

Summary
Hier präsentieren wir Ihnen eine Methode zur Messung direkt Transfer-RNA laden Ebenen aus gereinigtem Escherichia coli RNA sowie eine Möglichkeit zum relativen Pegel der Transfer-RNA oder jede andere kurze RNA über verschiedene Proben, die basierend auf den Zusatz von Spike-im Vergleich Zellen mit dem Ausdruck eines Referenz-Gens.
Abstract
Transfer-RNA (tRNA) ist ein wesentlicher Bestandteil der translationalen Maschinen in jedem Organismus. tRNAs binden und Aminosäuren auf das übersetzen Ribosom übertragen. Die relativen Pegel der verschiedenen tRNAs und das Verhältnis der Aminoacylated tRNA, total tRNA, bekannt als den Ladezustand sind wichtige Faktoren bei der Bestimmung der Genauigkeit und Geschwindigkeit der Übersetzung. Daher sind die Fülle und Ladestation tRNAs wichtige Variablen zu messen, wenn Protein-Synthese, zum Beispiel unter verschiedenen Stressbedingungen zu studieren. Hier beschreiben wir eine Methode zur Ernte tRNA und direkte Messung der relativen Fülle und den absoluten Ladezustand des spezifischen tRNA-Arten in Escherichia coli. Die tRNA geerntet wird dergestalt, dass die labile Bindung zwischen der tRNA und der Aminosäure gewahrt bleibt. Die RNA wird dann Gelelektrophorese und Northern Blot, welche Ergebnisse bei Trennung von der geladenen und ungeladenen tRNAs unterzogen. Die Ebenen der spezifische tRNAs in verschiedenen Proben können durch die Zugabe von Spike-in Zellen für die Normalisierung verglichen werden. Vor der Reinigung von RNA fügen wir 5 % der E. Coli -Zellen, die die seltene tRNASelC zu jeder Probe zu stossen. Die Menge der tRNA-Arten von Interesse in einer Probe wird dann auf die Höhe der tRNASelC in der gleichen Probe normalisiert. Zugabe von Spike-in Zellen vor RNA-Reinigung hat den Vorteil gegenüber Zusatz von gereinigtem Spike-in RNAs, die es auch Unterschiede in der Zelle Lyse Effizienz zwischen Proben ausmacht.
Introduction
Im folgenden präsentieren wir eine Methode zur Quantifizierung der spezifische tRNAs und Messung ihrer Aufladung von Nordbeflecken Ebenen. Die Methode basiert auf einer Technik, die zuerst von Varshney Et Al. entwickelt 1. durch Ernten von Zellen in Säure Trichloressigsäure (TCA) und halten die Proben bei 0 ° C während die RNA-Reinigung, die Esterbindung zwischen der tRNA und der Aminosäure ist konservierte2,3,4. Aminoacylated tRNAs unterschieden werden von ihren Nonacylated Pendants durch Gelelektrophorese und Nordbeflecken, zurückzuführen auf eine verminderte Beweglichkeit der Aminoacylated tRNA im Gel, verursacht durch die kovalent gebundene Aminosäure-5. Darüber hinaus präsentieren wir ein Protokoll für die Normalisierung der tRNA Mengen durch Zugabe von Spike-in Zellen überexprimiert selten verwendete tRNASelC 4.
tRNAs sind einige der am häufigsten vorkommende Moleküle in der Bakterienzelle und ein entscheidender Teil der Übersetzungsmaschinerie. tRNAs binden Aminosäuren und übertragen Sie sie auf die Übersetzungen Ribosomen. Die Bindung der Aminosäuren an tRNAs (Aminoacylation oder beim Laden) wird durch Aminoacyl-tRNA-Synthetasen erleichtert. Die relative Häufigkeit der verschiedenen geladenen tRNAs ist wichtig für die Treue der Proteinsynthese, zu gewährleisten, weil Unterrepräsentierung der Cognate aufgeladen tRNA für ein bestimmtes Boten-RNA (mRNA) Codon erhöht die Wahrscheinlichkeit, die ein nahe verwandtes tRNA wird liefern Sie irrtümlich die Aminosäure an die wachsende Polypeptidkette am Ribosom6. Die Bedeutung der geladenen tRNA spiegelt sich in der umfangreichen Antwort der E. Coli Zelle zu einem starken Einbruch in der Ladestation ein tRNA; die strenge Antwort. Während die strenge Antwort ist die Synthese von tRNAs, ribosomalen RNAs und die meisten mRNAs zu Gunsten der Transkription von spezifischen mRNAs zugeordnete Aminosäure Biosynthese und Stress überleben gesenkt, und die Wachstumsrate der Zellen sinkt dramatisch7 . Darüber hinaus hat neuere Arbeiten von uns und anderen gezeigt, dass E. Coli aktiv verschlechtert sich den Großteil seiner tRNA als Reaktion auf die betont, die dass die Übersetzung4,8, begrenzen darauf hindeutet, dass die Anpassung der tRNA-Ebenen wichtig für den Umgang mit diesen Belastungen. So werden zuverlässige Messungen der tRNA Häufigkeiten und Ebenen laden ein wichtiges Instrument für bakterielle Stressreaktionen vollständig zu verstehen.
E. coli ist allgemein verwendet, um auszudrücken rekombinantes Protein und wegen der Unterschiede in Codon Usage zwischen den Arten, suboptimale Ausdruck ist ein Problem, oft vor9. Dies kann durch Expression von zusätzlichen tRNAs benötigt, um die rekombinante mRNA10übersetzen umgangen werden. Messungen der tRNA laden Ebenen in dieser Stämme könnte führen Problembehandlung und Optimierung der Proteinexpression.
Diese Methode ermöglicht auch die Erkennung von "mischarging"; ein tRNA-Molekül-Aminoacylated mit einer Aminosäure nicht verwandtes. Aminoacylation von verschiedenen Aminosäuren verursachen eine tRNA mit leicht unterschiedlichen Geschwindigkeiten durch Polyacrylamid-Gele1,11migrieren. In einigen Fällen kann die Methode auch verwendet werden, verschiedene Änderung Muster auf ansonsten identischen tRNAs3unterscheiden.
Ein etablierter biochemische Verfahren für die Untersuchung der tRNA Ebenen laden ist Oxidation periodate. Die Methode beruht auf der Beobachtung, dass Aminoacylated, die tRNA geschützt periodate Oxidation und ungeladenen tRNA nicht. Nach periodate Oxidationsbehandlung das Nachladen von der tRNA verwendet, um den Ladevorgang Ebenen der geernteten RNA zu schätzen. Jedoch hat das Nachladen von mehreren tRNAs gezeigt, wodurch einige Ungenauigkeiten12Behandlung betroffen zu sein. Die Methode hier vorgestellten Maßnahmen laden direkt vom gereinigten RNA, also ohne irgendwelche Vorurteile von chemischen oder enzymatischen Reaktionen. Eine Einschränkung dieser Methode ist, dass nur ein tRNA-Arten in einer Zeit, so dass zwar die gleichen Northern blot abgestreift und reprobed für mehrere tRNAs erkannt wird, ist zeitaufwendig und etwas mühsam, die Daten auf viele tRNAs zu sammeln.
Ein verlässlicher Weg der Normalisierung Proben miteinander ist vital in Studien, wo das Ziel ist, die relativen Pegel eines Moleküls über verschiedene Proben zu vergleichen. Hier stellen wir eine Normalisierung Verfahren für RNA, wo eine kleine aliquoten von E. Coli Zellen überexprimiert seltene tRNASelC als Spike-in der experimentellen Proben vor RNA Reinigung hinzugefügt wird. Diese Methode eignet sich nicht nur bei der tRNAs Prüfung aber für relative Quantifizierung aller Arten von RNA als der experimentelle Aufbau ist, die keine endogene RNA zu trauen kann auf die gleiche zelluläre Konzentration in den Proben präsentieren. Zum Beispiel ist das Niveau eines "Housekeeping" RNA-wie ribosomaler RNA oft verwendet, wie ein endogener Verweis auf die relativen Mengen von einem anderen RNS zwischen verschiedenen vergleichen13Proben. Aber dies ist von geringem Nutzen, wenn die zelluläre Konzentration der Referenz RNA zwischen Proben variiert, wie bei ribosomaler RNA der Fall sein kann, tritt Probenahme während einer Belastung Antwort15,16,17 oder Eintritt in die stationäre Phase17. Die Zugabe von Spike-in Zellen, die experimentellen Proben vor RNA-Reinigung bietet eine solide und genaue Möglichkeit der Normalisierung unabhängig von der Probenahme-Einrichtung. Eine weitere Möglichkeit der Standardisierung ist der Zusatz von einem oder mehreren Spike-in RNAs, die experimentellen Proben nach RNA-Reinigung. Diese Methode berücksichtigt jedoch nicht für irgendwelche Unterschiede in RNA Erholung zwischen Proben.
Mit E. Coli Zellen die Overexpress Referenz RNA als Spike-in Zellen (siehe Abbildung 1) in einem Experiment auf E. Coli hat den Nachteil, das es ergibt sich darüber hinaus einer kleinen Menge von exogenen E. Coli total RNA, die Proben. Wir korrigieren für diese Ergänzung durch die Analyse einer Probe nur Spike-in Zellen parallel zu den experimentellen Proben (siehe die Northern blot in Abbildung 2, Gasse mit der Bezeichnung "SelC"). Das Protokoll dargestellt ist entwickelt für E. Coli k-12 aber dürfte für die meisten Bakterienarten anwendbar sein.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. Vorbereitung der Bakterienkulturen.
- Wachsen alle Kulturen in Erlenmeyerkolben mit einer Kultur/Kolben-Volumen-Verhältnis von höchstens 1/6. In einem typischen Versuchsaufbau zu wachsen, die Kulturen bei 37,0 ° C und schütteln mit 160 u/min in Morpholinepropanesulfonic Säure (MOPS) minimale mittlere 18 ergänzt durch die bevorzugte Kohlenstoffquelle, z. B. 0,2 % Glukose, für mindestens 10 aufeinander folgenden Generationen vor der RNA Ernte.
- Am Vortag RNA Ernte verdünnen eine entwachsen Kultur der gewünschten Sorte in MOPS minimaler Medium in einer Weise, die gewünschte OD 436 zum gewünschten Zeitpunkt am nächsten Tag zu erreichen. Berechnen Sie das Volumen der entwachsen Kultur, für die Impfung durch die Formel zu verwenden:
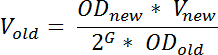
Hinweis: OD neue bezeichnet hier die gewünschte OD 436 nach der Inkubation V neue bezeichnet das Volumen der verdünnte Kultur, OD alten bezeichnet die OD-436 der entwachsen Kultur von verdünnt, G bezeichnet die Anzahl der Generationen die Kultur wachsen wird, von der Zeit der Verdünnung (t 1), der Zeit braucht man am Folgetag (t 2) und kann als berechnet werden: Zeitverschiebung (in Minuten) zwischen t 1 und t 2 geteilt durch die Generationszeit (in Minuten). Um stationäre Wachstum zum Zeitpunkt der Probenahme sicherzustellen, sollte G ≥ 10; V alte bezeichnet das Volumen der out-gewachsene Kultur, die auf frisches Medium übertragen werden sollte. Diese Gleichung ist eine Annäherung, die nicht für alle Lag-Phase berücksichtigt; ein Phänomen, das oft nach Verdünnung einer out-gewachsene Kultur in frische Medien beobachtet.- Wachstumsbedingungen variieren je nach dem Zweck des Versuchs, aber zur Begrenzung Zell-Zell-Variation in tRNA-Ebenen und die Reproduzierbarkeit der Ergebnisse, (empfohlen) zu erhöhen wachsen die Kulturen zu Staat vor der Probenahme zu stabilisieren 19.
2. Vorbereitung der Spike-in Zellen
Hinweis: die E. Coli-Stamm MAS1074, die die SelC gen tRNA SelC unter der Kontrolle einer Isopropyl β-D-1-Thiogalactopyranoside (IPTG) zum Ausdruck bringt- induzierbaren Promoter dient als der Spike-Stamm.
- Verdünnt ein entwachsen Kultur der MAS1074 zu einem OD-436 von 0,05 in MOPS minimaler Medium mit 100 µg/mL Ampicillin und 0,2 % Glukose ergänzt. Wachsen die Kultur bei 37 ° C bei 160 u/min schütteln.
Hinweis: Siehe auch die Diskussion für die Verwendung von eine alternative Referenz Belastung. - Bei OD 436 ~ 0.1, Hinzufügen einer Endkonzentration von 1 mM induzieren die Expression von tRNA SelC IPTG. Wachsen die Kultur zu OD 436 ~0.5 (~ 3-4 h des Wachstums) und den Kultur-Kolben in ein Eisbad verschieben. Die Spike-Zellkultur auf Eis halten, bis alle Proben zur Reinigung von RNA geerntet worden sind.
3. Ernte der experimentellen Proben.
- Pre-Warm (37 ° C), eine 100-mL-Erlenmeyerkolben begrenzt (oder ähnlich) mit 4 mL 10 % (w/V) Trichloressigsäure Säure (TCA) für jede Probe indem man sie in einem beheizten Wasserbad.
Hinweis: Vorsicht, zersetzt sich TCA bei Erhitzung produzieren toxische und korrosive Dämpfe wie Chlorwasserstoff und Chloroform. Die Lösung in Wasser ist eine starke Säure; Es reagiert heftig mit Basen und ist ätzend auf vielen Metallen. Angemessene Vorkehrungen getroffen werden, wenn diese Flüssigkeit mit. - Unmittelbar vor der Zellernte, Messung und Aufzeichnung der OD-436 der Kultur.
- Um die Zellen zu ernten, 4 mL Kultur auf eine vorgewärmte Kanne mit TCA übertragen.
- Mischen die Probe durch Schütteln der Flasche von hand für 5 S. Mixing mit TCA sofort alle enzymatische Aktivitäten deaktiviert und so die Ladestationen Ebenen der tRNAs bewahrt.
Hinweis: Wenn mehrere Proben gesammelt werden, halten Sie geerntete Proben in TCA auf Eis bis alle Proben gesammelt wurden.
4. Zugabe von Spike-in Zellen
Hinweis: Vorbereitung der Spike-in Zellen wird in Schritt2 beschrieben.
- Bei allen Proben für RNA Vorbereitung gesammelt worden, jede Probe 5 % Spitze-in Zellen (basierend auf OD) hinzufügen.
Hinweis: Das Volumen der Spitze-in Zellkultur benötigt wird nach folgender Formel berechnet:
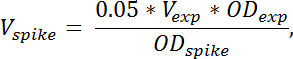
wo V Spike das Volumen der Spitze-in Zellkultur erforderlich bezeichnet, V EXP bezeichnet das Volumen der geernteten experimentelle Zellkultur (ohne TCA), OD exp bezeichnet die OD-436 der experimentellen Zellkultur zum Zeitpunkt der Ernte in TCA. OD Spike bezeichnet die OD-436 der Spike-in Zellkultur. Wenn mehrere Proben an verschiedenen ODs geerntet werden, sollte das Volumen der Spitze-in Zellen hinzugefügt, nach der Formel oben für jede Probe angepasst werden. - Quantifizierung des Beitrags aus den Spike-Zellen, die RNA-Spezies von Interesse, eine Probe nur mit Spike-in Zellen durch das Mischen von 4 mL Spike-in Zellen mit 4 mL TCA vorbereiten. RNA aus diesem Beispiel in der gleichen Weise wie für die anderen Proben vorbereiten.
- Optional eine weitere Flasche enthält nur die experimentelle Probe ohne Spike-in Zellen und aufzunehmen um Quantifizierung des Beitrags der experimentellen Probe zu der tRNA SelC Ebenen; die endogenen Ebenen der tRNA SelC in E. Coli k-12 sind verschwindend gering.
5. Vorbereitung der RNA
Hinweis: die RNA Vorbereitung Protokoll hier beschriebenen ist im Wesentlichen durch Varshney Et Al. beschrieben 1; Zur Vermeidung von Deacylation der Aminoacylated tRNAs während der Reinigung ist es wichtig, die Proben bei einem sauren pH-Wert und bei 0 ° C im Laufe der Reinigung zu halten. Sauberen sterile Tuben/Flaschen verwenden, um die Proben und Lösungen halten, und bereiten Sie alle Lösungen mit Reinstwasser (18,2 MΩ·cm Widerstand) Wasser.
- Pour 8 mL jeder Probe in ein Zentrifugenröhrchen mit Eis gekühlt. Zentrifugieren Sie Proben für 10 min bei 9.500 x g bei 4 ° c vollständig entfernen überstand und in 0,3 mL kalte 0,3 M Natriumacetat, pH 4,5 und 10 mM EDTA Aufschwemmen.
- Jede Probe zu einem kalten 1,5 mL Microcentrifuge Schlauch und 0,3 mL Phenol mit dem gleichen Puffer equilibriert.
Hinweis: Vorsicht, Phenol produziert giftige Dämpfe und ist stark ätzend auf die Haut. - Vortex jede Probe 10 X 15 s mit mindestens 1 min zwischen jedem Wirbel. Zwischen vortexen halten die Proben bei 0 ° c verwenden die häufige Pausen um sicherzustellen, dass die Proben kalt bleiben.
- Zentrifuge Proben für 15 min bei 20.000 x g bei 4 ° C. Transfer Röhren der Wasserphase (obere Phase) zu neuen 1,5 mL Microcentrifuge. Hinzufügen von 0,3 mL kaltes Phenol und Vortex 4 x 15 s wie zuvor.
- Zentrifuge für 10 min bei 20.000 x g bei 4 ° C. Transfer Röhren der Wasserphase zu neuen, kalten 1,5 mL Microcentrifuge mit 2,5 Mal dem Volumen von 96 % Ethanol (~ 750 µL). Überstürzen sich die RNA durch Inkubation der Röhren für 1 h bei-20 ° C oder über Nacht bei-80 ° c
- Zentrifuge Proben für 30 min bei 20.000 x g bei 4 ° c den Überstand verwerfen und sorgfältig fügen Sie 1 mL kaltem 70 % igem Ethanol ohne zu stören das RNA-Pellet. Sanft einmal umkehren, um die Innenseiten der Rohre mit Ethanol waschen.
Hinweis: Das Pellet kann schwierig sein, bei diesem Schritt sehen. - Zentrifuge für 10 min bei 20.000 x g bei 4 ° c vollständig entfernen den Überstand und das Pellet trocknen, bis kein Ethanol übrig ist. Das Pellet durch kräftig aufschütteln in 20 µL kalt 10 Natrium Acetat pH 4,5 und 1 mM EDTA Aufschwemmen.
Hinweis: Nicht übermäßig trocknen das Pellet wie es schwieriger wird, zu Aufschwemmen. - Optional, bewerten die Konzentration der RNA durch Messung der Absorption bei 260 nm.
- Optional, beurteilen die Qualität der gereinigten RNA durch Agarose-Gel-Elektrophorese- 20.
- Store Proben bei-80 ° c
Hinweis: Das Protokoll kann hier angehalten.
Hinweis: die Band von Aminoacylated zu unterscheiden tRNA von seinem Deacylated Gegenstück auf der Northern blot, ein chemisch Deacylated aliquoten wird durch alkalische Behandlung vorbereitet. In den meisten Fällen genügt die Deacylate ein einzelnes Beispiel für jedes Nordfleck.
- Tauwetter der RNA-Probe auf dem Eis. Übertragen Sie eine aliquote 4 µL RNA-Probe auf einen neuen Schlauch. Fügen Sie 46 µL Tris-HCl pH 9.0. 2 h bei 37 ° c inkubieren
- 15 µL von 0,3 M Natriumacetat bei pH 4.5 hinzufügen und fügen Sie anschließend 125 µL Ethanol 96 %. Überstürzen Sie RNA bei-20 ° C für 1 h
- Zentrifuge für 30 min bei 20.000 x g bei 4 ° c vollständig entfernen überstand und in 4 µL kalt 10 mM Natrium Acetat pH 4,5 und 1 mM EDTA Aufschwemmen.
Hinweis: Das Protokoll kann angehalten werden, wenn die Probe bei-80 gelagert ist ° c
7. Gel-Elektrophorese und Nordbeflecken
- Mix 4 µL Probe (unbehandelt oder chemisch Deacylated) mit 6 µL laden Puffer (0,1 M Natriumacetat (pH 5,0), 8 M Harnstoff, 0,05 % Bromophenol Blue und 0,05 % Xylol Cyanol).
Hinweis: Denken Sie daran, die Probe nur Spike-in Zellen, die chemisch Deacylated Probe (und die Probe ohne Spike-in Zellen, wenn man bereit war). - Laden die Proben auf ein 0,4 mm Dicke 6,5 % Polyacrylamid-Gel (19:1 Acrylam - Ide:bisacrylamide) mit 8 M Harnstoff in 0,1 M-Natrium-Acetat-Puffer (pH 5,0). 60 cm lange Gele für die korrekte Trennung der tRNA-Arten verwenden.
- Trennen die RNA durch Elektrophorese auf 10 V/cm Gel bei 4 ° C, bis die Bromophenol blue erreicht die Unterseite des Gels (~ 20 h).
- Bereich von Xylol Cyanol Farbstoff und 20 cm nach unten in Richtung der Bromophenol blau Farbstoff dient für das Beflecken (der Abstand zwischen zwei Farbstoffen sollte 25-30 cm). Trennen Sie die beiden Glasplatten vorsichtig, damit das Gel auf einer von ihnen bleibt. Ein dünnes Stück Filterpapier ist auf die Größe des gewünschten befleckende geschnitten und auf den Teil des Gels, die für das Beflecken verwendet werden platziert. Die Teile des Gels, die nicht durch Filterpapier abgedeckt sind abgeschnitten und weggeworfen. verwenden Sie das Filterpapier um zu heben Sie vorsichtig das Gel Stück Weg von der Glasplatte.
Hinweis: Das große Gel ist sehr zerbrechlich so pfleglich umgehen. - Eine positiv geladenen Nylon-Membran (z. B. Hybond N + Membran) wird auf das Gel gelegt und es ist Electroblotted bei 20 V für 90 min mit 40 mM Tris-Acetat (pH 8.1), 2 mM EDTA als Übertragung Puffers.
- Crosslink RNA an die Membran mit Hilfe von UV-Licht (0,12 J/cm 2).
Hinweis: Die Membran kann jetzt bei Raumtemperatur gelagert werden und das Protokoll kann hier angehalten werden.
8. Gestaltung und Überprüfung von Sonden
Hinweis: sorgfältige Gestaltung und Überprüfung von Northern Blot Sonden ist von entscheidender Bedeutung, um zuverlässige Ergebnisse zu erzielen und vermeiden unerwünschte Kreuz-Hybridisierung mit anderen RNA-Spezies.
- Verwendung kurzer synthetische Oligonukleotide als Sonden. Design die Sonde-Sequenz in einer Weise, dass es ist eine Ergänzung zu den einzigartigsten Teil der Sequenz von der tRNA von Interesse - das ist oft der Anticodon Loop.
Hinweis: Eine Liste von vorhergesagten tRNA-Sequenzen aus verschiedenen Organismen finden Sie in der genomischen tRNA Datenbank (GtRNAdb) 21. - Passen Sie die Länge der Sequenz zu einer prognostizierten Schmelztemperatur von 55-65 ° C.
- BLAST (grundlegende lokale Ausrichtung Search Tool) die gewählten Sequenz gegen die Genomsequenz der bakteriellen Belastung in das Experiment verwendet, um sicherzustellen, dass die Reihenfolge für die ausgewählten tRNA eindeutig ist, wie in 22 beschrieben.
Hinweis: In Tabelle 1 sind empfohlene Sonde Sequenzen für mehrere verschiedene tRNAs von E. Coli K12. Wenn die Sonde spezifisch für die tRNA von Interesse ist, nur zwei Bänder sind sichtbar auf der Northern Blot (geladenen und ungeladenen tRNA) und nur ein Band ist sichtbar auf der Spur mit der Deacylated Probe (ungeladenen tRNA). Wenn mehr als zwei Bänder vorhanden sind oder wenn weitere Validierung der Sonde Besonderheit gewünscht wird, siehe Diskussion.
9. Hybridisierung von Northern Blot
Hinweis: In den folgenden Schritten wird die Membran sequenziell mit Sonden ergänzen die gewünschte spezifische tRNAs, einschließlich einer Sonde für die Referenz hybridisiert tRNA SelC.
- Die Membran in einer Hybridisierung Röhre und 6 mL Hybridisierung Lösung (0,9 M NaCl, 0,05 M NaH 2 PO 4 (pH 7,7), 5 mM EDTA, 0,5 % (w/V) SDS, 100 mg/mL des geschert und denaturiert Hering Spermien-DNA und 5 X Denhardt ' s Lösung (100 X Denhardt ' s Lösung = 2 % 2 % Rinderserumalbumin und 2 % Polyvinylpyrrolidon 40 Ficoll)).
- Hybridisieren vor die Membran im Puffer für 1 h bei 42 ° C. hinzufügen 30 Pmol radioaktiven Oligo-DNA-Sonde 5 rotierende ' - End - beschriftet mit 32 P (wie von Sambrook Et Al. beschrieben vorbereitet 23)
Hinweis: Vorsicht, die energiereiche Beta-Emissionen aus 32 P können eine erhebliche Haut und Auge Dosis Gefahr präsentieren. Angemessene Vorkehrungen getroffen werden, wenn mit 32 s. radioaktiven Abfälle nach den entsprechenden institutionellen Sicherheitsprotokolle entsorgt werden sollen. - Inkubieren Sie die Sonde mit der Membran über Nacht drehen bei 42 ° C. Entfernen Sie die Sonde mit einer Einweg-10 mL-Pipette.
Hinweis: Wenn in einer Tiefkühltruhe die Sonde kann wiederverwendet werden. - In der Hybridisierung Röhre, waschen Sie die Membran kurz in 50 mL 0,3 M NaCl, 30 mM Natrium-Citrat, 0,1 % (w/V) SDS bei Raumtemperatur.
Hinweis: Diese ersten Wäsche enthält viel ungebundene Sonde und muss behandelt werden, als hochradioaktiver. - Wiederholen Schritt 9.4 einmal dann verschieben die Membran zu einem flachen Kunststoff-Container oder Behälter mit einem Deckel. Die Membran mit der Waschlösung (0,3 M NaCl, 30 mM Natrium-Citrat, 0,1 % (w/V) SDS) abdecken und 30 min bei Raumtemperatur inkubieren.
- Ändern die Waschlösung und die Wäsche weiter, bis ein befriedigendes Signal Rauschabstand (oft 3 bis 4 Änderungen der Waschlösung) erteilt worden ist. Verwenden Sie ein Geiger-Müller-Rohr zu erkennen, wie das Rauschen Signal-Verhältnis ändert sich durch die Messung der Strahlung von einem leeren Bereich der Membran und vergleichen Sie es mit der Gegend, wo die Sonde, speziell für tRNA hybridisiert hat.
- Um die Sonde aus der Membran nach der Belichtung Streifen können sicherstellen, dass (wichtig) die Membran nicht vor dem stripping Eingriff austrocknet. Legen Sie die Membran in eine dünne Plastiktüte oder wickeln sie eng mit Kunststoff wickeln und durch Klebeband oder Schweißen luftdicht zu verschließen. Legen Sie die versiegelte Membran auf einem Phosphorimaging Bildschirm.
Hinweis: Die erforderliche Belichtungszeit auf dem Phosphorimaging-Bildschirm wird bestimmt durch die spezifische Aktivität von der Sonde, die Menge an RNA fo sondiertR und die Hybridisierung Effizienz der Sonde und kann sehr unterschiedlich sein; von wenigen Minuten bis zu mehreren Tagen. Mit Erfahrung gibt die Intensität der Strahlung auf die Membran mit dem Geiger-Müller Schlauch entdeckt einen guten Indikator für die erforderliche Belichtungszeit. Für zuverlässige Quantifizierung ist es wichtig, dass der Bildschirm nicht überbelichtet ist. - Den Phosphorimaging-Bildschirm mit einem Laser-Scanner scannen und speichern Sie die Datei in " .gel " Format.
Hinweis: Ein high-Signal Rauschabstand ist notwendig für die zuverlässige Quantifizierung. Für eine tRNA sondieren sollte in zwei Bands führen; ein mit Aminoacylated tRNA (langsamer Migration) und eine mit Deacylated tRNA (schneller Migration). Siehe Abbildung 2.
10. Abisolieren der Membran
Hinweis: bevor die Membran für eine weitere tRNA sondiert werden kann, die früheren Sonde muss zunächst durch Entfernen der Membran entfernt werden.
- Hitze genug abstreifenden Puffer (15 mM NaCl, 1,5 mM-Natrium-Citrat, 0,1 % (w/V) SDS) zur Deckung der Membran und gießen es über die Membran. 15 min bei Raumtemperatur inkubieren.
- Wiederholen Sie Schritt 10.1 bis keine Strahlung detektiert werden, mit einem Geiger-Müller Schlauch; die Membran kann nun wieder hybridisiert die Schritte in Abschnitt 9 beschrieben werden.
11. Quantifizierung der tRNA und Ebenen laden
- Last der .gel-Datei in der Bildbearbeitung Software (siehe die Tabelle der Materialien). Zeichnen eine vertikale Linie entlang jeder Probe Spuren in einer Weise, dass es erstreckt sich über die Breite der Fahrspur der Großteil aber schließt die Kanten der Bands wo das Signal uneben, kann wie in Abbildung 3A gezeigt.
- Visualisieren zählt von jeder Bahn durch Klicken auf " Analyse "-> " Graph erstellen ". Die zwei Spitzen jeder Spur der geladenen und ungeladenen tRNA aus manuell zu definieren, wie in Abbildung 3 b.
- Jede Spitze zählt einzuholen, indem Sie auf " Analyse "-> " Revierbericht ", und notieren Sie den Bereich unter jedem der beiden Gipfel.
- Wenn der Fleck hat wurden sondiert für tRNA SelC und mindestens ein tRNA von Interesse, normalisieren die Ebenen der tRNA für tRNA SelC von Interesse.
- Berechnen Sie den Anteil der tRNA mit Ursprung aus den Spike-in Zellen in jeder Bahn und Subtrahieren von am Gesamtsignal in dieser Spur unter Verwendung der Gleichung:
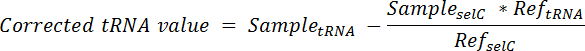
Hinweis: hier, Probieren Sie tRNA = das Signal eine einzige Spur einer Membran, die für die gewünschte tRNA; sondiert entnommen Probe SelC = das Signal erhalten von der gleichen Spur der gleichen Membran sondiert für tRNA SelC; Ref-tRNA = das Signal aus der Spur, enthält nur Spike-in Zelle RNA der gleichen Membran für die gewünschte tRNA; sondiert gewonnen Ref SelC = das Signal von der Fahrspur, enthält nur Spike-in Zelle RNA der gleichen Membran für tRNA SelC sondiert erhalten. - Zu normalisieren, teilen Sie den korrigierten tRNA-Wert von jeder Bahn durch die tRNA SelC Signal aus der gleichen Spur.
Hinweis: Der Beitrag der tRNA SelC Signal aus den ausgewählten Zellen vernachlässigbar ist (siehe Abbildung 2, Gasse mit der Bezeichnung "-SelC ").
- Berechnen Sie den Anteil der tRNA mit Ursprung aus den Spike-in Zellen in jeder Bahn und Subtrahieren von am Gesamtsignal in dieser Spur unter Verwendung der Gleichung:
- Berechnen die tRNA laden Ebene für jede Fahrspur dividiert das Signal der beiden Gipfeln aufgeladen tRNA mit der Summe des Signals aus Spitze (aufgeladenen + ungeladenen).
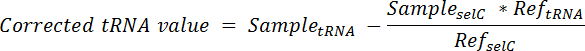
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Mithilfe des Verfahrens beschrieben hier, die Fülle und Aufladen der drei tRNAs in E. Coli k-12 vor und während der Aminosäure Hunger gemessen wurden.
Die Arginin-Auxotroph Stamm NF9154 (Thr Leu seinen ArgH Thi Mtl supE44) wurde zum mindestens zehn Generationen im MOPS minimaler Medium ergänzt mit 0,4 % Glycerin, 50 µg/mL Threonin, Leucin, Arginin und Histidin 5 µg/mL bei 37 ° C Schütteln bei angebaut 160 u / min. Arginin Hunger wurde durch Filtration von Kultur und Wiederfreisetzung in frischen MOPS Medium wie oben jedoch ohne Arginin eingeführt. Während Arginin Hunger Proteinsynthese wird stark reduziert, aber weiterhin auf niedrigem Niveau mit Arginin durch den Umsatz der vorhandenen Proteine freigesetzt. Proben zum Zeitpunkt geerntet wurden Punkte in Abb. 4angegeben. Nachdem alle Proben in TCA geerntet wurden, 5 % der MAS1074 Spike-in Zellen überexprimiert tRNASelC, wurde hinzugefügt, um jede Probe wie in Abbildung 1. RNA wurde gereinigt von den Proben durch Elektrophorese getrennt und auf eine Membran ausgelöscht, wie im Protokoll beschrieben. Die Membran war für tRNAArgVYZQ, tRNAGltTUVWund tRNAHisRtRNASelC, sondiert, wie in Abbildung 2dargestellt. Die Strahlung Signal von jeder Bahn wurde für jede der vier Sonden wie in Abbildung 3dargestellt und korrigiert und normalisierte wie in Abschnitt 11 beschrieben quantifiziert. Die Ergebnisse sind in Abbildung 4dargestellt. Abbildung 4A zeigt, dass die Ebenen aller getesteten tRNA-Arten schnell während Aminosäure Hunger, wie kürzlich berichtet4zu verringern. Abbildung 4 b zeigt, dass die tRNA laden Ebenen verhalten sich wie erwartet: geladenen Teil der Annahme von Arginin tRNA sinkt rasch nach dem Entfernen von Arginin aus das Wachstumsmedium während der Aufladung der anderen tRNAs leicht auf erhöhen verhungern weil tRNAs ansammeln, wenn ihre Rate der Entladung an die Ribosomen verringert sich aufgrund des Fehlens von Substrat für Protein Synthese4in Rechnung gestellt. Weiter in die Hunger-Zeit reduziert aufgeladen tRNA Bruchteil sich auf etwa vor verhungern infolge des Abbaus der Mehrheit der tRNA Pool (Abb. 4A), die Ergebnisse in einem größeren Ausmaß der Entladung von der restlichen tRNAs an die Ribosomen. Im Gegensatz dazu erhöht der geladene Anteil der tRNAArgVYZQ aus mindestens zwei Gründen; (1) Aktivierung der strengen Antwort bedeutet, dass die mRNA, tRNAArg, nicht in Rechnung gestellt wird der limitierende Faktor für die Übersetzung und (2) der gesamte Pool von tRNAArg ist reduziert (Abb. 4A) so einen größeren Teil des gesamten tRNAArg kann aufgeladen werden, indem die gleiche kleine Menge an Arginin. Dieses Experiment zeigt, wie die gleichzeitige Messung der tRNA Fülle und tRNA Ladestation Ebenen bietet qualitativ hochwertige Informationen über die Bedingungen für die Proteinsynthese der Zelle.

Abbildung 1: Zugabe von Spike-in Zellen zu Proben. In diesem Beispiel werden Proben aus einer einzigen Kultur von E. Coli NF915 in einer Zeitreihe vor und während der Aminosäure Hunger geerntet. Eine schematische Darstellung der Wachstumskurve der NF915 wird angezeigt, wo die blauen Punkte zeigen die Punkte der Probe Ernte. Die Y-Achsen sind Log-Skala. Proben von der experimentellen Kultur(en) werden geerntet, wie in Abschnitt 3 beschrieben. Eine schematische Darstellung der Wachstumskurve der Spike-Stamm MAS1074 zeigt sich auch, mit dem Punkt der Probe Ernte als einem blauen Punkt gekennzeichnet. Der Punkt der Figur soll veranschaulichen, dass die experimentellen Proben (von E. Coli NF915 in diesem Beispiel) eine aliquote Spike-in Zellen aus der gleichen Referenz-Kultur erhalten. Zu jeder Probe 5 % der Spike-in Zellen werden hinzugefügt, um die experimentelle Zellen, berechnet anhand der OD von der Zellkulturen (siehe Abschnitt 4). RNA ist anschließend gereinigt von allen Proben und Nordflecken verwendet. Bitte klicken Sie hier für eine größere Version dieser Figur.

Abbildung 2: Phosphor Imager Scan des Northern Blot sondiert für die angegebenen tRNAs. Die Membran enthält RNA aus E. Coli -Stamm NF915 geerntet zu den angegebenen Zeitpunkten vor und nach der Induktion von Arginin Hunger (Minuten). Darüber hinaus enthält die Membran zwei Fahrspuren von Proben, wo Spike-in Zellen weggelassen (-SelC DA und -SelC), und eine Spur mit RNA aus der Spike-in Zellen nur (SelC). Die Probe - SelC DA enthält chemisch Deacylated tRNA. Die beiden Bands aus der geladenen und ungeladenen tRNA-Arten sind klar voneinander getrennt. Hinweis: das vernachlässigbare Signal von tRNASelC in den Gassen ohne Spike-in Zellen hinzugefügt. Die gleichen Membran wurde sukzessive abgestreift und reprobed für die tRNAs auf der linken Seite angezeigt. Die Box Bereich in der oberen Blot gibt den Teil des Flecks, der für die nachfolgenden tRNA-Sondierungen angezeigt wird. Bitte klicken Sie hier für eine größere Version dieser Figur.

Abbildung 3. Quantifizierung der Northern Blot. tRNA Fülle wird mit Software gemessen. (A) A einspurige aus der Northern Blot in Abbildung 2 sondiert für tRNAArgVYZQgesehen. Die obere und untere Pfeil zeigen die Aminoacylated tRNA und der Deacylated tRNA, beziehungsweise. Die roten Linien, die parallel mit der Spur hinzugefügt und verwendet, um den Bereich für die Quantifizierung zu definieren. (B) Quantifizierung des Signals innerhalb der zwei gebrochene rote Linien gesehen in (A) dargestellt, wie ein Diagramm, wo die horizontale Achse zeigt den Abstand vom oberen Rand der Zeile in (A) (in Pixel), und die vertikale Achse zeigt die Intensität des Signals (in Punkten). Die beiden Spitzen mit Signal mit Ursprung aus der Aminoacylated tRNA und die Deacylated sind tRNA, bzw. manuell definiert. Die Daten, die zur weiteren Analyse exportiert werden ist der Wert, entspricht die Anbaufläche jedes von den beiden Gipfeln. Bitte klicken Sie hier für eine größere Version dieser Figur.

Abbildung 4. Repräsentatives Ergebnis: Quantifizierung der tRNA Häufigkeiten eind laden Ebenen während Arginin verhungern. Quantifizierung der Bands auf der Northern Blot in Abbildung 2dargestellt. (A) die Relative Fülle von tRNAGltTUVW (rot), tRNAArgVYZQ (blau) und tRNAHisR (grün). Die Ebene finden Sie unter null Minuten (Proben geerntet kurz vor dem Beginn der Hunger) wird auf 1 gesetzt. (B) Ebene die gleichen drei tRNAs wie in (A) aufladen. Bitte klicken Sie hier für eine größere Version dieser Figur.
| Sonde Spezifität | Sonde-Sequenz |
| tRNAargVYZQ | 5'-TCCGACCGCTCGGTTCGTAGC |
| tRNAgluTUVW | 5'-CCTGTTACCGCCGTGAAAGGG |
| tRNAhisR | 5'-CACGACAACTGGAATCACAATCC |
| tRNAleuPQVT | 5'-GTAAGGACACTAACACCTGAAGC |
| tRNAleuU | 5'-TATTGGGCACTACCACCTCAAGG |
| tRNAleuW | 5'-CTTGCGGCGCCAGAACCTAAATC |
| tRNAleuX | 5'-TATTTCTACGGTTGATTTTGAA |
| tRNAleuZ | 5'-AAAATCCCTCGGCGTTCGCGCT |
| tRNAlysQTVWYZ | 5'-TGCGACCAATTGATTAAAAGTCAAC |
| tRNAthrV | 5'-TGGGGACCTCACCCTTACCAA |
| tRNAtyrTV | 5'-TCGAACCTTCGAAGTCGATGA |
| tRNAselC | 5'-ATTTGAAGTCCAGCCGCC |
Tabelle 1. Tabelle der vorgeschlagenen Sonde Sequenzen für ausgewählte tRNAs in E. Coli k-12.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Dieses Protokoll beschreibt, wie Sie gleichzeitig messen Sie den Ladezustand des spezifischen E. Coli tRNAs und vergleichen Sie die relativen Pegel der tRNAs in verschiedenen Proben. Die kritischen Punkte des Protokolls sind 1) die Proben so zu behandeln, dass die zelluläre tRNA Ladestation Ebenen beibehalten werden, 2) tRNA Mengen so normalisieren, dass relative tRNA-Ebenen in verschiedenen Proben zuverlässig verglichen werden können, und (3) die sp zu gewährleisten, Ecificity der ausgewählten Sonden für die tRNAs von Interesse. Dieser Punkte werden im folgenden gesondert eingegangen.
tRNA-Reinigung
Die tRNAs werden aus E. Coli auf sanfte Weise gereinigt, die optimiert ist, um ihre zellulären Ladestation Niveaus beizubehalten. Es ist unsere Erfahrung, dass dieses Protokoll in erster Linie RNAs kürzer als 150 Auszüge nt, tRNA stellt somit einen großen Teil der gereinigten RNA. Aus diesem Grund empfiehlt es sich nicht um dieses Protokoll verwenden, um total RNA aus E. Colizu reinigen. Die hier vorgestellten Protokoll sollte auch für Erkennung und Quantifizierung von anderen RNAs wie kleine RNA (sRNA) ohne weitere Anpassungen geeignet sein. Weil das RNA-Extraktion-Protokoll für kurze RNA-Sequenzen bereichert sollte es auch Verwendung finden, auch wenn die sRNA untersuchte niedrigen geäussert wird.
Normalisierung von Spike-in Zellen
Um die Ebenen der tRNA über verschiedene Proben zu vergleichen, ist es notwendig, die tRNA-Ebenen auf das Niveau von ein Transkript zu normalisieren, die in die gleiche durchschnittliche Anzahl pro Zelle in alle Proben, um Probe zu Probe Variationen in RNA zu korrigieren ist Erholung und Gel geladen. Zu diesem Zweck spike wir die Proben mit 5 % des Referenzzellen, die die seltene tRNASelC vor RNA Reinigung overexpress. Die Spike-Methode ist vorzuziehen mit einer endogenen RNA als Referenz, weil es den Vergleich der tRNA über Bedingungen zulässt, wo keine endogenen RNAs mit Sicherheit bekannt sind, bei der gleichen zelluläre Konzentration zu finden. Mit E. Coli Zellen als Spike-in hat den Nachteil, dass 5 % zusätzliche E. Coli RNA hinzugefügt wird, alle experimentellen Proben, wo trägt sie auf das Signal der RNA des Interesses. Diese Vorspannung ist einschließlich einer Probe nur die Spike-in Zellen auf jeden Northern Blot und die RNA-Werte zu korrigieren, wie unter Schritt 11.4.1 zurückzuführen. Der Ausdruck der tRNA istSelC in Wildtyp-Zellen macht so niedrig im Vergleich zu anderen tRNAs (siehe Abbildung 2), daß es keinen signifikanten Unterschied in der Quantifizierung und somit vernachlässigt werden kann. Jedoch in den Referenz-Stamm MAS1074, wo SelC von einem T-7 -Förderer auf eine hohe Kopie Plasmid ausgedrückt wird, wir schätzen, dass mindestens 1,000-fold mehr tRNASelC in Anwesenheit von 1 mM IPTG produziert wird als der Wildtyp E. coli k-12 unter jeder Bedingung. MAS1074 hört sich nach zwei Generationen des vollen IPTG Induktion wachsen. Zu diesem Zeitpunkt nach Kultur Unterschiede in der Induktion der tRNASelC sind klein und kein Problem für dieses Protokoll wie alle Aliquote Spike-in Zellen für ein einziges Experiment stammen aus der gleichen induzierten Kultur der MAS1074. Als Alternative zu MAS1074 können Zellen aus dem Organismus, dessen RNA nicht mit der Hybridisierung Sonde Kreuzreaktion wird, als Spitze-in Zellen verwendet werden. Sulfolobus Solfataricus Zellen wurden als Spike-in für Experimente mit E. Coli tRNA4erfolgreich eingesetzt. E. Coli Spike-in Zellen sind jedoch voraussichtlich den Grad der Zelle lyse der experimentellen Zellen besser widerspiegeln, da es ungewiss ist, ob S. Solfataricus mit der gleichen Effizienz als lysen E. Coli. Außerdem wächst S. Solfataricus langweiliger, wie sie bei 85 ° C mit einer Generationszeit von ~ 16 h wachsen.
Eine Besonderheit des ausgewählten tRNAs
Wenn Aminoacylated und Deacylated tRNA erfolgreich wurde sollten gereinigte zwei Bands auf der nördlichen Membran erkannt werden. Der Abstand zwischen den beiden Bands werden unterscheiden sich je nach der spezifischen tRNA untersucht und kombinierte ergibt sich aus der Differenz zwischen Größe, polare Eigenschaften und Konformation, die eine bestimmte Aminosäure verursacht, wenn es eine besondere tRNA5 bindet . Zum Beispiel ist der Abstand zwischen der Aminoacylated und der Deacylated tRNA für tRNAArgVYZQ größer als bei tRNAGltTUVW, wie in Abbildung 2dargestellt. Aufgrund der großen Größe des Gels verwendet hier die RNA zu trennen ist die Auflösung hoch genug aufgeladen tRNA von ungeladenen zu unterscheiden, sondern auch die meisten verschiedene tRNAs von einander durch ihre Unterschiede in der Länge, Sequenz unterscheiden Zusammensetzung und Änderung Muster. Sogar Isoaccepting tRNAs können unterschieden werden, wie für das Leucin akzeptieren tRNAs2gezeigt. Beachten Sie, dass in dieser Studie nicht tRNALeuPQV von tRNALeuT, als der Unterschied in der Sequenz zu unterscheiden war ist jedoch nur ein G T Substitution.
Wenn mehr als zwei Bands auf der nördlichen Membran erscheinen möglicherweise eine Folge der Kreuzreaktionen der Sonde mit anderen RNAs. Erhöhung der Stringenz der Waschschritt (Schritt 9,5) durch Erhöhung der Temperatur kann in der Regel dieses Problem lösen. 30 min. bei 55 ° C zu waschen ist normalerweise ausreichend. Wenn Kreuz-Hybridisierung erhöhen die Stringenz waschen nicht entfernen, kann die Lösung sein, eine neue Sonde zu entwerfen, die komplementär zu einem anderen (oft teilweise überlappenden) Teil der tRNA-Sequenz ist. Weitere Bands werden auch Übertrag von einer vorherigen Sonde aufgrund unzureichender Abisolieren der Membran. Um dies zu verhindern, kontrollieren Sie ein Streifen indem man die Membran vor wieder zu sondieren. Wenn Bands auf dem Kontrolle-Scan angezeigt werden kann die Membran zusätzliche Abisolieren benötigen. Es ist in der Regel möglich, eine Membran Sonde mindestens 8-10 mal wieder, aber nach mehreren Sonden es kann schwer sein, vollständig entfernen. Wenn dies der Fall ist, kann die Membran für ein paar Monate vor Sondierung wieder als 32P zerfällt schnell gespeichert werden.
Weitere Überprüfungen der Sonde Besonderheit erhalten Sie durch die Durchführung einer Northern Blot mit Zellen, die unter einer Reihe von Bedingungen geerntet wo das Transkript des Interesses wird voraussichtlich in einer vorhersagbaren Weise betroffen sein. Beispielsweise sollten induzierbaren Ausdruck der tRNA an einem Plasmid oder Aminosäure Hunger für die cognate Aminosäure eine erhöhte relative Ausdruck Ebene oder ein niedriger Ladezustand führen bzw. der tRNA von Interesse.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Die Autoren haben nichts preisgeben.
Acknowledgments
Die Autoren danken Marit Warrer für ausgezeichnete technische Unterstützung. Diese Arbeit wurde unterstützt durch die Danish Council für unabhängige Forschung | Naturwissenschaften [1323-00343B] und der dänischen National Research Foundation [DNRF120].
Materials
| Name | Company | Catalog Number | Comments |
| Urea | Merck | 57-13-6 | Purity >99% |
| Polyacrylamide | Serva | 79-06-7 | |
| Bis (N,N'-Methylene-bis-acrylamide) | BIO-RAD | 161-0201 | |
| Trichloroacetic acid (TCA) | Sigma | 76-03-9 | |
| Phenol | Merck | 108-95-2 | |
| IPTG | |||
| Glucose | |||
| Sodium Acetate | |||
| EDTA | |||
| Ethanol | |||
| Tris | |||
| Hydrochloric acid | |||
| Sodium Chloride | |||
| NaH2PO4 | |||
| Sodium Citrate | |||
| SDS | |||
| Herring Sperm DNA | Sigma | 100403-24-5 | |
| BSA | |||
| Polivinylpyrrolidone | |||
| Ficoll | |||
| P32 γATP | Perkin Elmer | ||
| DNA oligos (probes) | TAG Copenhagen | ||
| Hybond N+ membrane | GE Healthcare | RPN203B | |
| Crosslinker | |||
| Electroblotter | BIO-RAD | ||
| Typhoon FLA 7000 Scanner | GE Healthcare | 28955809 | |
| Spectrophotometer | |||
| Hydridization oven | |||
| Geiger-Müller tube | |||
| Phosphor imager screen | GE Healthcare | ||
| Hybridization tube | |||
| Culture Flasks | |||
| 1.5 ml microcentrifuge tubes | |||
| 20 ml centrifuge tubes | |||
| Name | Company | Catalog Number | Comments |
| Software | |||
| ImageQuant | GE Healthcare | ||
| Excel | Microsoft |
References
- Varshney, U., Lee, C. -P., RajBhandary, U. L. Direct analysis of aminoacylation levels of tRNAs in vivo. Application to studying recognition of Escherichia coli initiator tRNA mutants by glutaminyl-tRNA synthetase. J Biol Chem. 266 (36), 24712-24718 (1991).
- Sorensen, M. A. Charging levels of four tRNA species in Escherichia coli Rel(+) and Rel(-) strains during amino acid starvation: a simple model for the effect of ppGpp on translational accuracy. J Mol Biol. 307 (3), 785-798 (2001).
- Krüger, M. K., Sørensen, M. A.
- Svenningsen, S. L., Kongstad, M., Stenum, T. S., Muñoz-Gómez, A. J., Sørensen, M. A. Transfer RNA is highly unstable during early amino acid starvation in Escherichia coli. Nucleic Acids Res. 45 (2), 793-804 (2017).
- Enriquez, J. A., Attardi, G. Evidence for aminoacylation-induced conformational changes in human mitochondrial tRNAs. Proc Natl Acad Sci U S A. 93 (16), 8300-8305 (1996).
- Parker, J. Errors and alternatives in reading the universal genetic code. Microbiol Rev. 53 (3), 273-298 (1989).
- Traxler, M. F., et al. The global, ppGpp-mediated stringent response to amino acid starvation in Escherichia coli. Mol Microbiol. 68 (5), 1128-1148 (2008).
- Zhong, J., et al. Transfer RNAs Mediate the Rapid Adaptation of Escherichia coli to Oxidative Stress. PLoS Genet. 11 (6), e1005302 (2015).
- Kane, J. F. Effects of rare codon clusters on high-level expression of heterologous proteins in Escherichia coli. Curr Opin Biotechnol. 6 (5), 494-500 (1995).
- Baca, A. M., Hol, W. G. Overcoming codon bias: a method for high-level overexpression of Plasmodium and other AT-rich parasite genes in Escherichia coli. Int J Parasitol. 30 (2), 113-118 (2000).
- Li, S., Pelka, H., Schulman, L. The anticodon and discriminator base are important for aminoacylation of Escherichia coli tRNA (Asn). J Biol Chem. 268 (24), 18335-18339 (1993).
- Rizzino, A. A., Freundlich, M. Estimation of in vivo aminoacylation by periodate oxidation: tRNA alterations and iodate inhibition. Anal Biochem. 66 (2), 446-449 (1975).
- Dittmar, K. A., Sorensen, M. A., Elf, J., Ehrenberg, M., Pan, T. Selective charging of tRNA isoacceptors induced by amino-acid starvation. EMBO Rep. 6 (2), 151-157 (2005).
- Zhou, K., et al. Novel reference genes for quantifying transcriptional responses of Escherichia coli to protein overexpression by quantitative PCR. BMC Mol Biol. 12 (1), 18 (2011).
- Jacobson, A., Gillespie, D. Metabolic events occurring during recovery from prolonged glucose starvation in Escherichia coli. J Bacteriol. 95 (3), 1030-1039 (1968).
- McCarthy, B. The effects of magnesium starvation on the ribosome content of Escherichia coli. Biochim Biophys Acta (BBA)-Specialized Section on Nucleic Acids and Related Subjects. 55 (6), 880-889 (1962).
- Zundel, M. A., Basturea, G. N., Deutscher, M. P. Initiation of ribosome degradation during starvation in Escherichia coli. Rna. 15 (5), 977-983 (2009).
- Piir, K., Paier, A., Liiv, A., Tenson, T., Maivali, U. Ribosome degradation in growing bacteria. EMBO Rep. 12 (5), 458-462 (2011).
- Schaechter, M., Maaloe, O., Kjeldgaard, N. O. Dependency on medium and temperature of cell size and chemical composition during balanced grown of Salmonella typhimurium. J Gen Microbiol. 19 (3), 592-606 (1958).
- Neidhardt, F. C., Bloch, P. L., Smith, D. F.
- Sambrook, J., Fritsch, E. F., Maniatis, T. Molecular cloning: a laboratory manual. , Cold spring harbor laboratory press. (1989).
- Sorensen, M. A., Jensen, K. F., Pedersen, S. High concentrations of ppGpp decrease the RNA chain growth rate. Implications for protein synthesis and translational fidelity during amino acid starvation in Escherichia coli. J Mol Biol. 236 (2), 441-454 (1994).
- Tian, C., et al. Rapid Curtailing of the Stringent Response by Toxin-Antitoxin Module-Encoded mRNases. J Bacteriol. 198 (14), 1918-1926 (2016).
