PBS עם 0.1% BSA להביא את הנפח הכולל עד 4 מ"ל. Aliquot לארבע צינורות 1.5 מ"ל עם 1 מ"ל כל אחד. דגירת התגובות ב 4 מעלות צלזיוס למשך 5 שעות תוך כדי סיבוב בעדינות.
- השתמש spheroplast חרוז יחס של כ -5: 1. הוסף 4 × 10 9 spheroplasts ו -8 × 10 8 חרוזים לצינור 15 מ"ל סטרילי.
- יחסי ציבורepare spheroplasts הנכנס חרוז עבור תגובת שרשרת פולימראז (PCR).
- מניח את צינורות תגובה פנורמי על המגנט במשך 3 דקות. הסר את supernatant ידי pipetting, ולשטוף את spheroplasts הנכנס חרוז ארבע פעמים עם קרח קר 1 × PBS עם BSA 0.1% באותו אופן כפי שמתואר צעדים 3.2.2.3 כדי 3.2.2.4. Resuspend את spheroplasts הנכנס החרוז בצינור אחד ב 25 μl של H המזוקק 2 O. אחסן את החרוזים ב -20 ° C או המשך לשלב 4.3.
- בצע PCR-פלסמיד שלם על spheroplasts הנכנס חרוז כדי להגביר את פלסמידים המכילים את הגנים scFvs נכנס חרוז.
- השג פריימרים עם הרצפים הבאים: 5'CCAACTCTTTTTCCGAAGGTAACTG3 '(קדימה פריימר) ו 5'TAGCTCTTGATCCGGCAAACAAA3' (פריימר ההפוכה).
הערה: אלה יחייבו מקצה לקצה על גדילים השני של פלסמיד pIMD (איור 2) ו נועדו לחשל את תכונה משותפת של pIMD, כך הגברה תתרחש קשררצף גרסת scFv. - Phosphorylate פריימרים.
הערה: ללא זירחון, מחדש קשירה לא תתרחש. Primers ניתן להזמין גם עם 5'-זירחון, ולא בשיטה זירחון זה בפרוטוקול זה.- בתוך צינור 0.5 מ"ל, להקים התגובה זירחון עבור PCR פריימר קדימה כמתואר בטבלה 1. חזור על תהליך זה עבור פריימר הפוכה.
- דגירת התגובות על 37 מעלות צלזיוס למשך 1 שעה. ואז דגירה אותם ב 65 מעלות צלזיוס למשך 20 דקות כדי לבטל את קינאז polynucleotide T4 (PNK). אחסן את פריימרים פוספורילציה ב -20 ° C.
- בצע PCR.
- בתוך צינור PCR, להכין את תגובת PCR, כמתואר בטבלה 2.
הערה: תגובות מרובות יכולות להיות מוכנות עבור תשואה גבוהה יותר. Spheroplasts בשימוש נכנס החרוז ניתן לאחסן ב -20 ° C. - מחממים את התגובות PCR ב 98 מעלות צלזיוס במשך 15 דקות בתוך Cycler תרמית על מנת להבטיח תמוגה מלא של spheroplasts. הסר את הצינורות מן Cycler תרמית, ולהוסיף 0.5 μl של פולימראז באיכות גבוהה לכל. חזור צינורות Cycler התרמית ולהפעיל באמצעות תוכנית המפורטים בטבלה 3.
- פינת מוצרי PCR על פי צורך. חנות ב -20 ° C או המשך לשלב 4.4.
- בתוך צינור PCR, להכין את תגובת PCR, כמתואר בטבלה 2.
- השג פריימרים עם הרצפים הבאים: 5'CCAACTCTTTTTCCGAAGGTAACTG3 '(קדימה פריימר) ו 5'TAGCTCTTGATCCGGCAAACAAA3' (פריימר ההפוכה).
טבלה 1. התגובה זירחון PNK (שלב 4.3.2.1).
| מֵגִיב | נפח (μl) |
| מזוקקים H 2 O | 15 |
| מאגר תגובת אנזים 10x T4 DNA | 2 |
| 100 מיקרומטר פריימר | 2 |
| קינאז T4 Polynucleotide (PNK) | 1 |
| מֵגִיב | נפח (μl) |
| מזוקקים H 2 O | 28.5 |
| חיץ פולימראז 5x באיכות גבוהה מאוד | 10 |
| פריימר 10 מיקרומטר פוספורילציה קדימה | 2.5 |
| פריימר 10 מיקרומטר פוספורילציה הפוכה | 2.5 |
| 40 מ"מ שילוב dNTP (10 מ"מ כל dNTP) | 1 |
| spheroplasts חרוז נכנס | 5 |
לוח 3. תוכנית ה- PCR (שלב 4.3.3.2).
| שלב | טמפרטורה (° C) | זמן (שניות: דקות) | מספר מחזורי |
| לפגל ראשוני | 98 | 00:30 | 1 |
| לְפַגֵל | 98 | 00:10 | 35 |
| רִכּוּך | 69 | 00:30 | |
| סיומת | 72 | 00:30 לכל kb | |
| סיומת סופית | 72 | 06:00 | 1 |
| לְהַחזִיק | 12 | אֵינְסוֹף | 1 |
- Re-circularize המוצרים PCR מחיטה פלסמיד, ולהשתמש במוצר ligated להפוך MC4100 E. קולי תאים.
- לטהר את מוצר ה- PCR על ידי הפעלת תגובת PCR על ג'ל agarose 23, מכתים את ה- DNA בתוך הג'ל 23, ושימושערכת ניקוי ג'ל לטהר את הפלסמיד לינארית לפי ההנחיות שסופקו על ידי היצרן. למדוד את ריכוז באמצעות ספקטרופוטומטר ב 260 ננומטר. אחסן את שבר מטוהרים ב -20 ° C עד הצורך, או המשך לשלב 4.4.2.
- Re-circularize פלסמיד מהמוצר PCR.
- כדי למנוע קשירת מולקולאריים של מוצר PCR, לבצע את תגובת הקשירה עם ריכוז נמוך 27 של 1 ng / μl של מוצר ה- PCR. חשב את נפח הדרושים להכנת התגובה קשירת 800 μl בריכוז הזה.
- הכן את תגובת הקשירה על קרח. בתוך צינור, מוסיפים את נפח מחושב בשלב 4.4.2.1 של מוצר ה- PCR, 80 μl של חיץ האנזים 10 × DNA, מזוקקים H 2 O עד 800 μl. הוסף 4 μl של האנזים T4 DNA, ומיד למקם צינורות ב 16 מעלות צלזיוס באמבט מים או Cycler תרמית. לדגור על 16 מעלות CO / N עבור 14 עד 18 שעות. אחסן את תגובות הקשירה הושלמו ב -20 ° Cעד צורך, או המשך לשלב 4.4.3.
- מניח את תגובת הקשירה על גוש חום 65 מעלות צלזיוס למשך 15 דקות כדי לחמם-לנטרל את אנזים DNA. ואז להשתמש ערכת ניקוי DNA קרום או microdialysis לדה-מלח הדנ"א ligated. חנות ב -20 ° C או המשך לשלב 4.4.4.
- השתמש במוצר קשירת חום מומת כולו מלוחים-דה להפוך MC4100 E. coli תאים 23. הכן מניות גליצרול, כמתואר שלב 1.3, של התאים המכילים את sublibrary סרק וכתוצאה מכך, ו aliquots חנות ב -80 ° C.
- חזור על שלב 4 בשלמותו באמצעות aliquot משלב 4.4.4 לעשות צילום פנורמי שני על sublibrary.
הערה: צילום פנורמי שני עוזר להעשיר את מרכיבי ספרייה אשר נקלטים היטב כדי האנטיגן היעד 19.
5. בצע מסך משני בשיטת Enzyme-linked immunosorbent Assay לזהות משובטים מבטיח עבור אפיון נוסף (איור 5) </ P>

איור 5. הקרנה משני מבוסס ELISA (שלב 5). (א) גרסות ספרייה מן sublibrary המועשר במהלך פנורמי הם מחוסנים בבארות בודדות של צלחת תרבות לצמיחת ביטוי. (ב) צלחת ELISA מצופה אנטיגן היעד. (ג) גרסות הספרייה מוקרנות באמצעות המסך המשני המבוסס ELISA כמתואר בפרוטוקול. לאחר הניתוח של נתונים המתקבלים המסך המשני, וריאנטים של עניין נבחרים ומאופיינים נוסף. מקבילים צעדי פרוטוקול מצוינים. אנא לחץ כאן כדי לצפות בגרסה גדולה יותר של דמות זו.
- להפשיר צינור אחד sublibrary סרק (משלב 4.4.4) וצלחת על צלחות אגר LB. דילולים פלייט מספרבריכוזים נמוכים מספיק כדי להבטיח מושבות בודדות (למשל, 10 2 - 10 6 דילולים -fold). דגירת הצלחות עבור 15 עד 18 שעות ב 37 מעלות צלזיוס. אחסן את הצלחות ב 4 ° C. או המשך לשלב 5.2.
- מושבות תרבות להשרות מן sublibrary סרק. בצע את כל השלבים בתנאים סטריליים. השתמש פיפטור רב ערוצים לצעדים מעורבים 96-גם צלחות.
- הוסף 200 μl של LB עם 20 מיקרוגרם / מ"ל ס"מ לבאר כל צלחת תרבות מסביב לתחתית 96-היטב.
- פיק מושבה בודדת מתוך הצלחת אגרה עם קצה פיפטה, למקם את קצה הבאר הראשונה של צלחת 96-היטב, ומערבבים בעדינות כדי לחסן. השתמש טיפ חדש עבור כל טוב. לחסן מושבה אחת לבאר כל אחד. כביקורת, לכלול לפחות שליטה עקרה אחד גם ללא מושבה המחוסנת.
- חזרו על שלבי 5.2.1 ו 5.2.2 לחסן מספר 96-גם צלחות.
- מניחים את הצלחות 96-היטב על שייקר microplate ב 310 סל"ד. לדגור על 376; צלזיוס למשך 20 עד 24 שעות כדי לבטא את scFvs.
- עבור כל צלחת תרבות המוכנה בשלב 5.2, שכבה אחת צלחת ELISA 96-היטב עם האנטיגן היעד.
- לדלל אנטיגן היעד מטוהרים ריכוז המתאים (למשל, 1 מיקרוגרם / מ"ל ל 4 מיקרוגרם / מ"ל) ב 1 × PBS על מנת להפוך את פתרון הציפוי. הפוך 5 מ"ל של תמיסת ציפוי על כל צלחת 96-היטב.
הערה: הריכוז המתאים תלוי אנטיגן ספציפי בשימוש ועשויים צריך להיות מותאם. - הוסף 50 μl של פתרון הציפוי היטב בכל צלחת 96-היטב גבוהה מחייב ברור קלקר ELISA. לטפוח בעדינות את הצלחת על פני שטח benchtop על מנת להבטיח כי כל פני השטח של כל אחד גם הוא מצופה. חזור על הפעולה עבור כל צלחת. דגירת הצלחות ב 4 ° CO / N.
- לדלל אנטיגן היעד מטוהרים ריכוז המתאים (למשל, 1 מיקרוגרם / מ"ל ל 4 מיקרוגרם / מ"ל) ב 1 × PBS על מנת להפוך את פתרון הציפוי. הפוך 5 מ"ל של תמיסת ציפוי על כל צלחת 96-היטב.
- לשכפל המושבות מהצלחות תרבות 96-גם על צלחות אגר.
- מניחים משכפל פוליסטירן סטרילי לתוך הבארות של צלחת תרבות לאסוף כמות קטנה של liquתְעוּדַת זֶהוּת. בזהירות להעלות למשכפל ולהעביר לצלחת אגר LB 15 ס"מ כך שכל טיפים נוגעים הצלחת. לאחר נוזל עבירה, להרים את המשכפל ישר למעלה. חזור על הפעולה עבור כל צלחת תרבות.
- לייבל הצלחת אגרה עם הכיוון הנכון כך שתוצאות מהמסך משני בצלחת 96-גם ניתן להתאים עם המושבה המשוכפלת הנכונה על הצלחת, אם אפיון נוסף הוא רצוי. לגדול ב 37 מעלות צלזיוס למשך 15 עד 18 שעות, ולאחר מכן לאחסן ב 4 ° C עד הצורך.
- בצע את המסך המשני ELISA.
- הכן את הפתרון חוסם על ידי ביצוע 2% (w / v) חלב יבש 1 × PBS. רוקן את פתרון הציפוי מצלחות ELISA. הוספת 100 μl של פתרון חסימת היטב כל אחד. לדגור על RT במשך שעה לפחות 2, או לחסום O / N ב 4 ° C.
- הכן את החיץ לשטוף ידי הוספת polysorbate 20 לריכוז סופי של 0.05% ב 1 × PBS. הפוך 250 מ"ל לכל צלחת ELISA.
- הוסף 20 μl שלחומר ניקוי התא תמוגה מרוכז היטב כל הצלחת התרבות מסביב לתחתית, הדגירה הצלחה התרבות על שייקר microplate ב RT במשך 15 עד 20 דקות. בגין תמוגה באותו הזמן כי חסימת צלחות ELISA תושלם כך תמוגה בכביסת השלב 5.5.4 יכולים להתבצע במקביל.
- רוקן את הפתרון חוסם מהצלחות ELISA. שטפו את ארבע פעמים ELISA צלחות חסמו עם 200 μl של חיץ לשטוף לכל טוב לכל לשטוף. רוקן את החיץ לשטוף מהבארות.
- העברת 50 μl מבארים כל צלחת תמוגה תא המתאים היטב של צלחת ELISA, באמצעות קצה חדש עבור כל טוב. דגירה את הצלחת ELISA ב RT עבור 1 עד 2 שעות.
- הכן את פתרון הנוגדן לזהות scFvs כבול.
- השתמש peroxidase חזרת (HRP), מצומדות נוגדן ראשוני הנקשרת תג epitope FLAG התמזגו scFvs הספרייה.
- לדלל את נוגדן הדילול המתאים לשימוש בתוך ELISA (ראה ספק & #39; s המלצות) ב 2% (w / v) חלב יבש polysorbate 0.05% 20 ב 1 × PBS. כן 5 מ"ל לכל צלחת.
- שטפו את צלחות ELISA ארבע פעמים עם חיץ לשטוף כמתואר שלב 5.5.4.
- הוסף 50 μl של פתרון נוגדנים היטב כל צלחת ELISA. דגירה של 1 עד 2 שעות ב RT.
- מכינים את המצע HRP ידי המסת o -phenylenediamine dihydrochloride (OPD) הטבליות מזוקקים H 2 O לכל פרוטוקול של היצרן, תוך הימנעות אור. הכן 20 מ"ל לכל צלחת ELISA.
- הכן 3 MH 2 SO 4 על ידי דילול H מרוכז 2 SO 4 עם H 2 O מזוקקים במידת הצורך. כן 5 מ"ל לכל צלחת ELISA.
זהירות: H 2 SO 4 היא חומצה חזקה. הקפד ללבוש ציוד מגן אישי מתאים. - שטפו את צלחות ELISA ארבע פעמים עם חיץ לשטוף, כמתואר שלב 5.5.4.
- דגירה צלחות ELISA עם substra HRPטה.
- הוסף 200 μl של המצע HRP היטב כל אחד. כדי למזער את חשיפה לאור, להוסיף למצע צלחת אחת ELISA בכל פעם, ועוטף ברדיד אלומיניום לפני שתמשיך הצלחת הבאה. דגירת הצלחות במשך 30 עד 60 דקות ב RT בחושך.
- אחרי 30 דק 'הראשון, בדוק את הצלחות המחשיך של המצע, דגירה עוד במידת הצורך לדמיין לפתח צבע.
- הוסף 50 μl של 3 MH 2 SO 4 היטב כל כדי להרוות את התגובה. בעזרת קצה שונה עבור כל טוב, לערבב את הפתרון בבארות ידי pipetting בעדינות מעלה ומטה בלי קצף. לקבלת עקביות ולמנוע רוויה, להוסיף את H 2 SO 4 במהירות ובזהירות לכל צלחות ELISA לפני ערבוב הפתרון עבור כל צלחת.
- מדוד את הספיגה של הפתרון בבארות של כל צלחת ב 492 ננומטר באמצעות קורא צלחת.
- לנתח את הנתונים הספיגים לזהות scFv וריאנטים exhib כיזה מבטיח אותות מחייבים ולאפיין scFvs מבטיחה אלה. בחר scFvs כי תערוכת אותות ספיגים גבוהים יותר מאשר אות הרקע וגבוהים האות הממוצע על כל צלחת.
הערה: הרמה הספיגה תהיה תלוי את המאפיינים של אנטיגן הנוגדן אנטי FLAG נלקחים בחשבון, ביחד עם הכח של וריאנטים scFv כי בודדו ההקרנה.
Representative Results
מנגנון בקרת איכות קיפול חלבונים התאי של מסלול טאט ב E. coli מגביל הובלה על פני קרום התא הפנימי לחלבונים כי מקופלים היטב בסביבת ציטופלסמית הצמצום. על ידי overexpressing שילוב של scFv לרצף אות ssTorA (רצף האות מן החלבון טורה, אשר מועבר באופן טבעי על ידי נתיב טאט 20), טרנסלוקציה תקועה, וכתוצאה מכך התצוגה של scFvs על הקרום הפנימי 19. לאחר הפרעה אנזימטית של הממברנה החיצונית, הנוגדנים המוצגים נעשים זמינים לסינון לפעילות מחייב אנטיגן. היכולת לנצל את מסלול טאט לתצוגה scFv הוצגה על ידי קרלסון et al. 19 (איור 6). הנוגדנים scFv scFv13 ו scFv13.R4 היו התמזגו או רצף יליד ssTorA או ssTorA שונה כי חסרה את צמד שאריות ארגינין-ארגינין מוכר על ידימסלול טאט. scFv13.R4 תוכנן על ידי מרטינו et al. מן scFv13 באמצעות ארבעה סיבובים של אבולוציה מכוונת, והוא ידוע לקפל גם בציטופלסמה 9. ScFv זה הוצג על הקרום הפנימי, אבל רק כאשר מבוטא היתוך רצף האות ssTorA הילידים (איור 6). מנגד, scFv13 אינו מקופל היטב cytoplasmically 9, ולכן הוא אינו מוצג היטב על הקרום הפנימי, ללא קשר רצף האות שאליו הוא התמזג. בנוסף, אם scFvs התבטא תאים חסרי חלבון TatC, מרכיב חיוני של מכונות טאט 20,28, תצוגה לא נצפתה ומראה את הקשר החשוב בין תצוגה פנימית-קרום ואת מסלול טאט. תוצאות אלו מראות כי חלבונים רק המכילים הפפטיד האות טאט וכי מקופלים כראוי בציטופלסמה מוצגים על הקרום הפנימי, מה שמאפשר הובלה דרך מסלול טאט לתפקד כמסך עבור fol התאי דינג.

איור 6. איתור של scFvs מוצג על הקרום הפנימי. Cytometry זרימה הניתוח בוצע כדי לזהות את התצוגה של scFv13 מקופל היטב היטב מקופל scFv13.R4 על הקרום הפנימי. scFvs היו התמזגו יליד ssTorA או ssTorA (KK), שבו זוג Arg-Arg ברצף ssTorA שונה כדי ליס-ליס. תגיות epitope FLAG בטרמינל C-על scFvs התגלו עם isothiocyanate והעמסת (FITC), מצומדות נוגדן אנטי הדגל. תאים ללא חלבון TatC (ΔtatC) ו ssTorA-scFv13 ללא תג FLAG נבחנו כקבוצת ביקורת. M מצביע על ערך הקרינה החציוני. הודפס מחדש בהפניה 19 באישורו. אנא לחץ כאן כדי לצפות בגרסה גדולה יותר של דמות זו.
o: keep-together.within-page = "1"> תצוגה פנימית-קרום יכול לבודד נוגדנים scFv בהצלחה עם רמות גבוהות של זיקה חלבון היעד ורמות גבוהות של מסיסות cytoplasmic. בנוסף, הסיבובים הבאים של אבולוציה מכוונת באמצעות תצוגה פנימי-קרום לשפר מאפייני נוגדן 19. כדי להמחיש זאת, ספריית PCR מועדת לטעויות מבוסס על scFv13, אשר יש רמה נמוכה של מחייב זיקה β-galactosidase, היה panned נגד-galactosidase β אנטיגן היעד באמצעות תצוגת פנורמי השיטה המתוארת בפרוטוקול. scFv 1-4 היה מבודד אחרי סיבוב אחד של mutagenesis פנורמי, והציג זיקה מחייבת גבוהה יותר בטא galactosidase מ scFv13 (איור 7 א) ועל רמה גבוהה יותר של מסיסות cytoplasmic (איור 7).
ספרייה חדשה, המבוססת על scFv 1-4, נעשתה באמצעות מועדת לטעויות PCR, פנורמי של ספרייה דור שני זה נגדβ-galactosidase נעשה באמצעות שינוי של הפרוטוקול המתואר. פנורמי נגד β-galactosidase לסיבוב השני של אבולוציה נעשה בנוכחות המטוהר, מסיס scFv 14 כמתחרה לשפר את הסיכוי של בידוד שיבוטים עם זיקה חזקה יותר מאשר scFv 1-4. לאחר סיבוב שני זה של mutagenesis פנורמי, scFv 2-1 ו scFv 2-3 בודדו באמצעות ההקרנה משני המבוססת ELISA. scFvs אלה לא הוצגו רק זיקה מחייבת גבוהה עבור β-galactosidase מ scFv13, אלא גם הציג יותר מחייב מאשר שיבוט בסיבוב הראשון scFv 1-4. scFv 2-1 הציג-galactosidase β מחייב להשוותו לזה של scFv13.R4 (איור 7 א). scFv 2-3 גם מראה עלייה נוספת מסיסות ציטופלסמית לעומת scFv 14, המדגיש את ההנדסה סימולטני של מסיסות אנטיגן מחייב. מאז זיקה וביטוי מסיס של scFvs מוקרנים על זמנית, זה אפשרי כי שנבחר scFv מלון מודמסיסות erate אבל versa מחייב או סגן גבוה. לדוגמא, scFv 2-1 יש ביטוי מסיס נמוך יותר scFv 2-3, אבל הוא מפגין זיקה מחייבת גבוהה יותר בטא galactosidase.

איור 7. יעד מחייב וביטוי cytoplasmic של scFv גרסאות מבודדים באמצעות התצוגה הפנימית-קרום. (א) scFvs התבטאו בציטופלסמה של E. קולי תאים (למשל., ללא רצף האות ssTorA) עם hexahistidine (6 × הזדרז) תג מטוהרים באמצעות עמודות ספין חומצה nitrilotriacetic ניקל. את הכריכה של scFvs מטוהרים בטא galactosidase נמדדה עם ELISA. scFvs המטוהר הועמס צלחות ELISA מצופה β-galactosidase, ואת scFvs כבול התגלה עם נוגדן אנטי 6 × הזדרז. הנתונים הם ממוצעים של שישה משכפל, ובר השגיאה מראה סטיית התקן של הממוצע.(ב) שברים מסיסים ובחלקם לא מסיסים של lysates תא מתאי המבטא את scFvs cytoplasmically נותחו על ידי כתם המערבי ומישש עם נוגדן אנטי 6 × הזדרז. ריכוז חלבון סה"כ שמש לנרמל את הטעינה של הדגימות. הודפס מחדש (א) ומותאמת (B) בהפניה 19 באישורו. אנא לחץ כאן כדי לצפות בגרסה גדולה יותר של דמות זו.
Discussion
הנדסת נוגדנים לפעילות ציטופלסמית היא משימה קשה בשל המילייה הצמצום של הציטופלסמה, אשר מעכבת את ההיווצרות של ייצוב אג"ח דיסולפיד 6,7. זה גורם לרוב נוגדנים להיות פעיל cytoplasmically אלא אם כן הם מהונדסים ליציבות מסיסה בציטופלסמה, בנוסף להיות מהונדס לכריכת זיקה. השיטות הקיימות של תצוגה הפאג, תצוגת משטח חיידקים, ושיטות תצוגה שמרים משטח כל להשתמש מסלול ההפרשה 14-16 עבור התצוגה של נוגדנים מהונדסים, אך שיטות אלו לא היו אמצעים דרושים כדי להנדס מתקפלים תאי. נוגדנים המהונדסים באמצעות תצוגה פנימי-קרום השתפרו יציבות cytoplasmic ו מסיס כי בקרת איכות קיפול של מסלול טאט מונעת טרנסלוקציה של נוגדנים מקופלים גרוע ובלתי יציב בציטופלסמה. שיטה זו מפשטת את התהליך החוזר והנשנה של נוגדנים תאיים הנדסית זיקהמסיסות nd, כמו שני הנכסים מתוכננים בצעד אחד. אמנם שיטה זו תוכננה עבור הנדסת נוגדנים עם מסיסות בסביבת תאיים הצמצום, זה יכול לחול גם על נוגדני הנדסה לתפקד בתנאים הלא צמצום, מאז החלבונים מהונדסים בשיטה זו לשמור המתקפלים שלהם בסביבת החמצון של periplasm.
למרות הטכניקה הזו מפשטת את תהליך הנדסת נוגדנים עם זיקה גבוהה מסיס cytoplasmic גבוהה, מספר מגבלות חשובות שיש להביא בחשבון בעת שימוש בפרוטוקול זה. כאשר מנתחים את אותות ELISA המסך המשניים לזהות מבטיח scFv וריאנטים, סף הבחנה בין הגירסות מעניינות פוטנציאל ואלה לא יכולים להפגין נאותים אנטיגן מחייב אינו צפוי להיות ברור עד לאחר מספר שיבוטים מתאפיינים נוספים. חשוב לחפש השתפר מחייב על הנוגדן האם; למרות זאת,אות גבוהה באופן חריג יכולה להיות מעידה על להיטות 29 או תופעות צבירת 30, אתגר אשר אינו ייחודית לגישת הקרנת תצוגה הפנימי-הקרום. מגבלה מפתח שיש לזכור בעת השימוש בפרוטוקול זה היא חוסר היכולת לשחזר spheroplasts לאחר צילום פנורמי, כמו שהם לא-ישימות (נתונים שלא פורסמו). זו מחייבת הגברת DNA וצעדים טרנספורמציה לשחזר את פלסמידים קידוד-נוגדן.
שלבים קריטיים כמה הפרוטוקול לאפשר ההנדסה סימולטני של קיפול ומחייבת של נוגדנים. להקרנה כדי להצליח, ספריית scFv שהוקרנה חייבת להיות מבוטאת היתוך הפפטיד אות ssTorA. ללא רצף זה, נוגדנים לא להיות מופנים אל מסלול טאט וכך לא יהיה translocated אל 19 periplasm. בנוסף, קיים הכרח כי תג epitope מסוף C הוא קבע את הנוגדנים כדי לאפשר זיהוי של נוגדנים המוצגים לפחמבחני דינג. אין ספק כי א coli זן נהג לבטא את scFvs חייב גם את מכונות מסלול טאט ההכרחיות, אבל זה נכון גם לגבי א הנפוץ קולי זנים.
עשיית שינויים בפרוטוקול זה ניתן לשפר את הפוטנציאל שלה לבודד נוגדנים עם התכונות הרצויות. צעד פנורמי תוסף עלול להסתיים לפני פנורמי נגד אנטיגן היעד כדי לרוקן את ספריית scFv של מרכיבים הלא רצוי. Spheroplasts הספרייה ניתן מודגרות עם חרוזים מגנטיים צופה BCCP לבד או מצופה עם חלבון שאינו רצוי, ואת spheroplasts אשר נקלט על ידי חרוזים אלה יכול להיות מושלך לפני הקרנת spheroplasts המאוגד הנותר לכריכה ליעד הרצוי. כפי שהוזכר נציג התוצאות, שיטה לשיפור הזיקה של scFv מבודדת היא לכלול מתחרה מסיס התגובה פנורמי להתחרות scFvs מוצג על spheroplasts. מכיוון comp המסיסetitor הוא פרוטאין טהור, אין DNA מוגבר ממנו, כך רצפים רק של scFvs מוצג על spheroplasts יהיו התאוששו תגובת PCR. בנוסף, שיטה זו יכולה להתארך עד הנדסת סוגים אחרים של נוגדנים או חלבונים מחייבים הלא נוגדן.
א תצוגה פנימית-קרום coli הנה פלטפורמה חזקה עבור נוגדני הנדסה עם זיקה גבוהה ורמות גבוהות של מסיסויות תאיות. שיטה זו מתאימה במיוחד עבור הנדסה יעילה של נוגדנים שנועדו לתפקד בסביבה התאית. נוגדנים תאיים אלה כבר נבדקים כמו רפויים פוטנציאליים במספר התחומים, לרבות מחלות ניווניות, סרטן, זיהומים נגיפיים 31. טכניקה זו תאפשר שימוש נרחב יותר של נוגדנים תאיים ככלי מחקר ורפואה בתחומים אלה בכל תחום אחר שבו לומד יעד חלבון באתרו הוא רצוי.
Materials
| Name | Company | Catalog Number | Comments |
| scFv library | Varies | A suitable scFv library should be obtained from a commercial or academic source. | |
| MC4100 E. coli cells | Coli Genetic Stock Center | 6152 | Cells need to be chemically competent or electrocompetent, depending on the selected transformation method. |
| Glycerol | Fisher Scientific | BP229-4 | |
| Difco dehydrated culture media LB Broth, Miller (Luria-Bertani) | BD | 244610 | |
| Chloramphenicol (Cm) | Fisher Scientific | BP904-100 | |
| Sodium chloride (NaCl) | Fisher Scientific | BP358-1 | |
| Potassium chloride (KCl) | Fisher Scientific | BP366-500 | |
| Sodium phosphate, dibasic (Na2HPO4) | Fisher Scientific | BP332-500 | |
| Potassium phosphate, monobasic (KH2PO4) | Fisher Scientific | BP362-500 | |
| Bovine serum albumin (BSA) | Fisher Scientific | BP9706-100 | |
| Sucrose | Fisher Scientific | BP220-1 | |
| Tris base | Fisher Scientific | BP1521 | |
| Ethylenediaminetetraacetic acid (EDTA), 0.5 M | Fisher Scientific | BP2482-500 | |
| Magnesium chloride (MgCl2) | Fisher Scientific | BP214-500 | |
| Lysozyme | Sigma Aldrich | L3790-10X1ML | |
| Vortex mixer | VWR | 97043-564 | |
| Bicine | Fisher Scientific | BP2646100 | |
| D-Biotin | Fisher Scientific | BP232-1 | |
| Isopropyl β-D-1-thiogalactopyranoside | Fisher Scientific | BP1755-1 | |
| BugBuster Master Mix (cell lysis detergent) | EMD Millipore | 71456 | |
| Vivaspin 2 MWCO, 3,000 daltons | GE Healthcare Sciences | 28932240 | |
| Target antigen | Varies | N/A | Purified target antigen may be purchased or produced/purified. |
| Dynabeads MyOne Streptavidin T1 | Invitrogen | 65601 | |
| Dynamag-2 magnet | Invitrogen | 12321D | |
| Tube rotator | VWR | 13916-822 | |
| PCR primers | IDT | N/A | Primer sequences are as described in the protocol. |
| 10x T4 DNA ligase reaction buffer | New England BioLabs | B0202S | |
| T4 Polynucelotide kinase (PNK) | New England BioLabs | M0201S | Make sure the T4 ligase buffer used in the primer phosphorylation reaction contains 1 mM ATP. |
| 5x Phusion HF buffer pack | New England BioLabs | B0518S | |
| Deoxynucleotide (dNTP) solution mix, 10 mM each dNTP | New England BioLabs | N0447L | |
| Phusion DNA polymerase | New England BioLabs | M0530S | Other high-fidelity polymerases may be used as an alternative, but the annealing temperature in Table 3 must be adjusted. |
| C1000 Touch thermal cycler with dual 48/48 fast reaction module | Bio-Rad | 185-1148 | |
| Agarose | Promega | V3121 | |
| SYBR Safe DNA gel stain | Invitrogen | S33102 | |
| Wizard SV gel and PCR clean-up system | Promega | A9281 | |
| T4 DNA ligase | New England BioLabs | M0202S | |
| Microdialysis membrane filter | EMD Millipore | VSWP04700 | |
| Agar | BD | 214030 | |
| 96-well polystyrene round-bottom cell culture plates | VWR | 10062-902 | |
| Costar general polystyrene assay plate lids | Corning | 3931 | |
| Microtitre plate shaker | VWR | 12620-926 | |
| Costar 96 well EIA/RIA Easy Wash clear flat bottom polystyrene high bind microplate | Corning | 3369 | |
| Bel-blotter polycarbonate 96-well replicating tool | Bel-Art Products | 378760002 | |
| Instant nonfat dry milk | Quality Biological | A614-1000 | |
| Tween 20 (polysorbate 20) | Fisher Scientific | BP337-500 | |
| PopCulture reagent (concentrated cell lysis detergent) | EMD Millipore | 71092-3 | |
| Monoclonal ANTI-FLAG M2-Peroxidase(HRP) antibody produced in mouse | Sigma Aldrich | A8592 | |
| SigmaFast OPD | Sigma Aldrich | P9187-50SET | |
| Sulfuric acid (H2SO4), 10 N solution | Fisher Scientific | SA200-1 | |
| Reynolds Wrap aluminum foil | VWR | 89079-075 | |
| BioTek Epoch microplate spectrophotometer | Fisher Scientific | 11120570 |
References
- Biocca, S., Pierandrei-Amaldi, P., Campioni, N., Cattaneo, A. Intracellular immunization with cytosolic recombinant antibodies. Biotechnology (NY). 12 (4), 396-399 (1994).
- Chen, S. Y., Bagley, J., Marasco, W. A. Intracellular antibodies as a new class of therapeutic molecules for gene-therapy. Hum. Gene Ther. 5 (5), 595-601 (2008).
- Gargano, N., Biocca, S., Bradbury, A., Cattaneo, A. Human recombinant antibody fragments neutralizing human immunodeficiency virus type 1 reverse transcriptase provide an experimental basis for the structural classification of the DNA polymerase family. J Virol. 70 (11), 7706-7712 (1996).
- Mhashilkar, A. M., et al. Inhibition of HIV-1 Tat-mediated LTR transactivation and HIV-1 infection by anti-Tat single chain intrabodies. Embo J. 14 (7), 1542-1551 (1995).
- Strube, R. W., Chen, S. Y. Characterization of anti-cyclin E single-chain Fv antibodies and intrabodies in breast cancer cells: enhanced intracellular stability of novel sFv-F-c intrabodies. J. Immunol. Meth. 263 (1-2), 149-197 (2002).
- Mössner, E., Koch, H., Plückthun, A. Fast selection of antibodies without antigen purification: adaptation of the protein fragment complementation assay to select antigen-antibody pairs. J. Mol. Biol. 308 (2), 115-122 (2001).
- Wörn, A., et al. Correlation between in vitro stability and in vivo performance of anti-GCN4 intrabodies as cytoplasmic inhibitors. J Biol Chem. 275 (4), 2795-2803 (2000).
- Knappik, A., Plückthun, A. Engineered turns of a recombinant antibody improve its in vivo folding. Protein Eng. 8 (1), 81-89 (1995).
- Martineau, P., Jones, P., Winter, G. Expression of an antibody fragment at high levels in the bacterial cytoplasm. J Mol Biol. 280 (1), 117-127 (1998).
- Steipe, B., Schiller, B., Plückthun, A., Steinbacher, S. Sequence statistics reliably predict stabilizing mutations in a protein domain. J. Mol. Biol. 240 (3), 188-192 (1994).
- Daugherty, P. S. Protein engineering with bacterial display. Curr Opin Struct Biol. 17 (4), 474-480 (2007).
- Lener, M., et al. Diverting a protein from its cellular location by intracellular antibodies. Eur. J. Biochem. 267 (4), 1196-1205 (2000).
- Lynch, S. M., Zhou, C., Messer, A. An scFv intrabody against the nonamyloid component of α-synuclein reduces intracellular aggregation and toxicity. J. Mol. Biol. 377 (1), 136-147 (2008).
- Gai, S. A., Wittrup, K. D. Yeast surface display for protein engineering and characterization. Curr. Opin. Struc. Biol. 17 (4), 467-473 (2007).
- Kieke, M. C., et al. Selection of functional T cell receptor mutants from a yeast surface-display library. Proc. Natl. Acad. Sci. USA. 96 (10), 5651-5656 (1999).
- Steiner, D., Forrer, P., Stumpp, M. T., Pluckthun, A. Signal sequences directing cotranslational translocation expand the range of proteins amenable to phage display. Nat. Biotechnol. 24, 823-831 (2006).
- Pugsley, A. P. The complete general secretory pathway in gram-negative bacteria. Microbiol. Rev. 57 (1), 50-108 (1993).
- Rapoza, M. P., Webster, R. E. The filamentous bacteriophage assembly proteins require the bacterial SecA protein for correct localization to the membrane. J. Bacteriol. 175 (6), 1856-1859 (1993).
- Karlsson, A. J., et al. Engineering antibody fitness and function using membrane-anchored display of correctly folded proteins. J. Molec. Biol. 416 (1), 94-107 (2012).
- DeLisa, M. P., Tullman, D., Georgiou, G. Folding quality control in the export of proteins by the bacterial twin-arginine translocation pathway. Proc Natl Acad Sci U S A. 100 (10), 6115-6120 (2003).
- Fisher, A. C., Kim, W., DeLisa, M. P. Genetic selection for protein solubility enabled by the folding quality control feature of the twin-arginine translocation pathway. Protein Sci. 15 (3), 449-458 (2006).
- Maynard, J., Georgiou, G. Antibody engineering. Annu Rev Biomed Eng. 2, 339-376 (2000).
- Green, M. R., Sambrook, J. Molecular Cloning: A Laboratory Manual. 1, Fourth, Cold Spring Harbor Laboratory Press. (2012).
- Milo, R., Jorgensen, P., Moran, U., Weber, G., Springer, M. BioNumbers--the database of key numbers in molecular and cell biology. Nucleic Acids Res. 38, D750-D753 (2010).
- Hermanson, G. T. Bioconjugate Techniques. , Third, Elsevier/Academic Press. (2013).
- Tayapiwatana, C., Chotpadiwetkul, R., Kasinrerk, W. A novel approach using streptavidin magnetic bead-sorted in vivo biotinylated survivin for monoclonal antibody production. J Immunol Methods. 317 (1-2), 1-11 (2006).
- Zhu, G., Song, L., Lippard, S. J. Visualizing inhibition of nucleosome mobility and transcription by cisplatin-DNA interstrand crosslinks in live mammalian cells. Cancer Res. 73 (14), 4451-4460 (2013).
- Bogsch, E. G., et al. An essential component of a novel bacterial protein export system with homologues in plastids and mitochondria. J. Biol. Chem. 273, 18003-18006 (1998).
- Julian, M. C., et al. Co-evolution of affinity and stability of grafted amyloid-motif domain antibodies. Protein Eng. Des. Sel. 28 (10), 339-350 (2015).
- Garber, K. Bispecific antibodies rise again. Nat. Rev. Drug. Discov. 13 (11), 799-801 (2014).
- Marschall, A. L., Dübel, S., Böldicke, T. Specific in vivo knockdown of protein function by intrabodies. MAbs. 7 (6), 1010-1035 (2015).
