Neben der typischen Watson-Crick-Doppelhelix können Nukleinsäuren aufgrund ihrer Guanin-reichen Sequenzen verschiedene Sekundärstrukturen annehmen, wie z.B. die alternative G-Quadruplex (G4)-Form. Die G4-Struktur basiert auf der Bildung von planaren Tetrameren, sogenannten G-Tetraden, in denen vier Guanine durch Hoogsteen-Wasserstoffbrückenbindungen wechselwirken. G-Tetraden werden durch monovalente Kationen, die im Zentrum des Guaninkerns koordiniert sind, gestapelt und weiter stabilisiert (Abbildung 1)1.
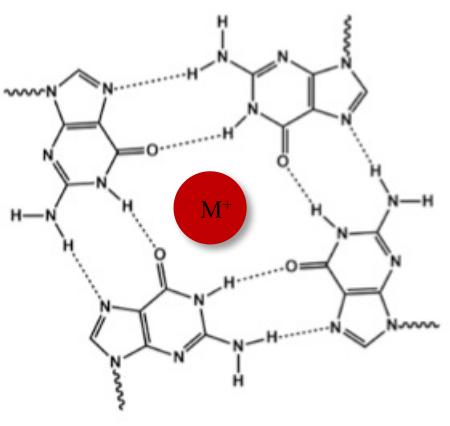
Abbildung 1: Schematische Darstellung einer G-Quadruplex-Struktur. (A) Schematische Darstellung einer G-Tetrade. Das planare Array wird durch Hoogsteen-Basenpaarung und durch ein zentrales Kation (M+) stabilisiert. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
Sequenzen mit vier oder mehr Durchläufen von mindestens zwei aufeinanderfolgenden Guanin-Nukleotiden sind potentielle G-Quadruplex-bildende Sequenzen (PQS), die sich in G-Quadruplex-Strukturen falten können. PQS befinden sich in vielen verschiedenen zellulären Kontexten, wie z.B. an Telomeren, Genpromotoren, ribosomaler DNA und Rekombinationsstellen, und sind an der Regulation vieler biologischer Prozesse beteiligt2. Daher ist die Identifizierung und experimentelle Validierung von G4s im menschlichen Genom, die derzeit hauptsächlich mit computergestützten Werkzeugen durchgeführt wird, ein biologisch relevantes Thema3. Um computergestützte Vorhersagen zu unterstützen oder unvorhergesehene G4-Strukturen zu erkennen, wird hier eine auf chemischer Kartierung basierende Methode zur Identifizierung der G4-Bildung in einem DNA-Template gezeigt, die die präzise Identifizierung von Guaninen ermöglicht, die die G-Tetradenstruktur bilden.
Der beschriebene chemische Mapping-Assay nutzt die unterschiedliche Reaktivität von Bis-3-Chlorpyperidinen (B-CePs) mit Guaninen nach der Bildung von G4-Strukturen. Aufgrund ihrer hohen Reaktivität mit Nukleophilen 4,5,6,7,8,9 sind B-CePs Nukleinsäure-Alkylanzien mit der Fähigkeit, sehr effizient mit der N7-Position von Guanin-Nukleotiden10 zu reagieren. Auf die Alkylierung folgt die Depurinierung und die Strangspaltung in einzel- und doppelsträngigen DNA-Konstrukten. Im Gegensatz dazu sind Guanine, die an der Bildung der G-Tetraden in G4-Anordnungen beteiligt sind, unempfindlich gegen die B-CeP-Alkylierung, da die N7-Positionvon Guanin an den Hoogsteen-Wasserstoffbrückenbindungen beteiligt ist. Diese spezifische Reaktivität von B-CePs ermöglicht nicht nur den Nachweis von G4-Strukturen, sondern auch die Identifizierung der Guanine, die die Tetrade bilden, da sie aus ihrem relativen Schutz vor Alkylierung im Vergleich zu Guaninen in einzel- und doppelsträngiger DNA abgeleitet werden können.
Das chemische Kartierungsprotokoll wird hier unter Verwendung von B-CeP 1 (Abbildung 2A) als Sonde für die Charakterisierung von Thrombin-bindendem Aptamer (TBA) vorgestellt, einer 15-mer-DNA, die in der Lage ist, die G4-Anordnung in Gegenwart von Kaliumkationen anzunehmen11,12. Die G4-Anordnung von TBA (G4-TBA) wird direkt mit zwei Kontrollen verglichen, nämlich TBA in der einzelsträngigen Form (ssTBA) und TBA, die zu ihrer komplementären Sequenz getempert wurde, um das doppelsträngige Konstrukt (dsTBA) zu bilden (Tabelle 1). Die Produkte der Sondierungsreaktionen werden durch hochauflösende Polyacrylamid-Gelelektrophorese (PAGE) auf Einzelnukleotidebene aufgelöst, indem einzelne Alkylierungsaddukte und DNA-Strangspaltungen an den alkylierten Guaninen lokalisiert werden. Die Visualisierung auf dem Gel wird durch Konjugation des TBA-Oligonukleotids mit einem Fluorophor an seinem 3′-Ende ermöglicht (Tabelle 1). Dieses Protokoll zeigt, wie TBA in seinen verschiedenen Konformationen (G4 und Kontrollen) gefaltet wird und wie Sondierungsreaktionen mit B-CePs gefolgt von PAGE durchgeführt werden.
