

Summary
Her presenterer vi en metode for å måle direkte overføring RNA lading nivåer fra renset Escherichia coli RNA samt en måte å sammenligne relative nivåer av overføring, eller noen andre kort RNA, på tvers av ulike eksempler basert på tillegg av spike celler uttrykke et gen som referanse.
Abstract
Overføre RNA (tRNA) er en viktig del av translasjonsforskning maskiner i en organisme. tRNAs binde og overføre aminosyrer å oversette ribosom. Den relative nivået av ulike tRNAs og forholdet mellom aminoacylated tRNA til totalt tRNA, kjent som lading, er viktige faktorer for å bestemme nøyaktighet og hastighet av oversettelse. Overflod og lading nivåer av tRNAs er derfor betydelig variabler å måle når studere proteinsyntese, for eksempel under ulike stress forhold. Her beskriver vi en metode for høsting tRNA og direkte måle både relative overflod og absolutt lading nivået av bestemte tRNA arter i Escherichia coli. TRNA er høstet slik at labil bånd mellom tRNA og dens aminosyre opprettholdes. RNA er så utsatt gel gelelektroforese og Nord blotting, som resulterer i separasjon av de ladet og uncharged tRNAs. Nivåene av bestemte tRNAs i ulike prøver kan sammenlignes på grunn av spike celler for normalisering. Før RNA rensing, legger vi til 5% av E. coli celler som overprodusere den sjeldne tRNAselC til hvert utvalg. Mengden av den tRNA arten av interesse i en prøve er deretter normalisert til mengden av tRNAselC i samme utvalget. Tillegg av spike celler før RNA rensing har en fordel tillegg av renset spike-i RNAs som det også står for noen forskjeller i cellen lysis effektivitet mellom eksempler.
Introduction
Nedenfor presenterer vi en metode for å kvantifisere bestemte tRNAs og måle deres lading nivåer av nordlige blotting. Metoden er basert på en teknikk utviklet av Varshney et al. 1. ved høste celler i Trekloredikksyre (TCA) og holde prøvene ved 0 ° C hele RNA rensing, ester bindingen mellom tRNA og aminosyren er bevart2,3,4. Aminoacylated tRNAs kan skilles fra sine nonacylated kolleger av gel gelelektroforese og nordlige blotting, grunn til en redusert mobilitet av aminoacylated tRNA i gel, forårsaket av covalently bundet aminosyre5. I tillegg presenterer vi en protokoll for å normalisere tRNA mengder av tillegg av spike celler overexpressing sjeldent brukte tRNAselC 4.
tRNAs er noen av de rikeste molekylene i bakteriell cellen og en helt avgjørende del av oversettelse maskiner. tRNAs binde aminosyrer og overføre dem til oversette ribosomene. Binding av aminosyrer til tRNAs (aminoacylation eller lading) er tilrettelagt av aminoacyl tRNA synthetases. Den relative overfloden av ulike ladet tRNAs er viktig for å sikre at gjengivelsen av proteinsyntese, fordi underrepresentation beslektet belastet tRNA for en gitt budbringer RNA (mRNA) codon øker sannsynligheten for at en nær beslektet tRNA vil feilaktig levere sin aminosyre voksende polypeptid kjeden på ribosom6. Betydningen av ladet tRNA gjenspeiles i omfattende svaret av E. coli cellen en alvorlig nedgang i lading nivåer av en tRNA; strenge responsen. Under strenge svaret syntese av tRNAs, ribosomal RNAs og de fleste mRNAs er senket for transkripsjon av bestemte mRNAs tilknyttet aminosyre biosyntesen og stress overlevelse, og veksten av cellene senkes dramatisk7 . Videre har siste arbeid og andre vist at E. coli aktivt forringer mesteparten av sin tRNA svar understreker at begrenser oversettelse4,8, tyder på at justering av tRNA kan være viktig for å takle slike påkjenninger. Dermed, pålitelige målinger av tRNA antall og lading nivåer vil være et viktig verktøy for å fullt ut forstå bakteriell stressresponser.
E.coli brukes vanligvis til å uttrykke rekombinante proteiner og på grunn av forskjeller i codon bruk mellom artene, suboptimal uttrykket er en problem ofte møter9. Dette kan circumvented uttrykk for flere tRNAs nødvendig å oversette de rekombinante mRNA10. Målinger av tRNA lading nivåer i slike stammer kan lede arbeidet med feilsøking og å optimalisere protein uttrykk.
Denne metoden kan også påvisning av "mischarging"; et tRNA molekyl aminoacylated med en ikke-beslektet aminosyre. Aminoacylation av ulike aminosyrer kan forårsake et tRNA overføre med litt annen fart gjennom polyakrylamid gels1,11. I noen tilfeller kan metoden også brukes til å skille annen endring mønstre på ellers identiske tRNAs3.
En annen etablert biokjemiske prosedyre for etterforskningen av tRNA lading nivåer er periodate oksidering. Metoden er avhengig av observasjon at periodate aminoacylated tRNA er beskyttet mot oksidering ikke og uncharged tRNA. Etter periodate oksidasjon behandling opplading av tRNA brukes til å anslå lading nivåene av de høstes. Imidlertid har opplading av flere tRNAs vist påvirkes av behandlingen gir noen unøyaktighet12. Metoden som presenteres her tiltak lading direkte fra renset RNA, dermed eksklusive eventuelle skjevheter fra kjemiske eller enzymatiske reaksjoner. En begrensning av denne metoden er den eneste tRNA arten er oppdaget på et tidspunkt, så selv om den samme nordlige blot kan være strippet og reprobed for flere tRNAs, det er tidkrevende og noe arbeidskrevende å samle inn data på mange tRNAs.
En pålitelig måte å normalisere prøver til hverandre er viktig i studier der målet er å sammenligne den relative nivået av et molekyl over forskjellige prøver. Her introduserer vi en normalisering prosedyre for RNA der en liten aliquot av E. coli celler overexpressing den sjeldne tRNAselC er lagt til som en spiker til alle eksperimentelle prøvene før RNA rensing. Denne metoden er nyttig ikke bare når undersøke tRNAs men for relativ kvantifisering av alle arter av når eksperimentelle oppsettet er slik at ingen endogene RNA kan være klarert å være tilstede på samme mobilnettet konsentrasjonen i alle prøvene. Nivået på en "rydde" RNA-lignende ribosomal RNA brukes for eksempel ofte som en endogen referanse sammenligne de relative mengdene av en annen mellom ulike eksempler13. Men dette er til liten nytte hvis mobilnettet konsentrasjonen av referanse RNA varierer mellom eksempler, som kan være tilfelle for ribosomal RNA hvis utvalg forekommer under en stress reaksjon15,16,17 eller under inn i stasjonære fase17. Tillegg av spike celler til de eksperimentelle prøvene før RNA rensing gir en solid og nøyaktig måte å normalisering uavhengig av prøvetaking. En annen måte for standardisering er én eller flere topp-i RNAs til de eksperimentelle prøvene etter RNA rensing. Men høyde denne metoden ikke for eventuelle forskjeller i RNA utvinning mellom eksempler.
Bruke E. coli celler som overexpress referansen RNA som spike celler (se figur 1) i et eksperiment på E. coli har ulempen at det resulterer i tillegg av en liten mengde eksogene E. coli totalt RNA til den prøver. Vi korrigere på denne ved å analysere et utvalg som inneholder bare spike celler parallelt med de eksperimentelle prøvene (se den nordlige blot i figur 2, lane kalt "selC"). Protokollen presenteres er utviklet for E. coli K-12 men sannsynligvis vil gjelde for de fleste bakterielle arter.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. forberedelse av bakteriekulturer.
- Vokse alle kulturer i Erlenmeyer flasker med en kultur/kolbe volum på minst 1/6. I en typisk eksperimentelle oppsett, vokse kulturer ved 37.0 ° C og rister på 160 rpm morpholinepropanesulfonic syre (MOPPER) minimal middels 18 med den foretrukne karbon kilden, for eksempel 0,2% glukose, i minst 10 etterfølgende generasjoner før RNA innhøsting.
- Dagen før RNA harvest, fortynne en Forvokst kultur for ønsket belastningen i MOPPER minimal medium slik at det vil komme den ønskede OD 436 på tiden dagen. Beregne volumet av Forvokst kultur bruke for vaksinering formelen:
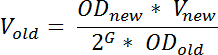
Merk: her OD nye angir den ønskede OD 436 etter inkubasjon V nye angir mengden utvannet kulturen, OD gamle betegner OD 436 Forvokst kultur utvannet fra, G angir antall generasjoner kulturen vil vokse, fra den klokkeslett fortynning (t 1) å det kreves dagen etter (t 2) og kan beregnes som: tid aldersforskjell (min) mellom t 1 og t 2 delt genereringen ta lang tid (i minutter). For å sikre stabil vekst ved prøvetaking, G bør være ≥ 10; V gamle angir mengden ut voksen kulturen som skal overføres til den friske mediet. Dette er en tilnærming som ikke står for alle lag fase; et fenomen ofte observert etter fortynning av en ut voksen kultur i frisk medier.- Vekst betingelsene varierer avhengig av formålet med eksperimentet, men hvis du vil begrense celle til celle variasjon i tRNA nivåer og øke reproduserbarhet av resultatene, (anbefales) vokse kulturer å stødig tilstand før prøvetaking 19.
2. Utarbeidelse av spike celler
Merk: E. coli belastningen MAS1074, som uttrykker selC genet koding tRNA selC under kontroll av en isopropyl β-D-1-thiogalactopyranoside (IPTG)- induserbart promoter brukes som topp-i belastningen.
- Dilute en Forvokst kulturen i MAS1074 til et OD 436 av 0,05 i MOPPER minimal medium med 100 µg/mL ampicillin og 0,2% glukose. Vokse kulturen på 37 ° C rister på 160 rpm.
Merk: Se diskusjon for bruk av en alternativ referanse belastning. - På OD 436 ~ 0.1, legge til IPTG en siste konsentrasjon av 1 mM til induserer uttrykk for tRNA selC. Vokse kulturen til OD 436 ~0.5 (~ 3-4 h vekst) og flytte kultur kolbe til en isbadet. Holde spiker i cellen kulturen på is inntil alle prøver for RNA rensing har vært høstet.
3. Høsting av eksperimentelle prøver.
- Pre varme (37 ° C) en avkortet 100 mL Erlenmeyer kolbe (eller lignende) som inneholder 4 mL 10% (w/v) Trekloredikksyre (TCA) for hver prøve ved å plassere den i en oppvarmet vannbad.
Merk: forsiktig, TCA dekomponerer ved oppvarming, produsere giftig og etsende gasser inkludert Hydrogenklorid og kloroform. Løsningen i vann er en sterk syre; den reagerer voldsomt med baser er skadelig for mange metaller. Riktige forholdsregler bør tas når du bruker denne væsken. - Umiddelbart før cellen harvest, måle og registrere OD 436 kultur.
- For å høste celler, overføre 4 mL kultur til en forvarmes kolbe med TCA.
- Blande utvalget av virvlende kolbe hånd for 5 s. blanding med TCA umiddelbart deaktiverer alle enzymatisk aktiviteter og dermed beholder lading nivåer av tRNAs.
Merk: Hvis flere eksempler samles, holde høstet prøver i TCA på is inntil alle prøver er samlet.
4. Tillegg av spike celler
Merk: utarbeidelse av spike celler er beskrevet i trinn 2.
- Når alle prøver for RNA forberedelse har blitt samlet, legge til 5% spike celler (basert på OD) til hvert utvalg.
Merk: Mengden spike i cellekultur som trengs er beregnet av formelen:
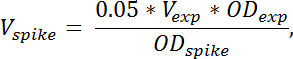
der V spike angir mengden spike i cellekultur som trengs, V exp Angir volumet av høstet eksperimentelle cellekultur (uten TCA), OD exp angir OD 436 eksperimentelle celle kultur ved harvest i TCA. OD spike angir OD 436 med spiker i cellekulturer. Hvis flere eksempler høstes ved ulike ODs, volumet av spike celler lagt bør justeres i henhold til formelen ovenfor for hvert utvalg. - å kvantifisere bidraget fra de spike cellene til RNA arter av interesse, forberede et utvalg som bare inneholder spike celler ved å blande 4 mL spike celler med 4 mL TCA. Forberede RNA fra dette eksemplet på samme måte som for andre prøvene.
- Eventuelt ta en annen kolbe som inneholder bare eksperimentelle prøven, uten spike celler, og inkluderer det å kvantifisere bidrag av eksperimentelle prøven til tRNA selC nivåer; endogene av tRNA selC i E. coli K-12 er negligibly lav.
5. Utarbeidelse av RNA
Merk: The RNA forberedelse protokoll beskrevet her er i hovedsak som beskrevet av Varshney et al. 1. For å unngå deacylation av aminoacylated tRNAs under rensingen er det viktig å holde prøvene surt pH og 0 ° C i løpet av rensing. Bruk ren sterilt rør/flasker til eksempler og løsninger, og forberede alle løsninger med ultrapure (18.2 MΩ·cm resistivitet) vann.
- Hell 8 mL hvert utvalg i en is-kjølt sentrifuge rør. Sentrifuge prøver i 10 min 9500 x g på 4 ° C. helt Fjern nedbryting og resuspend i 0,3 mL kaldt 0,3 M natrium acetate, pH 4.5 og 10 mM EDTA.
- Overføre hver prøve slik kalde 1,5 mL microcentrifuge og legge 0,3 mL fenol equilibrated med samme bufferen.
Merk: forsiktig fenol produserer giftige gasser og er svært skadelig for huden. - Vortex hver prøve 10 x 15 s med minst 1 minutt mellom hvert vortex. Mellom vortexing holde prøvene ved 0 ° C. Bruk hyppige pauser for å sikre at prøvene forblir kald.
- Sentrifuge prøver i 15 min 20.000 x g på 4 ° C. overføring vann fase (øvre fase) til nye 1,5 mL microcentrifuge rør. Legge til 0,3 mL kaldt fenol og vortex 4 x 15 s som før.
- Sentrifuge for 10 min 20 000 x g på 4 ° C. overføring vann fasen til ny, kald 1,5 mL microcentrifuge rør som inneholder 2,5 ganger volumet av 96% etanol (~ 750 µL). Utløse RNA av rugende rør 1t 20 ° C eller over natten på-80 ° C.
- Sentrifuge prøver for 30 min 20.000 x g på 4 ° C. forkaste nedbryting og legge nøye 1 mL kaldt 70% etanol uten å forstyrre RNA-pellets. Forsiktig inverteres gang for å vaske innsiden av rør med etanol.
Merk: Pellet kan være vanskelig å se på dette trinnet. - Sentrifuge for 10 min 20 000 x g på 4 ° C. helt fjerne nedbryting og air-dry pellet til ingen etanol er igjen. Resuspend pellet av kraftig vortexing i 20 µL kaldt 10 mM natrium acetate pH 4.5 og 1 mM EDTA.
Merk: Ikke over tørr pellet som det vil bli vanskelig å resuspend. - Eventuelt vurdere konsentrasjonen av RNA ved å måle absorbans ved 260 nm.
- Eventuelt vurdere kvaliteten av det renset av agarose gel geleelektroforese 20.
- Store samples ved-80 ° C.
Merk: Protokollen kan pauses her.
Merk: for å skille av aminoacylated tRNA fra motparten deacylated på nordlige blot, en kjemisk deacylated aliquot er utarbeidet av alkaliske behandling. For de fleste formål, er det tilstrekkelig å deacylate et enkelt eksempel for hver Nord blot.
- Tøvær RNA eksempel på is. Overføre en 4 µL aliquot av RNA utvalget til en ny tube. Legge til 46 µL Tris-HCl pH 9.0. Inkuber 2 h på 37 ° C.
- Legger til 15 µL av 0,3 M natrium acetate pH 4.5 og senere legger til 125 µL av 96% etanol. Utløse RNA på 20 ° C for 1 h.
- Sentrifuge for 30 min 20.000 x g på 4 ° C. helt fjerne nedbryting og resuspend i 4 µL kaldt 10 mM natrium acetate pH 4,5 og 1 mM EDTA.
Merk: Protokollen kan pauses her hvis prøven er lagret på-80 ° C.
7. Gel gelelektroforese og nordlige blotting
- blanding 4 µL utvalg (ubehandlet eller kjemisk deacylated) med 6 µL lasting buffer (0.1 M natrium acetate (pH 5.0), 8 M urea, 0,05% bromophenol blå og 0,05% xylen cyanol).
Merk: Husk å ta prøven som inneholder bare spike celler, de kjemisk deacylated eksempel (og prøven uten spike celler, hvis en var forberedt). - Laste eksemplene på en 0.4 mm tykk 6,5% polyakrylamid gel (19:1 acrylam - ide:bisacrylamide) som inneholder 8 M urea inne 0.1 M natrium acetate buffer (pH 5.0). Bruke 60 cm lang gels for riktig separasjon av tRNA arter.
- Separat RNA av geleelektroforese på 10 V/cm gel 4 ° C, inntil bromophenol blå når bunnen av gel (~ 20 h).
- Området fra xylen cyanol fargestoff og 20 cm ned bromophenol blått fargestoff brukes til blotting (avstanden mellom de to fargestoffer bør være 25-30 cm). Forsiktig skille de to glassplater, slik at gel på en av dem. Litt tynn filter papir er kuttet til størrelsen på området blotting og plasseres over delen av gel som skal brukes for blotting. Deler av gel som ikke dekkes av filter papir er kuttet og forkastet. bruke filter papir å nøye løfte gel stykket fra glassplaten.
Merk: Store gel er veldig skjør så håndteres med forsiktighet. - En positivt ladet nylon membran (f.eks Hybond N + membran) plasseres oppå gel og det er electroblotted på 20 V for 90 min bruker 40 mM Tris-acetate (pH 8.1), 2 mM EDTA som overføring buffer.
- Krysskobling RNA til membranen bruker UV-lys (0,12 J/cm 2).
Merk: Membranen kan nå oppbevares i romtemperatur og protokollen kan pauses her.
8. Design og verifisering av sonder
Merk: forsiktig design og verifisering av nordlige blot sonder er avgjørende for å få pålitelige resultater og unngå uønskede kryss-hybridisering med torvtak RNA.
- Bruk kort oligonucleotides syntetiske som sonder. Utforme sonde rekkefølgen på en måte at det er komplementære til den mest unike delen av en rekke tRNA rundt - dette er ofte anticodon loopen.
Merk: En liste over forventet tRNA sekvenser fra ulike organismer finnes i Genomic tRNA-databasen (GtRNAdb) 21. - Juster lengden på sekvensen hente en antatt smelter temperaturen på 55-65 ° C.
- Eksplosjon (grunnleggende lokale justering søkeverktøyet) valgt sekvens mot det Genova orden av bakterielle belastningen brukt i forsøket, kontrollere at sekvensen er unik for den valgte tRNA, som beskrevet i 22.
Merk: Tabell 1 viser foreslåtte sonde sekvenser for flere ulike tRNAs av E. coli K12. Dersom sonden er spesifikke for tRNA rundt, bare to band er synlig på nordlige blot (ladet og uncharged tRNA) og bare ett band er synlig i lane med deacylated prøven (uncharged tRNA). Hvis mer enn to band eller ytterligere validering av sonden spesifisitet er ønsket, se diskusjon.
9. Blanding av nordlige blot
Merk: I trinnene nedenfor membranen er sekvensielt hybridiserte med sonder utfyllende til de ønskede bestemte tRNAs, inkludert en sonde bestemt referansen tRNA selC.
- Sett membranen i en hybridisering rør og legge 6 mL hybridisering løsning (0.9 M NaCl, 0,05 M NaH 2 PO 4 (pH 7.7), 5 mM EDTA, 0,5% (w/v) SDS, 100 mg/mL av skåret og denaturert sild sperm DNA og 5 x Denhardt ' s løsning (100 x Denhardt ' s løsning = 2% bovin serum albumin, 2% polyvinylpyrrolidone 40 og 2% Ficoll)).
- Pre hybridize membranen i bufferen for 1t roterende på 42 ° C. Legg 30 pmol radioaktive oligo-DNA sonde 5 ' - slutt - merket med 32 P (forberedt som beskrevet av Sambrook et al. 23)
Merk: forsiktig, høy energi beta utslippene fra 32 P kan presentere en betydelig hud og øyne dose fare. Riktige forholdsregler bør tas når bruker 32 P. radioaktiv avfall bør avhendes i henhold til de aktuelle institusjonelle sikkerhet protokollene. - Ruge sonde med membran overnatting roterende på 42 ° C. Fjern sonden bruker en engangs 10 mL pipette.
Merk: Hvis lagret i en fryser proben kan gjenbrukes. - i hybridisering røret, vaske membranen i 50 mL av 0,3 M NaCl, 30 mM natriumsitrat, 0,1% (w/v) SDS ved romtemperatur.
Merk: Denne første vask inneholder mye ubundet sonde og må behandles som svært radioaktivt. - Gjenta trinn 9.4 en gang, så flytte membranen til en flat plast container eller brett inkludert lokk. Dekke membranen med vask løsning (0,3 M NaCl, 30 mM natriumsitrat, 0,1% (w/v) SDS) og Inkuber det i 30 min ved romtemperatur.
- Vask løsning og Fortsett vask til et tilfredsstillende signal til støyforhold er innhentet (ofte 3-4 endringer vaske løsning). Bruke en Geiger-Müller tube å oppdage hvordan støy til signal ratio endres ved å måle stråling fra et tomt område av membranen og sammenlign det med området der sonden har hybridiserte å tRNA.
- For å kunne fjerne sonden fra membranen etter eksponering, sikre at (viktig) membranen ikke tørke før stripping prosedyren. Plass membranen i tynne plastpose eller vikle det tett med plast brytes og forsegle den skal lufttett av bånd eller sveising. Plasser forseglet membranen på phosphorimaging skjerm.
Merk: Nødvendig eksponeringstid på skjermbildet phosphorimaging bestemmes til aktiviteten av sonden, mengden av RNA analysert forr og hybridisering effektiviteten av sonden, og kan variere. fra noen få minutter til flere dager. Med erfaring gir intensiteten av stråling på membranen med Geiger-Müller røret en god indikasjon på nødvendige eksponeringstid. For pålitelig kvantifisering, er det viktig at skjermen ikke er overeksponert. - Skanne phosphorimaging skjermen ved hjelp av en laser skanneren og lagre filen i " .gel " format.
Merk: En høy signal til støyforhold er nødvendig for pålitelig kvantifisering. Leter etter en tRNA skal resultere i to band; én inneholder aminoacylated tRNA (tregere migrasjon) og én inneholder deacylated tRNA (raskere overføring). Se figur 2.
10. Stripping membranen
Merk: før membranen kan bli analysert for en annen tRNA, tidligere sonden først fjernes ved stripping membranen.
- Varme nok stripping buffer (15 mM NaCl, 1,5 mM natriumsitrat, 0,1% (w/v) SDS) å dekke membranen og hell den over membranen. Inkuber det i 15 min ved romtemperatur.
- Gjenta trinn 10.1 til ingen stråling kan oppdages med en Geiger-Müller rør; membranen kan nå re hybridisert følge fremgangsmåten beskrevet i avsnitt 9.
11. Kvantifisere tRNA og lade nivåer
- laste filen .gel til avbilding programvare (se Tabell for materiale). Tegne en loddrett linje langs hver av prøven banene slik at det mesteparten av bredden på kjørefelt, men utelater kantene i band der signalet kan være ujevn, som vist i figur 3A.
- Effekter teller fra hver bane ved å klikke " analyse "-> " opprette grafen ". Definere manuelt de to toppene representerer de ladet og uncharged tRNA fra hver bane, som vist i figur 3B.
- Hente teller hver topp ved å klikke " analyse "-> " området rapporten ", og registrere området under hver av de to toppene.
- Da blot har blitt analysert for tRNA selC og minst ett tRNA av interesse, normalisere nivåene av tRNA av interesse for tRNA selC.
- Beregnes andelen av tRNA fra de spike cellene i hver kjørefelt og trekke fra det totale signalet i denne kjørefelt ved hjelp av formelen:
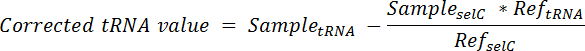
Merk: her, Prøve tRNA = signalet fra samme kjørefelt av en membran analysert for den ønskede tRNA; Eksempel selC = signalet fra samme kjørefelt av samme membranen analysert for tRNA selC; REF tRNA = signalet fra felt som inneholder bare topp-i cellen RNA av samme membranen analysert for den ønskede tRNA; REF selC = signalet fra felt som inneholder bare topp-i cellen RNA av samme membranen analysert for tRNA selC. - å normalisere, dele korrigert tRNA verdi fra hver lane tRNA selC signalet fra samme kjørefelt.
Merk: Bidrag på tRNA selC signalet fra samplet cellene er ubetydelig (se figur 2, lane merket "-selC ").
- Beregnes andelen av tRNA fra de spike cellene i hver kjørefelt og trekke fra det totale signalet i denne kjørefelt ved hjelp av formelen:
- Beregner tRNA lading nivå for hver bane ved å dele signalet fra toppen representerer ladet tRNA med summen av signalet fra begge topper (ladet + uncharged).
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Bruke fremgangsmåten beskrevet her, overflod og lading nivåer av tre tRNAs ble målt i E. coli K-12 før og under aminosyre sult.
Arginin auxotroph belastning NF9154 (thr leu hans argH thi mtl supE44) ble dyrket i minst ti generasjoner i MOPPER minimal medium med 0,4% glyserol, 50 µg/mL threonin, leucine, arginine og 5 µg/mL histidin på 37 ° C rister på 160 rpm. Arginin sult ble introdusert av filtrering av kultur og rørets i frisk MOPPER medium som ovenfor, men uten arginin. Arginin sult, proteinsyntese er sterkt redusert, men fortsetter på et lavt nivå bruker arginine utgitt av omsetningen av eksisterende proteiner. Prøvene ble høstet samtidig poeng angitt i Figur 4. Når alle prøver ble høstet i TCA, 5% av de MAS1074 pigg cellene overexpressing tRNAselC, ble lagt til hvert utvalg som vist i figur 1. RNA var renset fra prøvene, atskilt med geleelektroforese og strøket på en membran som beskrevet i protokollen. Membranen undersøkt for tRNAargVYZQ, tRNAgltTUVW, tRNAhisRog tRNAselC, som vist i figur 2. Stråling signalet fra hver bane ble kvantifisert for hver av de fire sonder som illustrert i Figur 3og korrigert og normalisert som beskrevet i Seksjon 11. Resultatene presenteres i Figur 4. Figur 4A viser at nivåene av alle testet tRNA arter raskt redusere under aminosyre sult, som nylig rapportert4. Figur 4B viser at tRNA lading nivåer oppfører seg som forventet: ladet brøkdel av arginin godta tRNA faller raskt på fjerning av arginin fra vekstmediet, mens lading nivåer av de andre tRNAs øke litt på sult fordi belastet tRNAs samle når timepris decharging på ribosomene reduseres på grunn av manglende substrat for protein syntese4. Videre inn i sult-perioden reduseres ladet tRNA brøken til ca før sult nivåer på grunn av nedbrytning av flertallet av tRNA bassenget (figur 4A), som resulterer i en større grad av decharging av de gjenværende tRNAs på ribosomene. Derimot øker ladet brøkdel av tRNAargVYZQ for minst to grunner. 1) aktivering av strenge svaret betyr at mRNA, ikke belastet tRNAarg, blir den begrensende faktoren for oversettelse og 2) den totale pool av tRNAarg er redusert (figur 4A) så en større del av den totale tRNAarg kan lades i samme lite arginin. Dette eksperimentet demonstrerer hvordan samtidige målinger av tRNA overflod og tRNA lading nivåer gir høy kvalitetsinformasjon om betingelsene for proteinsyntese i cellen.

Figur 1. Tillegg av spike celler smakebiter. I dette eksemplet høstes prøver fra en enkelt kultur av E. coli NF915 i en tid-serien før og under aminosyre sult. En skjematisk av vekstkurve av NF915 vises, der de blå prikkene indikerer poeng for eksempel. De y-akser er logaritmisk skala. Prøver av den eksperimentelle culture(s) høstes som beskrevet i kapittel 3. En skjematisk av vekstkurve av spike i belastningen MAS1074 vises også, med punktet for eksempel vises som en blå prikk. Poenget med figuren er å illustrere at alle eksperimentelle prøvene (av E. coli NF915 i dette eksemplet) mottar en aliquot med spike celler fra samme referanse kulturen. Til hver prøve, 5% av spike celler er lagt til eksperimentelle cellene, beregnes basert på OD i cellekulturer (se Seksjon 4). Deretter er RNA renset fra alle prøver og brukes til nordlige blots. Klikk her for å se en større versjon av dette tallet.

Figur 2. Fosfor imager scan av en nord klatt analysert for de angitte tRNAs. Membranen inneholder RNA fra E. coli belastningen NF915 høstes ved de angitte tidspunkt før og etter induksjon av arginin sult (minutter). I tillegg membranen inneholder to baner av prøvene der spike celler utelatt (-selC DA og -selC), og en fil som inneholder RNA fra topp-modulen cellene bare (selC). -SelC DA prøven behersker kjemisk deacylated tRNA. De to bandene som representerer ladet og uncharged tRNA arter skilles klart. Merk ubetydelig signalet fra tRNAselC i banene lagt uten spike celler. Samme membranen var suksessivt strippet og reprobed for tRNAs vises til venstre. Eske området i øvre blot viser delen av blot, som vises for den påfølgende tRNA probings. Klikk her for å se en større versjon av dette tallet.

Figur 3. Kvantifisering av nordlige blot. tRNA overflod måles ved hjelp av programvare. (A) A enkelt kjørefelt fra nordlige blot ses i figur 2 analysert for tRNAargVYZQ. Øvre og nedre pilen indikerer aminoacylated tRNA og deacylated tRNA, henholdsvis. De røde linjene kjører parallelt med lane er lagt og brukes til å definere området for kvantifisering. (B) kvantifisering av signal innenfor de to ødelagte røde linjene i (A) avbildet som en graf, hvor den vannrette aksen viser avstanden fra toppen av linjen i (A) (i piksler) og den loddrette aksen viser intensiteten av signalet (i teller). To topper som inneholder signal som stammer fra aminoacylated tRNA og deacylated tRNA, henholdsvis er definert manuelt. Dataene eksporteres for videre analyse er verdien som tilsvarer området under hver av de to toppene. Klikk her for å se en større versjon av dette tallet.

Figur 4. Representant resultat: kvantifisering av tRNA dyp end lading nivåer under arginine sult. Kvantifisering band på Nord blot vist i figur 2. (A) relative overflod av tRNAgltTUVW (rød), tRNAargVYZQ (blå) og tRNAhisR (grønn). Fant null minutter (eksempler høstet like før utbruddet av sult) er satt til 1. (B) lading nivået av de samme tre tRNAs som (A). Klikk her for å se en større versjon av dette tallet.
| Sonden spesifisitet | Sonden sekvens |
| tRNAargVYZQ | 5-TCCGACCGCTCGGTTCGTAGC |
| tRNAgluTUVW | 5-CCTGTTACCGCCGTGAAAGGG |
| tRNAhisR | 5-CACGACAACTGGAATCACAATCC |
| tRNAleuPQVT | 5-GTAAGGACACTAACACCTGAAGC |
| tRNAleuU | 5-TATTGGGCACTACCACCTCAAGG |
| tRNAleuW | 5-CTTGCGGCGCCAGAACCTAAATC |
| tRNAleuX | 5-TATTTCTACGGTTGATTTTGAA |
| tRNAleuZ | 5-AAAATCCCTCGGCGTTCGCGCT |
| tRNAlysQTVWYZ | 5-TGCGACCAATTGATTAAAAGTCAAC |
| tRNAthrV | 5-TGGGGACCTCACCCTTACCAA |
| tRNAtyrTV | 5-TCGAACCTTCGAAGTCGATGA |
| tRNAselC | 5-ATTTGAAGTCCAGCCGCC |
Tabell 1. Tabell over foreslåtte sonde sekvenser for valgte tRNAs i E. coli K-12.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Denne protokollen beskriver hvordan måle samtidig lading nivået av bestemte E. coli tRNAs og sammenligne den relative nivået av tRNAs i forskjellige prøver. Kritiske punkter i protokollen er 1) å håndtere prøvene slik at beholdes mobilnettet tRNA lading nivåene, 2) til å normalisere tRNA mengder slik at relativ tRNA nivåer i ulike prøver kan sammenlignes pålitelig, og 3) for å sikre sp ecificity av de valgte sonder for tRNAs av interesse. Disse punktene er adressert separat nedenfor.
tRNA rensing
TRNAs er renset fra E. coli på en skånsom måte som er optimalisert for å beholde sin mobil lading nivåer. Det er vår erfaring at denne protokollen primært ekstrakter RNAs kortere enn 150 nt, dermed tRNA utgjør en stor andel av det renset. Derfor anbefales det ikke å bruke denne protokollen til å rense totale RNA fra E. coli. Protokollen presenteres her bør også være egnet for deteksjon og kvantifisering av andre RNAs som liten RNA (sRNA) uten noen ytterligere tilpasninger. Fordi RNA utvinning protokollen beriker for kort RNA sekvenser bør den også finne bruk selv om sRNA under etterforskning uttrykkes ydmyk.
Normalisering av spike celler
Hvis du vil sammenligne nivåene av tRNA over forskjellige prøver, er det nødvendig å normalisere tRNA nivåene til nivået av en utskrift som finnes i den samme gjennomsnitt per celle i alle prøvene, for å korrigere for prøve å sample variasjoner i RNA utvinning og gel lasting. For dette formålet spike vi prøvene med 5% Referanse celler som overexpress den sjeldne tRNAselC før RNA rensing. Spike i metoden er å foretrekke over bruker en endogen RNA som referanse, fordi det tillater sammenligning av tRNA nivåer over betingelser der ingen endogene RNAs er kjent med sikkerhet på samme mobilnettet konsentrasjonen. Bruke E. coli celler som topp-in har ulempen at 5% ekstra E. coli RNA legges til alle eksperimentelle prøvene, der den bidrar til signalet av den rundt. Denne skjevhet er regnskapsføres etter inkludert et utvalg som inneholder bare de spike cellene på hver Nord blot, og korrigere RNA verdiene som beskrevet i trinn 11.4.1. Uttrykket av tRNAselC i vill type celler er så lavt sammenlignet med andre tRNAs (se figur 2) at det gjør ikke en betydelig forskjell i kvantifisering og dermed kan bli neglisjert. Men i referanse belastningen MAS1074, der selC er uttrykt ved en T7 promoter på en høy kopi plasmider, vi anslår at minst 1000 ganger mer tRNAselC er produsert i nærvær av 1 mM IPTG enn produsert av wildtype E. coli K-12 under noen omstendigheter. MAS1074 opphører etter to generasjoner av full IPTG induksjon. På dette punktet, kultur-til-kultur forskjeller i induksjon av tRNAselC er små, og ikke en bekymring for denne protokollen som alle dele spike celler for ett enkelt eksperiment er tatt fra samme indusert kultur MAS1074. Som et alternativ til MAS1074, kan celler fra en organisme som har RNA ikke vil cross-react med hybridisering proben brukes som topp celler. Sulfolobus solfataricus celler har med hell brukt som spiker for eksperimenter med E. coli tRNA4. Men E. coli spike celler er sannsynlig å mer nøyaktig gjenspeile graden av cellen lysis av eksperimentelle celler, som det er usikkert om S. solfataricus lyses med samme effektivitet som E. coli. Det er også mer kjedelig som de vokser på 85 ° C med en generasjon av ~ 16 h å voksende S. solfataricus .
Sikre spesifisitet av valgte tRNAs
Hvis både aminoacylated og deacylated tRNA har blitt skal renset to band registreres på den nordlige membranen. Avstanden mellom de to bandene vil variere avhengig av den bestemte tRNA under etterforskning, og er en kombinert konsekvens av forskjellen i størrelse, polar egenskaper og conformation som en bestemt aminosyre forårsaker når det binder en bestemt tRNA5 . For eksempel avstanden mellom aminoacylated og deacylated tRNA er større for tRNAargVYZQ enn det er for tRNAgltTUVW, som vist i figur 2. Av den store størrelsen på gel brukes her til å skille RNA, er oppløsningen høy nok til å skille ladet tRNA fra uncharged, men også å skille de fleste av de forskjellige tRNAs fra hverandre på grunn av deres forskjeller i lengde, sekvens komposisjon og modifikasjon mønstre. Selv isoaccepting tRNAs kan skilles som vist for leucine akseptere tRNAs2. Merk at i denne studien ikke var det mulig å skille tRNAleuPQV fra tRNAleuT, som forskjellen i sekvensen er imidlertid bare en G å T substitusjon.
Hvis mer enn to band vises på den nordlige membranen kan det være en konsekvens av kryssreaksjon av sonde med andre RNAs. Økende stringens av vask step (trinn 9.5) ved å øke temperaturen kan vanligvis løse dette. Vask 30 min ved 55 ° C er vanligvis tilstrekkelig. Økende vask stringens ikke fjerner kors-hybridisering, kan løsningen være å designe en ny sonde som er komplementære til en annen (ofte delvis overlappende) del av den tRNA sekvensen. Flere band kan også være carry-over fra en tidligere sonde, som følge av utilstrekkelig stripping av membranen. For å forhindre dette, gjør en stripe kontroll ved å utsette membranen før re undersøkelser. Hvis band vises på kontroll avsøke membranen må ekstra stripping. Er det vanligvis mulig å nytt undersøke en membran minst 8 - 10 ganger, men etter flere sonder kan det være vanskelig å kle helt. Hvis dette er tilfelle, kan membranen lagres for et par måneder før re undersøkelser som 32P henfall rask.
Ytterligere bekreftelse av sonden spesifisitet kan fås ved å utføre en nord blot med celler høstet under et sett med betingelser der transkripsjon av interesse er forventet å bli berørt på en forutsigbar måte. For eksempel bør induserbart uttrykk for tRNA fra plasmider eller aminosyre sult for beslektet aminosyre, resultere i et forbedret relative uttrykk nivå eller et lavere lading nivå, henholdsvis av tRNA rundt.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Forfatterne ikke avsløre.
Acknowledgments
Forfatterne takker Marit Warrer for utmerket kundestøtte. Dette arbeidet ble støttet av danske Rådet for uavhengig forskning | Naturvitenskap [1323-00343B] og danske National Research Foundation [DNRF120].
Materials
| Name | Company | Catalog Number | Comments |
| Urea | Merck | 57-13-6 | Purity >99% |
| Polyacrylamide | Serva | 79-06-7 | |
| Bis (N,N'-Methylene-bis-acrylamide) | BIO-RAD | 161-0201 | |
| Trichloroacetic acid (TCA) | Sigma | 76-03-9 | |
| Phenol | Merck | 108-95-2 | |
| IPTG | |||
| Glucose | |||
| Sodium Acetate | |||
| EDTA | |||
| Ethanol | |||
| Tris | |||
| Hydrochloric acid | |||
| Sodium Chloride | |||
| NaH2PO4 | |||
| Sodium Citrate | |||
| SDS | |||
| Herring Sperm DNA | Sigma | 100403-24-5 | |
| BSA | |||
| Polivinylpyrrolidone | |||
| Ficoll | |||
| P32 γATP | Perkin Elmer | ||
| DNA oligos (probes) | TAG Copenhagen | ||
| Hybond N+ membrane | GE Healthcare | RPN203B | |
| Crosslinker | |||
| Electroblotter | BIO-RAD | ||
| Typhoon FLA 7000 Scanner | GE Healthcare | 28955809 | |
| Spectrophotometer | |||
| Hydridization oven | |||
| Geiger-Müller tube | |||
| Phosphor imager screen | GE Healthcare | ||
| Hybridization tube | |||
| Culture Flasks | |||
| 1.5 ml microcentrifuge tubes | |||
| 20 ml centrifuge tubes | |||
| Name | Company | Catalog Number | Comments |
| Software | |||
| ImageQuant | GE Healthcare | ||
| Excel | Microsoft |
References
- Varshney, U., Lee, C. -P., RajBhandary, U. L. Direct analysis of aminoacylation levels of tRNAs in vivo. Application to studying recognition of Escherichia coli initiator tRNA mutants by glutaminyl-tRNA synthetase. J Biol Chem. 266 (36), 24712-24718 (1991).
- Sorensen, M. A. Charging levels of four tRNA species in Escherichia coli Rel(+) and Rel(-) strains during amino acid starvation: a simple model for the effect of ppGpp on translational accuracy. J Mol Biol. 307 (3), 785-798 (2001).
- Krüger, M. K., Sørensen, M. A.
- Svenningsen, S. L., Kongstad, M., Stenum, T. S., Muñoz-Gómez, A. J., Sørensen, M. A. Transfer RNA is highly unstable during early amino acid starvation in Escherichia coli. Nucleic Acids Res. 45 (2), 793-804 (2017).
- Enriquez, J. A., Attardi, G. Evidence for aminoacylation-induced conformational changes in human mitochondrial tRNAs. Proc Natl Acad Sci U S A. 93 (16), 8300-8305 (1996).
- Parker, J. Errors and alternatives in reading the universal genetic code. Microbiol Rev. 53 (3), 273-298 (1989).
- Traxler, M. F., et al. The global, ppGpp-mediated stringent response to amino acid starvation in Escherichia coli. Mol Microbiol. 68 (5), 1128-1148 (2008).
- Zhong, J., et al. Transfer RNAs Mediate the Rapid Adaptation of Escherichia coli to Oxidative Stress. PLoS Genet. 11 (6), e1005302 (2015).
- Kane, J. F. Effects of rare codon clusters on high-level expression of heterologous proteins in Escherichia coli. Curr Opin Biotechnol. 6 (5), 494-500 (1995).
- Baca, A. M., Hol, W. G. Overcoming codon bias: a method for high-level overexpression of Plasmodium and other AT-rich parasite genes in Escherichia coli. Int J Parasitol. 30 (2), 113-118 (2000).
- Li, S., Pelka, H., Schulman, L. The anticodon and discriminator base are important for aminoacylation of Escherichia coli tRNA (Asn). J Biol Chem. 268 (24), 18335-18339 (1993).
- Rizzino, A. A., Freundlich, M. Estimation of in vivo aminoacylation by periodate oxidation: tRNA alterations and iodate inhibition. Anal Biochem. 66 (2), 446-449 (1975).
- Dittmar, K. A., Sorensen, M. A., Elf, J., Ehrenberg, M., Pan, T. Selective charging of tRNA isoacceptors induced by amino-acid starvation. EMBO Rep. 6 (2), 151-157 (2005).
- Zhou, K., et al. Novel reference genes for quantifying transcriptional responses of Escherichia coli to protein overexpression by quantitative PCR. BMC Mol Biol. 12 (1), 18 (2011).
- Jacobson, A., Gillespie, D. Metabolic events occurring during recovery from prolonged glucose starvation in Escherichia coli. J Bacteriol. 95 (3), 1030-1039 (1968).
- McCarthy, B. The effects of magnesium starvation on the ribosome content of Escherichia coli. Biochim Biophys Acta (BBA)-Specialized Section on Nucleic Acids and Related Subjects. 55 (6), 880-889 (1962).
- Zundel, M. A., Basturea, G. N., Deutscher, M. P. Initiation of ribosome degradation during starvation in Escherichia coli. Rna. 15 (5), 977-983 (2009).
- Piir, K., Paier, A., Liiv, A., Tenson, T., Maivali, U. Ribosome degradation in growing bacteria. EMBO Rep. 12 (5), 458-462 (2011).
- Schaechter, M., Maaloe, O., Kjeldgaard, N. O. Dependency on medium and temperature of cell size and chemical composition during balanced grown of Salmonella typhimurium. J Gen Microbiol. 19 (3), 592-606 (1958).
- Neidhardt, F. C., Bloch, P. L., Smith, D. F.
- Sambrook, J., Fritsch, E. F., Maniatis, T. Molecular cloning: a laboratory manual. , Cold spring harbor laboratory press. (1989).
- Sorensen, M. A., Jensen, K. F., Pedersen, S. High concentrations of ppGpp decrease the RNA chain growth rate. Implications for protein synthesis and translational fidelity during amino acid starvation in Escherichia coli. J Mol Biol. 236 (2), 441-454 (1994).
- Tian, C., et al. Rapid Curtailing of the Stringent Response by Toxin-Antitoxin Module-Encoded mRNases. J Bacteriol. 198 (14), 1918-1926 (2016).
