

Summary
Aquí presentamos un método para medir directamente el ARN de transferencia carga niveles de purificada de Escherichia coli RNA como una forma de comparar niveles relativos de ARN de transferencia, o cualquier otro RNA corto, a través de diferentes muestras basadas en la adición de espiga en células expresan un gen de referencia.
Abstract
ARN de transferencia (tRNA) es una parte esencial de la maquinaria traduccional en cualquier organismo. ARNt se unen y transferir los aminoácidos al ribosoma traduciendo. Los niveles relativos de tRNAs distintos y la proporción de aminoacylated tRNA al tRNA total, conocido como el nivel de carga, son factores importantes en la determinación de la exactitud y velocidad de traducción. Por lo tanto, la abundancia y niveles de carga de tRNAs son variables importantes a medir en el estudio de la síntesis de proteínas, por ejemplo en diversas condiciones de estrés. Aquí, describimos un método para cosechar tRNA y medición directa de la abundancia relativa y el nivel absoluto de la carga de la especie específica del ARNt en Escherichia coli. El tRNA se recolecta de tal manera que se conserve el vínculo inestable entre el ARNt y su aminoácido. El RNA entonces se somete a electroforesis en gel y Northern Blot, que resulta en la separación de los tRNAs cargados y descargados. Los niveles de tRNAs específicos en diferentes muestras se pueden comparar debido a la adición de espiga en las células para la normalización. Antes de la purificación del ARN, agregamos 5% de células de e. coli que sobreproducción la tRNA raraselC a cada muestra. La cantidad de las especies de tRNA de interés en una muestra luego se normaliza la cantidad de tRNAselC en la misma muestra. Además de la espiga en las células antes de la purificación del ARN tiene la ventaja sobre adición de purificada spike en RNAs que también explica las diferencias en la eficiencia de la lisis de células entre las muestras.
Introduction
A continuación, presentamos un método para cuantificar el ARNt específico y medir su carga niveles por borrar norteño. El método se basa en una técnica desarrollada por Varshney et al. 1. recolección de las células en ácido tricloroacético (TCA) y manteniendo las muestras a 0 ° C a lo largo de la purificación de RNA, el enlace éster entre el tRNA y el aminoácido es conservado2,3,4. TRNAs Aminoacylated pueden distinguirse de sus contrapartes nonacylated por electroforesis y blotting norte, debido a una movilidad disminuida de la aminoacylated tRNA en el gel, causado por el ácido amino covalentemente5. Además, presentamos un protocolo para normalizar la cantidad de tRNA por adición de spike en células overexpressing el tRNA raramente usadoselC 4.
tRNAs son algunas de las moléculas más abundantes en la célula bacteriana y una parte esencial de la maquinaria de traducción. tRNAs unen aminoácidos y transfieren a los ribosomas traduciendo. La Unión de aminoácidos para tRNAs (aminoacilación o carga) facilita el aminoacil tRNA sintetasas. La abundancia relativa de diferentes tRNAs cargados es importante para asegurar la fidelidad de la síntesis de proteínas, debido a insuficiente representación del cognado cargado que tRNA para un codón determinado ARN mensajero (ARNm) aumenta la probabilidad de que un tRNA cerca de cognado entregar erróneamente su aminoácido a la cadena polipeptídica creciente en el ribosoma6. La importancia del tRNA cargado se refleja en la amplia respuesta de la célula de e. coli a una severa caída en los niveles de carga de un tRNA; la respuesta estricta. Durante la respuesta estricta disminuye la síntesis de ARNt, ARN ribosómico y mRNAs mayoría a favor de la transcripción de los mRNAs específicos asociados con aminoácido biosíntesis y estrés de supervivencia, y disminuye drásticamente la tasa de crecimiento de las células7 . Además, trabajos recientes y otros ha demostrado que e. coli activamente degrada a la mayoría de su tRNA en respuesta a las tensiones que limitan la traducción4,8, sugiriendo que puede ser el ajuste de los niveles de tRNA importante para hacer frente a dichas tensiones. Así, mediciones fiables de abundancias del tRNA y los niveles de carga será una herramienta importante para comprender plenamente las respuestas de estrés bacteriano.
E. coli se utiliza comúnmente para expresar proteínas recombinantes y debido a las diferencias en la utilización de codones entre especies, expresión óptima es un problema a menudo9. Esto puede eludirse por expresión de tRNAs adicionales necesarios para traducir el mRNA recombinante10. Las medidas de tRNA carga niveles en estas cepas podrían guiar los esfuerzos de solución de problemas y ayudar a optimizar la expresión de la proteína.
Este método también permite la detección de "confusiones"; un aminoacylated de molécula de ARNt con un aminoácido no cognado. Aminoacilación por diferentes aminoácidos puede causar un tRNA migrar con velocidades ligeramente diferentes a través de geles de poliacrilamida1,11. En algunos casos, el método también puede utilizarse para distinguir patrones de modificación diferentes tRNAs idéntico3.
Otro había establecido procedimiento bioquímico para la investigación del tRNA carga niveles es periodato de oxidación. El método se basa en la observación que aminoacylated tRNA está protegido de periodato de oxidación y tRNA descargada no es. Después periodato de tratamiento de la oxidación la recarga de los tRNA se utiliza para estimar los niveles de carga del ARN cosechado. Sin embargo, la recarga de varios tRNAs se ha demostrado para ser afectado por el tratamiento proporcionando alguna inexactitud12. El método presentado aquí medidas de carga directamente del RNA purificado, excluyendo así cualquier sesgo de reacciones químicas o enzimáticas. Una limitación de este método es que sólo una especie de tRNA se detecta a la vez, aunque el mismo Northern blot puede ser despojado y reprobed para varios tRNAs, es lento y algo laborioso recopilar datos sobre muchos tRNAs.
Una manera confiable de normalización de las muestras entre sí es vital en los estudios donde el objetivo es comparar los niveles relativos de una molécula a través de diferentes muestras. Aquí presentamos un procedimiento de normalización para RNA donde una pequeña alícuota de e. coli las células overexpressing el tRNA raraselC se agrega como un punto en a todas las muestras experimentales antes de la purificación del ARN. Este método es útil no sólo al examinar tRNAs pero para la cuantificación relativa de cualquier especie de ARN cuando la configuración experimental es tal que no RNA endógeno puede confiar que presentan en la misma concentración celular en todas las muestras. Por ejemplo, el nivel de una "limpieza" del ARN ribosómico RNA-como a menudo se utiliza como una referencia endógena para comparar las cantidades relativas de otro ARN entre diferentes muestras13. Pero esto es de poca utilidad si la concentración celular de la RNA de referencia varía entre las muestras, como puede ser el caso de ARN ribosómico si muestreo ocurre durante un estrés respuesta15,16,17 o entrada en fase estacionaria17. La adición de spike en células en las muestras experimentales antes de purificación de RNA proporciona una forma sólida y precisa de normalización independiente de la configuración de muestreo. Otra forma de normalización es la adición de uno o más punto en RNAs a las muestras experimentales después de la purificación de RNA. Sin embargo, este método no tiene en cuenta para las diferencias en la recuperación de RNA entre muestras.
Usando células de e. coli que sobrexpresan la referencia RNA como spike-en las células (ver figura 1) en un experimento de e. coli tiene el inconveniente que resulta además de una pequeña cantidad de exógena e. coli total de RNA para la muestras. Se corrigen para esta incorporación mediante el análisis de una muestra que contiene solamente spike en células en paralelo con las muestras experimentales (véase el Northern blot en carril figura 2, con la etiqueta "selC"). El protocolo que se presenta está desarrollado para e. coli K-12 pero es probable que sea aplicable para la mayoría de las especies bacteriana.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. preparación de culturas bacterianas.
- Crecer todos cultivos en matraces Erlenmeyer con una relación de volumen de cultura/frasco de a lo más 1/6. En una típica configuración experimental, crecen las culturas a 37,0 ° C y agitación a 160 rpm en el morpholinepropanesulfonic ácido (MOPS) mínimo media 18 complementado con la fuente de carbono preferido, por ejemplo 0.2% glucosa, por lo menos 10 generaciones consecutivas antes de la cosecha de RNA.
- El día antes de la cosecha de RNA, diluir un pequeño cultivo de la cepa deseada en medio mínimo de fregonas de tal manera que llegará el deseado OD 436 en el tiempo deseado al día siguiente. Calcular el volumen del pequeño cultivo a utilizar para la inoculación por la fórmula:
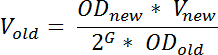
Nota: aquí, OD nuevo denota el deseado OD 436 después de la incubación, nuevo V denota el volumen del cultivo diluido, OD antigua denota OD 436 de la cultura pequeño diluida de, G denota el número de generaciones, crecerá la cultura, de la tiempo de dilución (t 1) a la vez es necesario el día después (t 2) y puede calcularse como: tiempo de diferencia (en minutos) entre t 1 y t 2 dividido por el tiempo de generación (en min). Para asegurar el crecimiento del estado estacionario en el momento del muestreo, debe ser G ≥ 10; Antigua V denota el volumen de la cultura fuera crecido que debe transferirse a medio fresco. Esta ecuación es una aproximación que no tiene en cuenta en cualquier fase de retraso; un fenómeno observado con frecuencia después de la dilusión de una cultura fuera cultiva en medios frescos.- Condiciones de crecimiento variará dependiendo de la finalidad del experimento, pero para limitar la variación de célula a célula en niveles de tRNA y para aumentar la reproducibilidad de los resultados, (recomendado) crecen las culturas constante estado antes de la toma de muestras para 19.
2. Preparación de spike en células
Nota: la cepa de e. coli MAS1074, que expresa el gene de selC codifica tRNA selC bajo el control de un isopropilo β-D-1-tiogalactopiranósido (IPTG)- promotor inducible se utiliza como la tensión de pico en.
- Diluir una cultura pequeño de MAS1074 a un OD 436 de 0.05 en MOPS mínimo suplementado con 100 μg/mL ampicilina y 0,2% de glucosa. Crece el cultivo a 37 º C agitando a 160 rpm.
Nota: Vea la discusión para el uso de una cepa de referencia alternativos. - En OD 436 ~ 0,1, agregar IPTG hasta una concentración final de 1 mM para inducir la expresión de tRNA selC. Crecer la cultura OD 436 ~0.5 (~ 3-4 h de crecimiento) y mueva el frasco de cultivo a un baño de hielo. Conservar el cultivo en punto celular en hielo hasta que todas las muestras para la purificación de RNA han sido cosechadas.
3. Cosecha de muestras experimentales.
- Pre-caliente (37 ° C) una tapa matraz Erlenmeyer de 100 mL (o similar) que contiene 4 mL 10% (w/v) el ácido tricloroacético (TCA) para cada muestra, colocándola en un baño de agua caliente.
Nota: PRECAUCIÓN, TCA se descompone al calentarse, produciendo vapores tóxicos y corrosivos, incluyendo cloruro de hidrógeno y el cloroformo. La solución en agua es un ácido fuerte; Reacciona violentamente con bases y es corrosivo para muchos metales. Debe tenerse precaución adecuada cuando se utiliza este líquido. - Inmediatamente antes de la cosecha celular, mida y registre el OD 436 de la cultura.
- Para cosechar las células, transferir 4 mL de cultivo a un matraz precalentado que contiene TCA.
- Homogeneizar la muestra agitando el matraz con la mano 5 s. mezcla con TCA inmediatamente desactiva las actividades enzimáticas de todo y así conserva los niveles de carga de los tRNAs.
Nota: Si se recogen varias muestras, mantenga las muestras cosechadas en TCA en hielo hasta que todas las muestras han sido recogidas.
4. Además de spike en células
Nota: preparación de spike en células se describe en el paso 2.
- Cuando todas las muestras para la preparación de RNA han sido recogido, añadir 5% aumento en las células (basadas en OD) a cada muestra.
Nota: El volumen de cultivo de spike en celular necesitada se calcula por la fórmula:
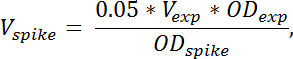
donde V punto denota el volumen de la cultura de spike en la célula es necesitada, V exp denota el volumen del cultivo de la cosecha celular experimental (sin TCA), OD exp denota OD 436 de la cultura de célula experimental en el momento de la cosecha en TCA. OD punto denota OD 436 de la cultura en punto de la célula. Si varias muestras son cosechadas en diferentes Sao, el volumen de espiga en añadido debe ajustarse según la fórmula anteriormente para cada muestra. - Para cuantificar la contribución de las células de espiga a la especie de RNA de interés, preparar una muestra que sólo contenía células de spike mezclando células de espiga 4 mL con 4 mL TCA. Preparar el RNA a partir de esta muestra en la misma forma que las otras muestras.
- Opcionalmente, tomar otro frasco que contenga sólo la muestra experimental, sin espiga en las células y para cuantificar la contribución de la muestra experimental que el tRNA incluyen niveles de selC; los niveles endógenos de tRNA selC en E. coli K-12 son insignificante bajo.
5. Preparación del ARN
Nota: el ARN preparación protocolo descrito aquí es esencialmente la descrita por Varshney et al. 1; Para evitar deacylation de los tRNAs aminoacylated durante la purificación es importante mantener las muestras a pH ácido y a 0 ° C durante el curso de la purificación. Utilizar frascos de tubos limpios estériles para mantener las muestras y las soluciones y preparar todas las soluciones con alta pureza (18.2 MΩ·cm resistencia) agua.
- Echar 8 mL de cada muestra en un tubo de centrífuga bien fría. Centrifugar las muestras durante 10 minutos a 9.500 g de x en el sobrenadante 4 de quitar ° de C. completamente y vuelva a suspender en frío acetato de sodio de 0,3 M 0,3 mL, pH 4.5 y 10 mM EDTA.
- Transferir cada muestra a un tubo de microcentrífuga de 1,5 mL frío y añadir fenol 0.3 mL equilibrada con el mismo buffer.
Nota: PRECAUCIÓN, fenol produce vapores tóxicos y es altamente corrosivo a la piel. - Vortex cada muestra 10 x 15 s con al menos 1 minuto entre cada vórtice. Entre Vortex mantener las muestras a 0 ° C. Use pausas frecuentes para asegurar que las muestras frías.
- Muestras de centrifugar 15 min a 20.000 x g a 4 ° C. transferencia fase líquida (fase superior) de microcentrífuga de 1,5 mL nuevos tubos. Agregar 0,3 mL fenol frío y vortex 4 x 15 s como antes.
- Centrifugar por 10 min a 20.000 x g a 4 ° C. transferencia la fase agua de microcentrífuga de 1,5 mL nuevo, frío tubos conteniendo 2,5 veces el volumen de 96% de etanol (~ 750 μL). Precipitar el ARN por incubar los tubos durante 1 hora a-20 ° C o durante la noche a -80 ° C.
- Muestras de centrifugar por 30 min a 20.000 x g a 4 ° C. desechar el sobrenadante y añadir cuidadosamente etanol frío al de 70% 1 mL sin perturbar el pellet de RNA. Invierta suavemente una vez para lavar el interior de los tubos con etanol a.
Nota: La pelotilla puede ser difícil de ver en este paso. - Centrifugar por 10 min a 20.000 x g a 4 ° C. completamente Quite el sobrenadante y secar el precipitado hasta que no quede etanol. Resuspender el precipitado por Vortex vigorosamente en 20 μl frío 10 sodio acetato pH 4.5 y 1 mM EDTA.
Nota: No seque demasiado la pastilla pues sería difícil suspender. - Opcionalmente, evaluar la concentración de ARN mediante la medición de absorbancia a 260 nm.
- Opcionalmente, evaluar la calidad del ARN purificado por gel de agarosa electroforesis 20.
- Muestras de tienda a -80 ° C.
Nota: El protocolo puede ser una pausa aquí.
Nota: para distinguir la banda de aminoacylated tRNA de su homólogo desacilada en el norte blot, una químicamente desacilada alícuota se prepara por tratamiento alcalino. Para la mayoría de los casos, es suficiente para deacylate una sola muestra para cada mancha blanca /negra norteña.
- Deshielo del ARN muestra en hielo. Transferir una alícuota de 4 μL de la muestra de RNA a un tubo nuevo. Añadir 46 μl Tris-HCl, pH 9.0. Incubar por 2 h a 37 ° C.
- Añadir 15 μl de acetato de sodio de 0,3 M a pH 4,5 y posteriormente añadir 125 μl de etanol al 96%. Precipitar el ARN a-20 ° C por 1 h.
- Centrifugar por 30 min a 20.000 x g a 4 ° C. completamente eliminar el sobrenadante y resuspender en 4 μL frío 10 mM sodio acetato pH 4.5 y 1 mM EDTA.
Nota: El protocolo puede ser una pausa aquí, si la muestra se almacena a -80 ° C.
7. Gel de electroforesis y blotting norte
- Mix 4 μL de muestra (no tratada o desacilada químicamente) con 6 μl de tampón de carga (0,1 M acetato de sodio (pH 5.0), 8 M urea, azul de bromofenol 0.05% y 0.05% xileno cyanol).
Nota: No olvide incluir la muestra que contiene sólo spike-en las células, la química desacilada muestra (y la muestra sin espiga en las células, si uno se preparó). - Cargar las muestras en un 0,4 mm espesor 6,5% gel de poliacrilamida (19:1 acrylam - ide:bisacrylamide) que contiene urea de 8 M en tampón de acetato de sodio de 0.1 M (pH 5.0). Usar geles largo 60 cm de separación adecuada de las especies de tRNA.
- Se separa el ARN por electroforesis en gel de V/cm 10 a 4 ° C, hasta el azul de bromofenol alcanza la parte inferior del gel (20 h).
- La zona del tinte de cyanol xileno y 20 cm hacia abajo hacia el azul de bromofenol tinte se utiliza para borrar (la distancia entre los dos tintes debe ser 25-30 cm). Separar cuidadosamente las placas de dos vidrio, para que el gel permanece en uno de ellos. Un pedazo fino de papel de filtro se corta con el tamaño de la zona deseada de Blot y colocado encima de la parte del gel que se utiliza para borrar. Las partes del gel que no están cubiertas por papel de filtro cortadas y desechadas. Utilice el papel de filtro para levantar cuidadosamente la pieza de gel de la placa de cristal.
Nota: El gel grande es muy frágil así que manejar con cuidado. - Una membrana de nylon cargada positivamente (por ejemplo, la membrana Hybond N +) se coloca en la parte superior del gel y se electroblotted en 20 V durante 90 minutos con 40 mM Tris acetato (pH 8,1), 2 mM EDTA como tampón de transferencia.
- De la reticulación del RNA a la membrana usando luz UV (0,12 J/cm 2).
Nota: La membrana ahora puede ser almacenada a temperatura ambiente y el protocolo se puede detener aquí.
8. Diseño y verificación de sondas
Nota: cuidado diseño y verificación de sondas de Northern blot es crucial para obtener resultados confiables y evitar los no deseado Cruz-hibridación con otras especies de RNA.
- Uso cortos oligonucleótidos sintéticos como sondas. Diseño de la secuencia de la sonda de forma que es complementaria a la parte más singular de la secuencia de los tRNA de interés - esto es a menudo el lazo anticodón.
Nota: Una lista de secuencias de ARNt prevista de varios organismos puede encontrarse en el tRNA genómica (GtRNAdb) de la base de datos 21. - Ajustar la longitud de la secuencia para obtener una prevista fusión temperatura de 55-65 ° C.
- BLAST (Basic Local búsqueda herramienta de alineación) la secuencia de los elegidos contra la secuencia del genoma de la cepa bacteriana utilizada en el experimento, para comprobar que la secuencia es única para el tRNA seleccionado, como se describe en 22.
Nota: La tabla 1 enumera secuencias sonda sugerido para varios tRNAs diferentes de e. coli K12. Si la sonda es específica para el tRNA de interés, sólo dos bandas son visibles en el Northern blot (tRNA cargado y sin carga) y solamente una venda es visible en el carril con la muestra de desacilada (tRNA sin cargar). Si hay más de dos bandas o si además se desea validación de especificidad de la sonda, ver debate.
9. Hibridación de la mancha blanca /negra norteña
Nota: en los pasos siguientes, la membrana es secuencialmente cruzado por hibridación con sondas complementarias a los tRNAs específicos deseados, incluyendo una sonda específica de la referencia de tRNA selC.
- Colocar la membrana en un tubo de hibridación y añadir 6 mL de solución de hibridación (NaCl de 0,9 M, 0,05 M NaH 2 PO 4 (pH 7.7), 5 mM EDTA, 0,5% (p/v) de SDS, 100 mg/mL de deforma y desnaturaliza el ADN de esperma de arenque y 5 x Denhardt ' solución de s (100 x Denhardt ' solución s = 2% albúmina de suero bovino y polivinilpirrolidona 2% 40 2% Ficoll)).
- Pre-hibridar la membrana en el buffer durante 1 hora, girando en el sondeo ° C. Añadir 30 pmol radiactivos oligo-ADN 42 5 ' - final - marcado con 32 P (preparado según lo descrito por Sambrook et al. 23)
Nota: PRECAUCIÓN, las emisiones de alta energía beta de 32 P pueden presentar una sustancial piel y ojo dosis peligro. Debe tomarse PRECAUCIÓN adecuada cuando usando 32 residuos radiactivos P. debe eliminarse de acuerdo con los protocolos de seguridad institucional. - Incubar la sonda con la membrana rotando durante la noche a 42 ° C. retirar la sonda con una pipeta desechable 10 mL.
Nota: Si almacena en un congelador de la sonda puede ser reutilizada. - En el tubo de hibridación, lavar la membrana poco en 50 mL de 0,3 M de NaCl, 30 mM citrato de sodio, 0.1% (p/v) de SDS a temperatura ambiente.
Nota: Este primer lavado contiene sonda mucho independiente y debe manejarse como altamente radiactivo. - Repetir paso 9.4 una vez, luego mueva la membrana a un envase plano de plástico o una bandeja como una tapa. Cubrir la membrana con la solución de lavado (0,3 M NaCl, 30 mM citrato de sodio, 0.1% (p/v) de SDS) e incubar durante 30 min a temperatura ambiente.
- Cambiar la solución de lavado y continuar el lavado hasta que se cuente con una satisfactoria relación señal a ruido (a menudo 3-4 cambios de solución de lavado). Utilice un tubo Geiger-Müller para detectar cómo cambia el relación señal ruido mediante la medición de la radiación de un área vacía de la membrana y se compara con la zona donde la sonda ha cruzado por hibridación a tRNA.
- Para poder pelar la punta de prueba de la membrana después de la exposición, asegúrese de que (importante) la membrana no seque antes el procedimiento de desmontaje. Colocar la membrana en una bolsa de plástico delgada o envoltura herméticamente con plástico envolver y sellar para ser hermético por cinta o soldadura. Coloque la membrana sellada en una pantalla de phosphorimaging.
Nota: El tiempo de exposición necesario en la pantalla phosphorimaging es determinada por la actividad específica de la sonda, la cantidad de RNA sondeado for y la eficiencia de hibridación de la sonda y puede varían grandemente; a unos pocos minutos a varios días. Con la experiencia, la intensidad de la radiación detectada en la membrana con el tubo de Geiger-Müller da una buena indicación del tiempo de exposición requerido. Para la cuantificación confiable, es importante que la pantalla está sobreexpuesta no. - Exploración de la pantalla de phosphorimaging utilizando un escáner láser y guarde el archivo en " .gel " formato de.
Nota: Una alta relación señal a ruido es necesaria para la cuantificación confiable. Sondeo de un tRNA debe dar lugar a dos bandas; aminoacylated contiene un tRNA (migración más lenta) y desacilada contiene un tRNA (migración más rápida). Vea la figura 2.
10. Remover la membrana
Nota: antes de la membrana puede ser sondada por otro tRNA, la sonda anterior debe retirar primero la pelando de la membrana.
- Calor suficiente buffer extracción (15 mM de NaCl, 1, 5 mM citrato de sodio, 0.1% (p/v) de SDS) para cubrir la membrana y viértala sobre la membrana. Incubar por 15 min a temperatura ambiente.
- Repetir paso 10.1 hasta que ninguna radiación puede ser detectado con un tubo Geiger-Müller, la membrana se pueden volver a hibridizada siguiendo los pasos descritos en la sección 9.
11. Cuantificación de tRNA y los niveles de carga
software- carga el archivo .gel en la proyección de imagen (véase la Tabla de materiales). Trace una línea vertical a lo largo de cada uno de los carriles de la muestra de tal manera que abarca más de la anchura del carril, pero excluye los bordes de las bandas donde la señal puede ser desigual, como se muestra en la Figura 3A.
- Visualiza cuentas de cada carril haciendo clic " Análisis "-> " crear gráfico ". Definir manualmente los dos picos que representan a la cargada y el tRNA descargado de cada carril, como se muestra en la figura 3B.
- Obtener cuentas de cada pico haciendo clic " Análisis "-> " informe de área " y registrar el área bajo cada uno de los dos picos.
- Cuando la mancha se han sondeado para tRNA selC y por lo menos un ARNt de interés, normalizar los niveles de la tRNA de interés para tRNA selC.
- Calcular la proporción del tRNA que se origina en las células de espiga en cada carril y restar el total de la señal en ese carril mediante la ecuación:
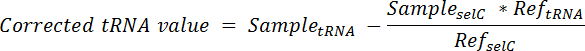
Nota: aquí, TRNA de la muestra = la señal obtenida de un solo carril de una membrana sondeado para el tRNA deseada; Muestra selC = la señal obtenida desde el mismo carril de la misma membrana sondeado para tRNA selC; Ref tRNA = la señal obtenida desde el carril que contiene ARN sólo spike en la célula de la misma membrana sondada para el tRNA deseada; Ref selC = la señal obtenida desde el carril que contiene ARN sólo spike en la célula de la misma membrana sondada para tRNA selC. - Para normalizar, divida el valor corregido del tRNA de cada carril por la señal de selC tRNA del mismo carril de.
Nota: La contribución a la señal de selC tRNA de las células de la muestras es insignificante (véase la figura 2, carril marcado "-selC ").
- Calcular la proporción del tRNA que se origina en las células de espiga en cada carril y restar el total de la señal en ese carril mediante la ecuación:
- Calcular el tRNA nivel para cada carril de carga dividiendo la señal de pico que representa el tRNA cargado con la suma de la señal de ambos picos (cargado + uncharged).
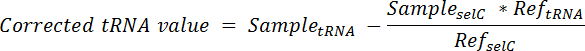
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Utilizando el procedimiento descrito aquí, la abundancia y carga se miden los niveles de tres tRNAs en e. coli K-12 antes y durante el hambre de aminoácidos.
El auxotroph arginina cepa NF9154 (leu thr su argH thi mtl supE44) se cultivaba durante al menos diez generaciones en MOPS medio mínimo suplementado con 0.4% de glicerol, 50 treonina μg/mL, leucina, arginina e histidina 5 μg/mL a 37 º C agitando en el 160 rpm. Hambre de arginina se introdujo por la filtración de la cultura y la resuspensión en medio de MOPS fresco como sobre pero sin arginina. Durante hambre de la arginina, la síntesis de proteínas se reduce fuertemente, pero continúa en un nivel bajo con arginina liberada por el volumen de negocios de proteínas existentes. Las muestras fueron cosechadas en el momento puntos indicaron en la figura 4. Después de todas las muestras fueron cosechadas en TCA, 5% de las MAS1074 spike en células overexpressing tRNAselC, se añaden a cada muestra, como se muestra en figura 1. RNA fue purificada a partir de las muestras, separada por electroforesis y borrado en una membrana, como se describe en el protocolo. La membrana fue sondada para tRNAargVYZQ, tRNAgltTUVW, tRNAhisRy tRNAselC, como se ve en la figura 2. La señal de radiación de cada carril se cuantificó para cada una de las cuatro sondas como ilustrado en la figura 3y corregido y normalizada como se describe en la sección 11. Los resultados se presentan en la figura 4. Figura 4A muestra que los niveles de todas las especies probadas de tRNA disminuyen rápidamente durante hambre de aminoácidos, como recientemente divulgado4. Figura 4B se muestra que el tRNA carga niveles se comportan como se esperaba: la fracción cargada de la tRNA arginina-aceptar cae rápidamente sobre la eliminación de la arginina del medio de crecimiento, mientras que los niveles de carga de los otros tRNAs aumentan ligeramente a hambre porque cobraban tRNAs se acumulan cuando su tasa de decharging en los ribosomas disminuye debido a la falta de sustrato para la síntesis de proteínas4. Más en el período de inanición, la fracción de tRNA cargado disminuye a niveles de aproximadamente la inanición debido a la degradación de la mayoría de la piscina de tRNA (Figura 4A), que se traduce en una mayor tasa de decharging de los tRNAs restantes en los ribosomas. Por el contrario, aumenta la fracción cargada de los ARNtargVYZQ por al menos dos razones; 1) activación de la respuesta estricta significa que mRNA, no cargado tRNAarg, se convierte en el factor limitante para la traducción y 2) la piscina total del tRNAes menor (Figura 4A) así que una mayor fracción de la tRNA totalarg puede ser cargado por la misma pequeña cantidad de arginina. Este experimento demuestra cómo las mediciones simultáneas de tRNA abundancia y niveles de carga de tRNA proporciona información de alta calidad sobre las condiciones para la síntesis de proteínas dentro de la célula.

Figura 1. Además de espiga en las células para muestras. En este ejemplo, muestras de una sola cultura de e. coli NF915 se cosechan en una serie de tiempo antes y durante el hambre de aminoácidos. Se muestra un esquema de la curva de crecimiento de NF915, donde los puntos azules indican los puntos de recolección de la muestra. Los ejes son escala logarítmica. Muestras de la cultura experimental se cosechan como se describe en la sección 3. También se muestra un esquema de la curva de crecimiento de la espiga en la cepa MAS1074, con el punto de recolección de la muestra indicada como un punto azul. El punto de la figura es ilustrar que todas las muestras experimentales (de e. coli NF915 en este ejemplo) reciban una parte alícuota de espiga en las células de la misma cultura de referencia. A cada muestra, 5% de aumento en las células se agregan a las células experimentales, calculadas basado en el OD de las culturas de célula (véase capítulo 4). Posteriormente, RNA es purificado de todas las muestras y utiliza para Norte blot. Haga clic aquí para ver una versión más grande de esta figura.

Figura 2. Exploración de imágenes de fósforo de un Northern blot sondeado para los tRNAs indicado. La membrana contiene RNA de la cepa de e. coli NF915 cosechadas en los momentos indicados antes y después de la inducción de la inanición de arginina (minutos). Además, la membrana contiene dos carriles de las muestras donde se han omitido spike en células (-selC DA y - selC), y un carril que contiene RNA de la espiga en células única (selC). La muestra - selC DA contiene químicamente desacilada tRNA. Se separan claramente las dos bandas que representan las especies cargadas y descargadas del tRNA. Nota la señal insignificante de tRNAselC en los carriles sin espiga en las células. La misma membrana sucesivamente fue despojada y reprobed de los tRNAs indicada a la izquierda. El área de caja en el blot superior indica la parte de la mancha, que se muestra para los posteriores sondeos de tRNA. Haga clic aquí para ver una versión más grande de esta figura.

Figura 3. Cuantificación de Northern blot. abundancia de tRNA se mide utilizando el software. (A) A simple pista de Northern blot vista en la figura 2 sondeado para tRNAargVYZQ. La flecha superior e inferior indican el aminoacylated tRNA y la desacilada tRNA, respectivamente. Las líneas rojas paralelas con el carril se agregan y se utiliza para definir el área para la cuantificación. (B) cuantificación de la señal dentro de las dos líneas rojas rotas en (A) se representa como un gráfico, donde el eje horizontal indica la distancia desde la parte superior de la línea en (A) (en píxeles), y el eje vertical muestra la intensidad de la señal (en cuenta). Los dos picos con señales procedentes de la aminoacylated tRNA y la desacilada tRNA, respectivamente, son definidos manualmente. Los datos que se exportan para su posterior análisis están el valor correspondiente al área bajo cada uno de los dos picos. Haga clic aquí para ver una versión más grande de esta figura.

Figura 4. Resultado representativo: cuantificación de la abundancia de tRNA und niveles de carga durante hambre de la arginina. Cuantificación de las bandas en el Northern blot que se muestra en la figura 2. (A) la relativa abundancia de tRNAgltTUVW (rojo), tRNAargVYZQ (azul) y tRNAhisR (verde). El nivel encontrado en cero minutos (muestras cosechadas justo antes de la aparición del hambre) se establece en 1. (B) nivel de la misma tres tRNAs como en (A) de la carga. Haga clic aquí para ver una versión más grande de esta figura.
| Especificidad de la sonda | Secuencia de la sonda |
| tRNAargVYZQ | 5'-TCCGACCGCTCGGTTCGTAGC |
| tRNAgluTUVW | 5'-CCTGTTACCGCCGTGAAAGGG |
| tRNAhisR | 5'-CACGACAACTGGAATCACAATCC |
| tRNAleuPQVT | 5'-GTAAGGACACTAACACCTGAAGC |
| tRNAleuU | 5'-TATTGGGCACTACCACCTCAAGG |
| tRNAleuW | 5'-CTTGCGGCGCCAGAACCTAAATC |
| tRNAleuX | 5'-TATTTCTACGGTTGATTTTGAA |
| tRNAleuZ | 5'-AAAATCCCTCGGCGTTCGCGCT |
| tRNAlysQTVWYZ | 5'-TGCGACCAATTGATTAAAAGTCAAC |
| tRNAthrV | 5'-TGGGGACCTCACCCTTACCAA |
| tRNAtyrTV | 5'-TCGAACCTTCGAAGTCGATGA |
| tRNAselC | 5'-ATTTGAAGTCCAGCCGCC |
Tabla 1. Tabla de secuencias sonda sugerido para tRNAs seleccionado en e. coli K-12.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Este protocolo describe cómo medir simultáneamente el nivel de carga de concreto e. coli tRNAs y comparar los niveles relativos de los tRNAs en diferentes muestras. Son los puntos críticos del protocolo 1) manejar las muestras de tal manera que se mantienen los niveles de carga celular tRNA, 2) para normalizar la cantidad de tRNA de tal manera que pueden compararse confiablemente niveles relativos del tRNA en diferentes muestras y 3) para el sp ecificity de las sondas seleccionadas para los tRNAs de interés. Estos puntos se abordan por separado a continuación.
purificación del tRNA
Los tRNAs son purificadas de e. coli de una manera suave que está optimizada para conservar sus niveles de carga celulares. Es nuestra experiencia que este protocolo principalmente extractos de RNAs menos 150 nt, así tRNA constituye una gran fracción del ARN purificado. Por esta razón, se recomienda no utilizar este protocolo para purificar RNA total de e. coli. El protocolo presentado aquí también debe ser adecuado para la detección y cuantificación de otros RNAs como RNA pequeño (sRNA) sin cualquier más adaptaciones. Porque enriquece el protocolo de extracción de RNA de secuencias cortas de RNA debe encontrar uso incluso si el sRNA bajo investigación humilde se expresa.
Normalización por spike en células
Para comparar los niveles de los tRNA en diversas muestras, es necesario normalizar los niveles de ARNt a los niveles de una transcripción que está presente en el mismo número promedio por la célula en todas las muestras, con el fin de corregir variaciones de muestra a muestra en el ARN recuperación y carga de gel. Para ello, nos spike las muestras con 5% de las células de referencia que sobreexpresan el tRNA raraselC antes de purificación de RNA. Es preferible el método de punto sobre utilizando un RNA endógeno como referencia, porque permite la comparación de niveles de tRNA en condiciones donde no RNAs endógenos son conocidos con certeza se encuentra en la misma concentración celular. Usando células de e. coli como la espiga en tiene el inconveniente que el 5% extra de e. coli RNA se agrega a todas las muestras experimentales, donde contribuye a la señal del RNA de interés. Este sesgo es contablemente como una muestra que contiene sólo las espiga en las células en cada mancha blanca /negra norteña y corregir los valores de RNA como se describe en el paso 11.4.1. La expresión del tRNAselC en células de tipo salvaje es tan baja en comparación con otros tRNAs (ver figura 2) ese él no hace una diferencia significativa en la cuantificación y por lo tanto puede ser descuidada. Sin embargo, en la cepa de referencia MAS1074, donde selC se expresa por un promotor de7 T en un plásmido de alto copia, estimamos que al menos 1,000-fold más tRNAselC se produce en presencia de 1 mM IPTG que el producido por wildtype E. coli K-12 bajo cualquier condición. MAS1074 deja de crecer después de dos generaciones de inducción completa de IPTG. En ese momento, las diferencias de cultura a cultura en inducción de tRNAselC son pequeñas, y no un motivo de preocupación para este protocolo como todas las alícuotas de spike en células de un solo experimento se toman de la misma cultura inducida de MAS1074. Como alternativa a MAS1074, las células de un organismo cuyo RNA no que cruz-reaccionan con la sonda de hibridación pueden utilizarse como punto en las células. Células de Sulfolobus solfataricus se han utilizado con éxito como spike para experimentos con e. coli tRNA4. Sin embargo, spike en células de e. coli son capaces de reflejar con mayor precisión el grado de lisis celular de las células experimentales, ya que es incierto si S. solfataricus descompone mediante lisis con la misma eficiencia como e. coli. También, crece S. solfataricus es más tedioso ya que crecen a 85 ° C con un tiempo de generación de h ~ 16.
Garantizar la especificidad de las tRNAs
Si aminoacylated y desacilada tRNA ha sido con éxito deben detectarse purificados dos bandas en la membrana del norte. La distancia entre las dos bandas será diferente dependiendo el tRNA específico bajo investigación y es una consecuencia combinada de la diferencia de tamaño, propiedades polares y conformación que un aminoácido particular causa cuando se une un tRNA particular5 . Por ejemplo, la distancia entre el aminoacylated y el desacilada tRNA es mayor para tRNAargVYZQ que es para tRNAgltTUVW, como se ve en la figura 2. Como consecuencia del gran tamaño del gel utilizado aquí para separar el ARN, la resolución es lo suficientemente alta como para distinguir tRNA cargado de sin cargar sino también para distinguir la mayoría de los tRNAs diferentes unos de otros debido a sus diferencias en longitud, secuencia composición y patrones de modificación. Incluso isoaccepting tRNAs se pueden distinguir como se muestra para la leucina aceptar tRNAs2. Sin embargo, nota que en este estudio no fue posible distinguirleuPQV de tRNA tRNAleuT, como la diferencia en la secuencia es solamente una substitución de G a T.
Si aparecen más de dos bandas en la membrana del Norte podría ser una consecuencia de la reacción cruzada de la sonda con otros RNAs. Aumentar el rigor de la etapa de lavado (paso 9.5) aumentando la temperatura normalmente puede resolver esto. Lavado 30 min a 55 ° C es suficiente. Si aumentar el rigor de lavado no quita la hibridación cruzada, la solución puede ser diseñar una nueva sonda que es complementaria a una parte (a menudo parcialmente superpuesta) diferente de la secuencia de tRNA. Bandas adicionales también pueden ser remanente de un sondeo anterior, como resultado de insuficiente extracción de la membrana. Para evitar esto, hacer un control de tira exponiendo la membrana antes de volver a sondear. Si aparecen bandas en la exploración del control la membrana puede requerir desmontaje adicional. Normalmente es posible volver a sondear una membrana al menos 8 - 10 veces pero después de varias puntas de prueba puede ser difícil desnudar completamente. Si este es el caso, la membrana puede almacenarse por unos meses antes de volver a sondear como 32P se descompone rápido.
Posterior verificación de la especificidad de la sonda puede obtenerse mediante la realización de un norte blot con células cosechadas bajo un conjunto de condiciones donde se espera que la transcripción de interés se verá afectada de manera predecible. Por ejemplo, expresión inducible de la tRNA de un plásmido, o hambre de aminoácidos para el aminoácido cognado, debe resultar en un nivel de mayor expresión relativa o en un nivel inferior de carga, respectivamente, del tRNA de interés.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Los autores no tienen nada que revelar.
Acknowledgments
Los autores agradecen a Marit Warrer excelente asistencia técnica. Este trabajo fue financiado por el Consejo Danés de investigación independiente | Ciencias naturales [1323-00343B] y la Fundación Nacional Danés de investigación [DNRF120].
Materials
| Name | Company | Catalog Number | Comments |
| Urea | Merck | 57-13-6 | Purity >99% |
| Polyacrylamide | Serva | 79-06-7 | |
| Bis (N,N'-Methylene-bis-acrylamide) | BIO-RAD | 161-0201 | |
| Trichloroacetic acid (TCA) | Sigma | 76-03-9 | |
| Phenol | Merck | 108-95-2 | |
| IPTG | |||
| Glucose | |||
| Sodium Acetate | |||
| EDTA | |||
| Ethanol | |||
| Tris | |||
| Hydrochloric acid | |||
| Sodium Chloride | |||
| NaH2PO4 | |||
| Sodium Citrate | |||
| SDS | |||
| Herring Sperm DNA | Sigma | 100403-24-5 | |
| BSA | |||
| Polivinylpyrrolidone | |||
| Ficoll | |||
| P32 γATP | Perkin Elmer | ||
| DNA oligos (probes) | TAG Copenhagen | ||
| Hybond N+ membrane | GE Healthcare | RPN203B | |
| Crosslinker | |||
| Electroblotter | BIO-RAD | ||
| Typhoon FLA 7000 Scanner | GE Healthcare | 28955809 | |
| Spectrophotometer | |||
| Hydridization oven | |||
| Geiger-Müller tube | |||
| Phosphor imager screen | GE Healthcare | ||
| Hybridization tube | |||
| Culture Flasks | |||
| 1.5 ml microcentrifuge tubes | |||
| 20 ml centrifuge tubes | |||
| Name | Company | Catalog Number | Comments |
| Software | |||
| ImageQuant | GE Healthcare | ||
| Excel | Microsoft |
References
- Varshney, U., Lee, C. -P., RajBhandary, U. L. Direct analysis of aminoacylation levels of tRNAs in vivo. Application to studying recognition of Escherichia coli initiator tRNA mutants by glutaminyl-tRNA synthetase. J Biol Chem. 266 (36), 24712-24718 (1991).
- Sorensen, M. A. Charging levels of four tRNA species in Escherichia coli Rel(+) and Rel(-) strains during amino acid starvation: a simple model for the effect of ppGpp on translational accuracy. J Mol Biol. 307 (3), 785-798 (2001).
- Krüger, M. K., Sørensen, M. A. Aminoacylation of hypomodified tRNAGlu in vivo. J. Mol. Biol. 284 (3), 609-620 (1998).
- Svenningsen, S. L., Kongstad, M., Stenum, T. S., Muñoz-Gómez, A. J., Sørensen, M. A. Transfer RNA is highly unstable during early amino acid starvation in Escherichia coli. Nucleic Acids Res. 45 (2), 793-804 (2017).
- Enriquez, J. A., Attardi, G. Evidence for aminoacylation-induced conformational changes in human mitochondrial tRNAs. Proc Natl Acad Sci U S A. 93 (16), 8300-8305 (1996).
- Parker, J. Errors and alternatives in reading the universal genetic code. Microbiol Rev. 53 (3), 273-298 (1989).
- Traxler, M. F., et al. The global, ppGpp-mediated stringent response to amino acid starvation in Escherichia coli. Mol Microbiol. 68 (5), 1128-1148 (2008).
- Zhong, J., et al. Transfer RNAs Mediate the Rapid Adaptation of Escherichia coli to Oxidative Stress. PLoS Genet. 11 (6), e1005302 (2015).
- Kane, J. F. Effects of rare codon clusters on high-level expression of heterologous proteins in Escherichia coli. Curr Opin Biotechnol. 6 (5), 494-500 (1995).
- Baca, A. M., Hol, W. G. Overcoming codon bias: a method for high-level overexpression of Plasmodium and other AT-rich parasite genes in Escherichia coli. Int J Parasitol. 30 (2), 113-118 (2000).
- Li, S., Pelka, H., Schulman, L. The anticodon and discriminator base are important for aminoacylation of Escherichia coli tRNA (Asn). J Biol Chem. 268 (24), 18335-18339 (1993).
- Rizzino, A. A., Freundlich, M. Estimation of in vivo aminoacylation by periodate oxidation: tRNA alterations and iodate inhibition. Anal Biochem. 66 (2), 446-449 (1975).
- Dittmar, K. A., Sorensen, M. A., Elf, J., Ehrenberg, M., Pan, T. Selective charging of tRNA isoacceptors induced by amino-acid starvation. EMBO Rep. 6 (2), 151-157 (2005).
- Zhou, K., et al. Novel reference genes for quantifying transcriptional responses of Escherichia coli to protein overexpression by quantitative PCR. BMC Mol Biol. 12 (1), 18 (2011).
- Jacobson, A., Gillespie, D. Metabolic events occurring during recovery from prolonged glucose starvation in Escherichia coli. J Bacteriol. 95 (3), 1030-1039 (1968).
- McCarthy, B. The effects of magnesium starvation on the ribosome content of Escherichia coli. Biochim Biophys Acta (BBA)-Specialized Section on Nucleic Acids and Related Subjects. 55 (6), 880-889 (1962).
- Zundel, M. A., Basturea, G. N., Deutscher, M. P. Initiation of ribosome degradation during starvation in Escherichia coli. Rna. 15 (5), 977-983 (2009).
- Piir, K., Paier, A., Liiv, A., Tenson, T., Maivali, U. Ribosome degradation in growing bacteria. EMBO Rep. 12 (5), 458-462 (2011).
- Schaechter, M., Maaloe, O., Kjeldgaard, N. O. Dependency on medium and temperature of cell size and chemical composition during balanced grown of Salmonella typhimurium. J Gen Microbiol. 19 (3), 592-606 (1958).
- Neidhardt, F. C., Bloch, P. L., Smith, D. F. Culture medium for enterobacteria. J Bacteriol. 119 (3), 736-747 (1974).
- Sambrook, J., Fritsch, E. F., Maniatis, T. Molecular cloning: a laboratory manual. , Cold spring harbor laboratory press. (1989).
- Sorensen, M. A., Jensen, K. F., Pedersen, S. High concentrations of ppGpp decrease the RNA chain growth rate. Implications for protein synthesis and translational fidelity during amino acid starvation in Escherichia coli. J Mol Biol. 236 (2), 441-454 (1994).
- Tian, C., et al. Rapid Curtailing of the Stringent Response by Toxin-Antitoxin Module-Encoded mRNases. J Bacteriol. 198 (14), 1918-1926 (2016).
