

Summary
Zelluläre Gesamt-RNA ein Template für die Untersuchung schlechte kurzfristige Änderungen in der RNA-Synthese und Zerfall sowie die Kinetik der RNA-Prozessierung. Hier beschreiben wir metabolische Markierung der neu transkribierten RNA mit 4-Thiouridin gefolgt von Thiol-spezifische Biotinylierung und Aufreinigung von neu transkribierten RNA ermöglicht, diese Einschränkungen zu überwinden.
Abstract
Die Entwicklung der gesamten Transkriptom-Microarrays und Sequenzierung der nächsten Generation revolutioniert unser Verständnis der Komplexität der zellulären Genexpression. Zusammen mit einem besseren Verständnis der molekularen Mechanismen beteiligt sind genaue Messungen der zugrundeliegenden Kinetik immer wichtiger. Hier stehen diese leistungsstarken Methoden wesentliche Einschränkungen aufgrund intrinsischen Eigenschaften der Vorlage Proben sie studieren, dh gesamten zellulären RNA. In vielen Fällen Veränderungen in zelluläre Gesamt-RNA entstehen entweder zu langsam oder zu schnell, um die molekularen Mechanismen und deren Kinetik mit ausreichender Auflösung darzustellen. Darüber hinaus werden der Beitrag der Änderungen in der RNA-Synthese, Verarbeitung und Zerfall nicht ohne weiteres unterschieden.
Wir haben vor kurzem entwickelte hochauflösende Genexpressionsanalysen, diese Einschränkungen zu überwinden. Unser Ansatz basiert auf metabolische Markierung der neu transkribierten RNA mit 4-thiouri Basisdine (daher auch als 4SU-Tagging bezeichnet) durch rigorose Reinigung neu transkribiert RNA mit Thiol-spezifische Biotinylierung und Streptavidin-beschichteten magnetischen Kügelchen gefolgt. Es ist für einen breiten Bereich von Organismen einschließlich Vertebraten, Drosophila und Hefe. Wir haben erfolgreich angewendet 4SU-Tagging, um Echtzeit-Kinetik der Transkriptionsfaktor Aktivitäten zu untersuchen, liefern präzise Messungen der RNA Halbwertszeiten und erhalten neue Einblicke in die Kinetik der RNA-Prozessierung. Schließlich können Computermodelle eingesetzt, um eine integrierte, umfassende Analyse der zugrunde liegenden molekularen Mechanismen zu erzeugen.
Introduction
Gene Expression Profiling ist ein wichtiges Instrument verwendet, um zelluläre Prozesse und die damit verbundenen komplexen Interaktion Netzwerk studieren. Studien zur mRNA waren typischerweise die Methode der Wahl, um grundlegende Einblicke in die zugrunde liegenden molekularen Mechanismen zu erhalten. Die Entwicklung der gesamten Transkriptom-Microarrays 1 und, in jüngerer Zeit, der nächsten Generation Sequenzierung von RNA (RNA-seq) 2-4 angeheizt diesen Ansatz. Während diese Technologien revolutioniert unser Verständnis der Komplexität der zellulären Genexpression, stehen sie wesentliche Einschränkungen aufgrund ihrer intrinsischen Eigenschaften der Vorlage Probe, dh gesamten zellulären RNA. Erstens, kurzfristige Veränderungen in Gesamt-RNA Ebenen nicht übereinstimmen Änderungen in Transkriptionsraten, sind aber von Natur aus abhängig von der RNA-Halbwertszeit der jeweiligen Transkripte. Während eine fünffache Induktion eines kurzlebigen Transkript, zB Codierung für einen Transkriptionsfaktor, wird leicht nachweisbar sein in Gesamt-RNAinnerhalb einer Stunde die gleiche Induktion einer langlebigen Transkript, z. B. Codierung für ein metabolisches Enzym, bleiben nahezu unsichtbar. Darüber hinaus, auch eine komplette Abschaltung (> 1.000-fach Down-Regulation) in der Transkription von durchschnittlich Gen mit einer RNA-Halbwertszeit von fünf Stunden nehmen Sie einfach fünf Stunden seines gesamten RNA-Spiegel von nur zweifache verringern . Daher begünstigt Analyse von Gesamt-RNA die Erkennung von Up-Regulation von kurzlebigen Transkripte, von denen viele für Transkriptionsfaktoren und Gene mit regulatorischen Funktionen 5 codieren. Darüber hinaus wird die tatsächliche kinetische Kaskade Regelung verdunkelt und primären Signalwege nicht von sekundären unterschieden werden. Beide wiederum zu erheblichen Verzerrungen in stromabwärts bioinformaticsanalysen führen. Zweitens können Veränderungen in Gesamt-RNA Levels nicht auf Änderungen in der RNA-Synthese oder Zerfall zugeschrieben werden. Messungen der Letztere erfordern Zelle invasive Ansätze, z. B. Blockierung transcriptizur Verwendung von Actinomycin D 6, und erweiterte Überwachung der laufenden RNA Verfall im Laufe der Zeit. Bei einer mittleren mRNA Halbwertszeit in Säugerzellen von 5 - 10 h 5,7, wird mRNA-Spiegel der meisten Gene nur um weniger als das Doppelte nach mehreren Stunden der transkriptionellen Verhaftung verringert. Diese eher kleine Unterschiede führen in grob ungenaue Messungen von mRNA Halbwertszeiten für die meisten zellulären Genen aufgrund der exponentiellen Natur der zugrunde liegenden mathematischen Gleichungen. Schließlich, während RNA-seq der gesamten zellulären RNA ergab, dass etwa die Hälfte unserer Gene unterliegen alternatives Spleißen Veranstaltungen 8, den zugrunde liegenden Kinetik sowie die dynamischen Mechanismen Führung Gewebe-und Kontext-spezifische Regulation der RNA-Prozessierung noch schwer abzuschätzen sind. Darüber hinaus verarbeitet der Beitrag der RNA zu differentiellen Genexpression, insbesondere für nicht-kodierenden RNAs, bleibt zu bestimmen. Insgesamt stellen diese Einschränkungen Haupthindernisse fürbioinformatische kinetische Modellierung der zugrunde liegenden molekularen Mechanismen.
Vor kurzem haben wir eine Methode entwickelt, genannt hochauflösende Genexpressionsanalysen, um diese Probleme zu überwinden 5,7,9. Es basiert auf metabolische Markierung der neu transkribierten RNA unter Verwendung von 4-Thiouridin (4SU-Tagging), ein natürlich vorkommendes Uridinderivat basiert und bietet direkten Zugang zu neu transkribierten Transkripte mit minimaler Störung des Zellwachstums und der Genexpression (siehe Abbildung 1) 5, 10-12. Exposure von eukaryotischen Zellen zu 4SU Ergebnisse in seine schnelle Aufnahme, Phosphorylierung 4SU-Triphosphat und Einbau in neu transkribierten RNA. Nach Isolierung der gesamten zellulären RNA ist die 4SU-markierten RNA-Fraktion Thiol-spezifisch biotinylierte Erzeugen einer Disulfidbrücke zwischen Biotin und den neu transkribierten RNA. 'Total zellulären RNA' kann dann quantitativ in markierte ('neu transkribiert') und unmarkierten ('pre-existin getrennt werdeng ') RNA mit hoher Reinheit unter Verwendung von Streptavidin-beschichteten magnetischen Kügelchen. Schließlich wird markierten RNA aus den Perlen durch Hinzufügen eines Reduktionsmittels (z. B. Dithiothreitol) Spalten der Disulfidbrücke und Lösen der neu transkribierten RNA aus den Perlen zurückgewonnen.
Neu transkribiert RNA zeigt die transkriptionelle Aktivität von jedem Gen während des Zeitrahmens 4SU Exposition. 4SU-Tagging in der Zeitskala von Minuten stellt somit eine Momentaufnahme der eukaryotischen Genexpression und eine ideale Vorlage für down-stream bioinformatischen Analysen (z. B. Promotor-Analyse). In Fällen, in denen stationären Bedingungen angenommen werden kann, die Verhältnisse neu transkribiert / total neu transkribiert / unmarkiertem und unmarkierten / Gesamt-RNA liefern nicht-invasiven Zugang zu präzisen RNA Halbwertszeiten 7,13. Darüber hinaus ist es wichtig, dass die neu transkribierten RNA nach nur 5 min von 4SU-Tagging (5 min 4SU-RNA) gereinigt beachten ist jünger als 15 und 60 min 4SU-RNA.Bei der Durchführung sowohl ultra-kurz und zunehmend länger 4SU-Tagging in einem experimentellen Setting mit RNA-seq kombiniert, werden die Kinetik der RNA-Prozessierung bei Nukleotid Auflösung 9 offenbart. Schließlich erlauben Zeitverlauf Analysen neu transkribiert und Gesamt-RNA mit Computermodellen kombiniert eine integrative Analyse der RNA-Synthese und Zerfall 14.
Zusammenfassend kann dieser Ansatz zur direkten Analyse der Dynamik der RNA-Synthese, Verarbeitung und Zersetzung in eukaryotischen Zellen. Es gilt in allen wichtigen Modellorganismen einschließlich Säugetiere, Insekten (Drosophila), Amphibien (Xenopus) und Hefe 5,15,16. Es ist direkt kompatibel mit Microarray-Analyse 5,17, RNA-seq 9,13,14, und gilt in vivo 12,15. Hier haben wir ausführlich die Methodik zu beschriften, zu isolieren und zu reinigen, neu transkribierten RNA in kultivierten Säugetierzellen. Darüber hinaus Potentiometeral Probleme und Fallstricke werden diskutiert.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. Metabolische Markierung mit 4-Thiouridin
Machen Sie einen detaillierten Plan des Versuchsaufbaus / schedule, z. B. wenn die 4SU auf Zellkultur hinzufügen und wenn die Proben zu ernten. Plan für mindestens 5 min zwischen jeder Bedingung. Nur behandeln Zellen einer Bedingung zu einer Zeit. Griff max. 3 - 5 Gerichte zu einem bestimmten Zeitpunkt. Griff Zellen möglichst schnell auf Veränderungen der Temperatur und CO 2-Gehalt zu minimieren. Vermeiden, dass die Zellen dem Licht nach 4SU zugesetzt, da dies zu einer Vernetzung der 4SU-markierten RNA an zelluläre Proteine führen kann.
Beginn der Kennzeichnung
- Thaw 4-Thiouridin (4SU) kurz vor der Verwendung und Pipette erforderliche Menge an 4SU für jede Bedingung in ein steriles Falcon-Röhrchen.
- Nehmen Sie die erforderliche Menge an Zellkulturmedium (5 ml pro 10 cm Schale) von den Gerichten und in den 4SU-haltigen Falcon-Röhrchen und gründlich mischen. Entfernen und entsorgen Sie die restlichen Mittel aus den Gerichten. <li> Übernehmen 4SU-haltigem Medium zurück zu den Gerichten.
Ende der Etikettierung
- Entfernen Zellkulturmedium aus Zellen. 5 ml Trizol zu jeder Platte. Für komplexe Experimente mit mehreren Zeitpunkten oder Bedingungen, wird dieser Schritt am besten durch zwei Personen, ein Entfernen des Mediums, die andere Zugabe Trizol und Ernte des Lysats getan.
- Inkubation für 5 min bei Raumtemperatur für die komplette Zell-Lyse.
- Verwenden Sie eine 10 ml Pipette, um die Platte sorgfältig spülen mit dem zusätzlichen Trizol. Dies hilft vollständige Zelllyse und Probe Erholung. Gehen Sie vorsichtig mit Trizol ist extrem gefährlich, wenn immer in Kontakt mit der Haut oder den Augen! Haben Gegenmittel für Phenol Verbrennungen an der Hand (z. B. Polyethylenglykol 300 oder 400 in der industriellen Brennspiritus (70:30)). Übertragen Proben Polypropylenröhrchen. Bitte beachten Sie, dass Standard-Falcon-Röhrchen nicht widerstehen diese hohen g-Kräfte). Die Proben können bei -20 ° C für mindestens einen Monat bis Gesamt-RNA aus der is hergestellt.
2. RNA Vorbereitung mit modifizierten Trizol Protocol
- 1 ml Chloroform (0,2 ml pro ml Trizol) und kräftig schütteln für 15 sec. Inkubieren bei Raumtemperatur für 2 - 3 min.
- Zentrifuge bei 13.000 × g für 15 min bei 4 ° C.
- Übertragen wässrige obere Phase (mit dem RNA) in ein neues 15 ml Polypropylen-Röhrchen.
- In der Reaktion ½ Volumen der beiden RNA Niederschlag Puffer und Isopropanol (zB auf 3 ml Überstand 1,5 ml RNA Niederschlag Puffer und 1,5 ml Isopropanol).
- Gut mischen. Inkubieren bei Raumtemperatur für 10 min.
- Zentrifuge bei 13.000 × g für 10 min bei 4 ° C. Überstand verwerfen.
- Spin down kurz (5.000 × g für 30 sec) und restliches Isopropanol mit 200 ul Pipette.
- In ein gleiches Volumen 75% Ethanol und schütteln, bis das Rohr Pellet löst. Vermeiden Sie brechen sie in viele kleine Stücke, da dies, kann die Entfernung von Rückständenl Ethanol schwierig.
- Zentrifuge bei 13.000 × g für 10 min bei 4 ° C. Überstand verwerfen.
- Spin down RNA kurz und entfernen restliche Ethanol mit einer 200 ul Pipette. Wiederholen Sie Schritt und entfernen restliche Ethanol mit einem 20 ul Pipette. Nach diesen beiden Schritten sollten keine weiteren Trocknung der Pellets vorgenommen werden.
- In 100 ul H 2 O pro 100 ug erwartet RNA-Ausbeute und gut mischen durch Auf-und Abpipettieren 5-6 mal, um das Lösen der RNA zu unterstützen.
- Auflösen und denaturieren RNA durch Erhitzen auf 65 ° C für 10 min (Shaker) und sofort auf Eis stellen.
- Messen RNA-Konzentration bei 260 nm mit einem Spektralphotometer NanoDrop, nach den Anweisungen des Herstellers. Diese RNA kann bei -80 ° C für mindestens einen Monat gelagert werden.
3. Thiol-spezifische Biotinylierung von Neu transkribierten RNA
- Beginnen Sie mit 60 - 80 ug Gesamt zellulären RNA.
- Bilden Kennzeichnung Reaktion. Pipette in der folgendenBestellung (pro ug RNA):
- 1 ul 10x Biotinylierung Buffer
- 7 ul RNA (mit 1 ug RNA in Nuklease-freiem H 2 O verdünnt)
- 2 ul Biotin-HPDP (1 mg / ml DMF)
Fügen Sie immer die Biotin-HPDP letzte und sofort mischen durch Pipettieren. Falls die Biotin ausfällt, kann DMF-Gehalt bis zu einer Endkonzentration von 40% gesteigert werden.
- Inkubieren bei Raumtemperatur für 1,5 h bei Drehung.
- Fügen Sie dem gleichen Volumen Chloroform. Gründlich mischen. Inkubieren 2 - 3 Minuten, bis die Phasen zu trennen beginnen und Blasen beginnen zu verschwinden.
- Zentrifuge bei 20.000 × g für 5 min bei 4 ° C. Vorsichtig übertragen die obere wässrige Phase in ein neues Röhrchen.
- Wiederholen Sie die Schritte 3.4 und 3.5 einmal. Sie können diesen Schritt in 2 ml Phase Lock Gel Schwere Rohre führen zum Verlust der RNA zu reduzieren.
- RNA Niederschlag: Add 1/10 das Volumen von 5 M NaCl und ein gleiches Volumen vonIsopropanol auf die Wasserphase.
- Zentrifuge bei 20.000 × g für 20 min bei 4 ° C. Überstand verwerfen.
- In ein gleiches Volumen 75% Ethanol, Zentrifuge bei 20.000 × g für 10 min bei 4 ° C, Überstand verwerfen.
- Spin kurz und entfernen Restethanol mit 200 ul Pipette.
- Spin kurz und entfernen restliche Ethanol mit 20 ul Pipette.
- Nicht in die RNA zu trocknen. Re-suspend es in 50 - 100 ul H 2 O (~ 1 ul pro 1 ug Eingang RNA). Gut mischen durch Auf-und Abpipettieren 5 - 6 mal.
- Prüfen RNA-Qualität durch elektrophoretische Analyse auf RNA-Abbau auszuschließen.
4. Dot-Blot-Analyse von 4SU-Einbau (Optional)
4SU Einarbeitung kann leicht durch Dot-Blot-Analyse von biotinylierten RNA bestimmt werden. Dies ist ein optionaler Schritt, Fehlersuche und Abschätzung der 4SU Einbauraten relativ zu einem biotinylierten DNA-Oligo-Steuerung ermöglicht. Für diesen Test wir rIhr Kommentar zu Verwendung iodoacetyl-Biotin statt Biotin-HPDP für Biotinylierung 4SU-markierte RNA in Schritt 3.2. Dies führt zu einer irreversiblen Biotinylierung 4SU-RNA. Daher Spalte-Methoden (z. B. RNeasy) kann zur Rückgewinnung von viel kleineren Mengen von biotinylierten RNA (z. B. 5 ug) verwendet werden. Während RNA biotinyliert mit Biotin-HPDP eignet sich auch für diesen Assay ist das resultierende Signal schwächer und das Signal-Rausch-Verhältnis schlechter (Abb. 3).
- Verfolgen Sie das Protokoll für 4SU-Kennzeichnung und Isolierung von Gesamt-RNA zellulären wie in den Abschnitten 1 und 2 beschrieben.
- Biotinylieren 4SU-markierte RNA, wie in Kapitel 3 beschrieben ersetzt Biotin-HPDP mit iodoacetyl-Biotin und führen zwei Chloroformextraktionen vollständig zu entfernen übermäßige iodoacetyl-Biotin-Reste.
- Recover biotinylierten RNA durch Isopropanol / Ethanol-Fällung wie beschrieben oder unter Verwendung einer Säule-basierten Ansatz (z. B. RNeasy) bei kleinen Mengen von RNA (<10 pg) Verwendet.
- Inkubieren Sie die Zeta Membran in Nuklease-freiem Wasser mit rockigen für 10 min.
- Nehmen Sie die Membran aus der Nuklease-freies Wasser und entfernen Sie überschüssige Flüssigkeit, indem Membran zwischen zwei saubere Papierhandtücher und fest andrücken. Air-Trocknen der Membran für 5 min in schöner Punkten führen.
- Für jede Probe, bereiten 20 ul 200 ng / ul RNA mit eiskaltem Dotblot Bindungspuffer (10 mM NaOH, 1 mM EDTA). Bewerben 5 ul dieser Verdünnung (dh 1 ug RNA) sowie drei weiteren 10-fache Verdünnungen (dh 100, 10 und 1 ng RNA, respectively) zum Zeta Membran durch Pipettieren. Pipettieren durch einen leeren Rack Pipettenspitzen verwendet werden, um gleichmäßig verteilt Abstand zu liefern. Alternativ können Sie einen Dot-Blot gemäß den Anweisungen des Herstellers.
- Anwenden 5 ul der Biotin-markierten DNA-Oligo in Konzentrationen von 20 ng / ul bis 20 pg / ul (dh 100 bis 0,1 ng Oligo) als positive control der Membran durch Pipettieren. Verwenden Sie einen biotinylierten, 4SU-naive Probe als negative Kontrolle.
- Luft trocknen die Membran für 5 min.
- Inkubieren der Membran für 30 min in 40 ml Blockierungspuffer unter Schütteln.
- Inkubieren der Membran mit 10 ml 1:1000 Streptavidin-Meerrettich-Peroxidase für 15 min (5 ml PBS + 5 ml 20% SDS + 10 ul Streptavidin-Meerrettich-Peroxidase)
- Waschen Membran zweimal in 40 ml PBS + 10% SDS (20 ml PBS + 20 ml 20% SDS) für 5 min.
- Waschen Membran zweimal in 40 ml PBS + 1% SDS (38 ml PBS + 2 ml 20% SDS) für 5 min.
- Waschen Membran zweimal in 40 ml PBS + 0,1% SDS (40 ml PBS + 200 ul 20% SDS) für 5 min.
- Entfernen Sie überschüssige Flüssigkeit, indem Membran zwischen zwei saubere Papierhandtücher und Druck auf sie fest.
- Visualisieren Membran-gebundenen HRP mit ECL den Anweisungen des Herstellers.
- Legen Sie die Membran in Folie / Beutel, Luftblasen entfernen und Inkubation für 2 min im Dunkeln.
- Expose MembranFilm für 1 - 5 min.
5. Trennung von markierten und unmarkierten RNA unter Verwendung von Streptavidin-beschichteten magnetischen Kügelchen
- Wärme Waschpuffer (3 ml pro Probe) bis 65 ° C in einem Wasserbad inkubiert.
- Bereiten Sie frischen 100 mM Dithiothreitol (DTT) in Nuklease-freiem H 2 O. Tun Sie dies durch Dekantieren 15 - 30 mg DTT Pulver in ein sauberes 50 ml Falcon-Röhrchen auf der ultra-feinen Waage gelegt. Wiegen und fügen erforderliche Menge an Nuklease-freiem H 2 O.
- Wärme biotinylierte RNA-Proben auf 65 ° C für 10 min zur Denaturierung und sofort auf Eis stellen.
- Platz μMacs Spalten in die magnetische Halterung. Wir empfehlen, nicht mehr als 12 Proben in einer Zeit (6 - 8 Proben sind optimal) zu verarbeiten.
- Pre-Abgleichtransistor Miltenyi Säulen mit 1 ml Waschpuffer Raumtemperatur. Dies dauert etwa 15 min.
- Inzwischen werden 100 ul Streptavidinbeads zu 50 - 100 ul biotinylierten RNA. Inkubieren bei Raumtemperatur für 15 min mit der Drehung. < li> Wenn eine der Spalten nicht eingeleitet hat Ablassen von nun dies kann durch leichten Druck auf die Spitze der Säule mit einem behandschuhten Finger erleichtert werden. Sobald die Strömung hat begonnen die Säulen leicht abfließen kann.
- Übernehmen Sie die RNA / Perlen an den Säulen. Verwerfen Sie den Durchfluss durch es sei denn, Sie wollen die unmarkierten RNA-Fraktion (siehe Abschnitt 7) zu erholen.
- Waschen dreimal mit 0,9 ml 65 ° C Waschpuffer (1 ml Pipettenspitzen schrumpfen beim Pipettieren Puffer bei 65 ° C).
- Dreimal mit 0,9 ml Waschpuffer Raumtemperatur.
- Pipette 700 ul Puffer RLT (RNeasy MinElute Cleanup Kit, Qiagen) in neue 2-ml-Röhrchen und legen Sie sie unter den Spalten.
- Eluieren neu transkribierten RNA in die RLT-Puffer durch Zugabe von 100 ul 100 mM DTT zu den Spalten.
- Führen Sie eine zweite Elution Runde 3 min später in das gleiche Rohr durch das Hinzufügen eines weiteren 100 ul 100 mM DTT.
6. Wiederherstellung der neu transkribierten RNA
NHALT "> mit dem RNeasy MinElute Cleanup (Qiagen) nach Protokoll des Herstellers weiter. Elute in 25 ul Nuklease-freiem H 2 O. Measure RNA-Konzentrationen mit einem Spektralphotometer Nanodrop. Um den Bedarf auftauen und wieder einfrieren RNA vor dem Absenden zu vermeiden es zu einem High-Throughput-Assay, empfehlen wir die Vorbereitung unmittelbar nach dem neu transkribierten RNA cDNA gereinigt wird. Verwenden Sie 2,5 ul der neu transkribierten RNA in 20 ul cDNA-Synthese-Mix für cDNA-Synthese nach den Anweisungen des Herstellers. Führen qRT-PCR-Kontrollen unter Verwendung von 1 : 10 Verdünnungen der cDNA mix Shop RNA bei -80 ° C..7. Rückgewinnung von unbeschriftet, Unbound RNA (Optional)
Bei der ungebundenen RNA wiederhergestellt werden muss, sammeln und kombinieren die Flow-Through (nach Zugabe der RNA-Streptavidinbeads Lösung für die Spalten) und die erste Wäsche für anschließende Fällung. In der Regel genügt es zur Ausfällung nur 50% des ungebundenen RNA als this enthält> 80% des Ausgangsmaterials.
- Fügen Sie dem gleichen Volumen Isopropanol (kein Salz muss hinzugefügt wie der Waschpuffer enthält bereits 1 M NaCl werden).
- Zentrifuge bei 20.000 × g für 20 min bei 4 ° C. Überstand verwerfen.
- In ein gleiches Volumen 75% Ethanol, Zentrifuge bei 20.000 × g für 10 min bei 4 ° C, Überstand verwerfen.
- Spin kurz und entfernen Restethanol mit 200 ul Pipette.
- Spin kurz und entfernen restliche Ethanol mit 20 ul Pipette.
- Nicht in die RNA zu trocknen. Resuspendieren in 100 ul H 2 O. Gut mischen durch Auf-und Abpipettieren 5 - 6 mal. Inkubation bei 65 ° C für 10 min unter Schütteln und übertragen direkt zu Eis.
- Prüfen RNA-Qualität durch elektrophoretische Analyse auf RNA-Abbau auszuschließen.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
1. Ausgangsmaterial und erwartete Renditen
Nach 1 Std. (hr) von 4SU-Exposition neu transkribierten RNA entspricht etwa 1-4% der gesamten zellulären RNA. Dies wird niedriger sein Wachstum verhaftet Zellen, da sie nicht mehr synthetisieren RNA zur Rechenschaft für das Zellwachstum / Replikation. Als Kennzeichnung für 1 Stunde, empfehlen wir der Testdurchführung mit 60 - 80 ug Gesamt-RNA. Beginnend mit weniger als 30 ug Gesamt-RNA Ergebnisse in kleinen RNA-Pellets, die schwer nach der Biotinylierung Schritt sehen sind und somit kann leicht verloren gehen. Eingangs-RNA-Spiegel kann bis zu 150 ug für sehr kurze Dauer der Kennzeichnung (- 10 min Beispiel 5) erhöht werden. Wenn die Dauer der RNA-Markierung von 1 Stunde bis 5 Minuten der Beitrag der kurzlebigen Intron-Sequenzen in RNA transkribiert neu steigt von ~ 60% ~ 80% 9 verkürzt. Wie Introns wesentlich länger zu codierenden Sequenzen sowie 5'-und 3'-UTRs verglichen werden, die Menge an neu transkribiertRNA, die nach kurz-oder sogar ultra-kurzen 4SU-Tagging gereinigt werden kann, nicht linear sinken. Als solche erhalten wir> 0,5% der Gesamt-RNA nach 5 min von 4SU-Tagging in nicht-haftenden menschlichen B-Zell-Linien 9. Es sollte jedoch beachtet werden, dass eine höhere Konzentration von 4SU und etwas länger Dauern Kennzeichnung erforderlich sein, um ähnliche 4SU Einbauraten in anhaftenden Zellen erreichen werden. Während selbst eine geringe 4SU-Einbaurate ermöglicht effiziente Erfassung und Reinigung von großen, Uridin-reiche Transkripte, sehr kurze Transkript mit niedrigen Uridingehalt (zB miRNAs) sind wahrscheinlich, um die Reinigung zu entkommen, auch wenn hohe 4SU Konzentrationen (> 1 mM). In NIH-3T3 Maus-Fibroblasten, die mit 1 hr von 200 pM 4SU Exposition neu transkribierten RNA mit etwa einem 4SU Rückstand pro 50 - 100 Nukleotiden (nt) 5. Dies sollte ermöglichen hocheffiziente Rückgewinnung Transkripte> 500 - 1000 nt lang. Dementsprechend haben wir nur eine untergeordnete beobachtet TranskriptgrößeBias bei der Etikettierung für 1 h mit 200 uM 4SU in beiden Maus-Fibroblasten und humanen B-Zellen 7. Während 1 h von 200 pM 4SU ergaben sich keine signifikanten Veränderungen in der zellulären Transkripte Ebenen in Maus-Fibroblasten, verlängerte Exposition von Zellen gegenüber ≥ 200 pM 4SU führen führt zwar zu einer messbaren Wachstum Defizit innerhalb von 24 h (unveröffentlichte Daten). Deshalb sollten sowohl die Dauer der Kennzeichnung und der Arbeitnehmer 4SU-Konzentration minimiert werden, um ektopische oder toxische Wirkungen zu vermeiden. Ein einfacher Weg, um die minimale 4SU-Konzentration für eine effiziente Verwertung der neu transkribierten RNA erforderlich zu bestimmen, ist neu transkribierten RNA nach 4SU-Kennzeichnung mit steigenden Konzentrationen von 4SU (zB 50-1600 uM) reinigen. Wie in den 2A und 2B gezeigt, die Verwertung von neu transkribierten RNA für 1 Stunde in primären humanen Fibroblasten markiert deutlich erhöht von 50 bis 200 pM 4SU aber dann begann Plateau.
2. DotBlot Quantifizierung 4SU Incorporation (optional)
In manchen Fällen kann es von Interesse sein, die Menge an 4SU Einarbeitung in Gesamt-RNA zu messen. Dies wird am besten durch Dot-Blot-Analyse an den biotinylierten RNA unter Verwendung eines Streptavidin-Konjugat durchgeführt. Aufgrund seiner chemischen Natur Iodacetyl-Biotin ist reaktiven Thiolgruppen als Biotin-HPDP was in der Biotinylierung von nahezu allen 4SU Rückstände in neu transkribierten RNA. Es ist wichtig zu beachten, dass, wie Biotin-HDPD, Iodacetyl-Biotin nicht wasserlöslich ist und damit effizient durch Chloroform-Extraktion, wie für Biotin-HPDP durchgeführt entfernt. Daher kann identischen Reaktionsbedingungen und Konzentrationen als bei der Verwendung von Biotin-HPDP eingesetzt werden. Allerdings ist iodoacetyl-Biotin nicht umkehrbar. Es kann daher nicht für die Reinigung von neu transkribierten RNA in Spalte Ansätze verwendet werden. Während die Verwendung von Iodacetyl-Biotin ermöglicht 4SU-Einbau quantifizieren, betrachten Biotin-HPDP basierte Messungen sowohl4SU-Einbau und Biotinylierung Effizienz. Unter Verwendung der zwei Biotinylierungsreagenzien zu derselben Probe ermöglicht die Messung der Biotinylierung von RNA-eingebautes 4SU. Biotinylierung Effizienz von Biotin-HPDP für 4SU-markierten RNA scheint etwa um das Dreifache kleiner als der Iodacetyl-Biotin anzeigt, dass nur etwa ein Drittel der Reste in 4SU neu transkribierten RNA tatsächlich von Biotin-HPDP (Abbildung 3) biotinyliert. Durch den Vergleich der Probe Signalintensitäten mit dem biotinylierten Oligo-DNA kann Biotinylierung Dichten gemessen werden. Für die meisten Säugetier-Zelllinien ein positives Signal sollte noch in 10 ng biotinylierte RNA nach 1 h von 200 pM 4SU Kennzeichnung nachweisbar. Ein schwaches Signal Hintergrund ist in der Regel nachweisbar für die höchste Konzentration (1 ug) von unmarkiertem RNA.
3. Reinigung von Neu transkribierten RNA
Rückgewinnung von neu transkribierten RNA ist sehr Quanquantitative. Wenn Sie mit der gleichen RNA-Konzentration gestartet kann man erwarten, dass die gleichen Mengen an neu transkribierten RNA für alle Proben zu erhalten. 260 nm (Anwesenheit von Detergenzien aus den Waschpuffern abgeleitet), die mit OD 260 Messungen stören kann - wie viele Spalten-basierte Assays kann Sammlung von neu transkribierten RNA mit dem RNeasy Kit MinElute in zusätzliche Absorption bei 230 führen. Dies führt zu einem geringeren Ausmaß beobachtet, wenn man eine neue 2-ml-Röhrchen für jede Zentrifugationsschritt. Dennoch sollten alle unverhältnismäßig hohen OD-Messungen (> 2-fach größer als die anderen Proben) mit Vorsicht betrachtet werden, insbesondere wenn OD 260/280 Verhältnisse <1,7 sind. Für Down-Stream-Analysen ist es daher oft am besten, um die gleiche Menge an RNA-Template Lautstärke für alle Proben zu verwenden. In Fällen, in denen die Erträge der markierten RNA sind niedriger als erwartet Check für Anzeichen von RNA-Abbaus durch elektrophoretische Analyse. Neu transkribiert RNA enthält deutlich größere Mengen von großen, ungespleißte Transkripte mit den typischen rRNA-Banden viel weniger prominent (Abbildung 4).
4. Quantifizierung der neu transkribierten RNA
Schließlich können Einbauraten 4SU in neu transkribierten RNA beispielhaft direkt durch spektrophotometrische Analyse auf dem Absorptionsmaximum des 4SU bei 330 nm und die OD 330/260 Verhältnis 5,18 basierend quantifiziert werden. Dies erfordert> 3 ug markierte RNA in einem kleinen Volumen (10 - 20 ul) konzentriert durch Isopropanol / Ethanol-Fällung. Um zu vermeiden, verlieren die kleinen RNA-Pellet Co-Fällung mit 30 ug Nuklease-freies Glykogen (Fermentas, # R0551) durchgeführt werden soll. Ein weiterer Höhepunkt ist sichtbar bei 330 nm spiegelt die Einbaurate von 4SU in neu transkribiert RNA (Abbildung 5).
/ Files/ftp_upload/50195/50195fig1highres.jpg "/>
Abbildung 1. . Zeit bei der Vorbereitung der gesamten zellulären RNA gefolgt - Grundsatz der metabolischen Markierung mit 4-Thiouridin (4SU) 4SU wird, um Zellen für die gewünschte (120 min 5) gegeben. Nach thiolspezifischen Biotinylierung wird gesamten zellulären RNA in 4SU-markierten, neu transkribierten RNA und unmarkierten, bereits bestehenden RNA unter Verwendung von Streptavidin-beschichteten magnetischen Kügelchen getrennt. Neu transkribiert RNA wird von den Perlen mit einem Reduktionsmittel, das die Disulfidbrücken, die die neu transkribierten RNA Link zu den Perlen spaltet erholt. Klicken Sie hier, um eine größere Abbildung anzuzeigen .
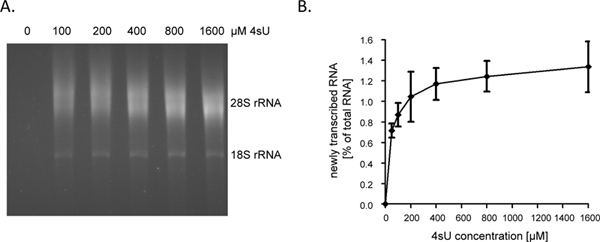
Abbildung 2. Rückgewinnung von neu transkribierten RNA nach steigenden Konzentrationen von 4SU. (A) Primäre humane Vorhaut-Fibroblasten (HFF) wurden mit 100 inkubiert, 200, 400, 800 oder 1.600 uM 4SU. Neu transkribierten RNA wurde aus 50 ug gesamten zellulären RNA gereinigt und einer elektrophoretischen Analyse. Wie erwartet, wurde eine konzentrationsabhängige Erhöhung wieder neu transkribierten RNA beobachtet, begann Plateau bei höheren Konzentrationen. (B) Mengen gereinigten neu transkribierten RNA quantifiziert wurden mit dem ImageJ 1.45s Software. Kombinierte Daten von vier unabhängigen Experimenten über die Höhe der neu transkribierten RNA erholt nach verschiedenen Konzentrationen von 4SU-Kennzeichnung von entweder 50 bis 800 pM 4SU (n = 2) oder 100 -. 1.600 uM 4SU (n = 2) dargestellt Klicken Sie hier um eine größere Abbildung anzuzeigen .
upload/50195/50195fig3.jpg "alt =" 3 "fo: content-width =" 4.5in "fo: src =" / files/ftp_upload/50195/50195fig3highres.jpg "/>
Abbildung 3. Schätzung 4SU Einbau in 4SU-markierten RNA-Dot-Blot-Analyse unter Verwendung. Gesamt-RNA aus NIH-3T3-Fibroblasten oder menschliche Vorhaut-Fibroblasten (HFF) mit 200 uM 4SU für eine Stunde inkubiert isoliert. Keine 4SU wurde zu einem Gericht als negative Kontrolle aufgenommen. Für beide HFF Kontakt gehemmt (n = nicht-wachsende Zellen) und wachsende Zellen (y) wurden eingeschlossen. RNA wurde unter Verwendung von Trizol-Reagenz und anschließend an Biotin konjugiert-HPDP oder Iodacetyl-Biotin. Jeder Probe wurde auf 200 ng / ul und 5 ul dieser Verdünnung (dh 1 ug RNA), sowie drei weiteren 10fache Verdünnungen (dh 100, 10 und 1 ng RNA bezeichnet) eingestellt waren entdeckt auf einem Stück Zeta Membran. 5 ul Verdünnungen von Biotin-markierten DNA-Oligo wurden auf der Membran als positive Kontrollen an konzen platziertIonen im Bereich von 20 ng / ul bis zu 20 pg / ul (dh 100 bis 0,1 ng, respectively). Biotin Dichte wurde sondiert mit einem Streptavidin-Meerrettichperoxidase-Konjugat.
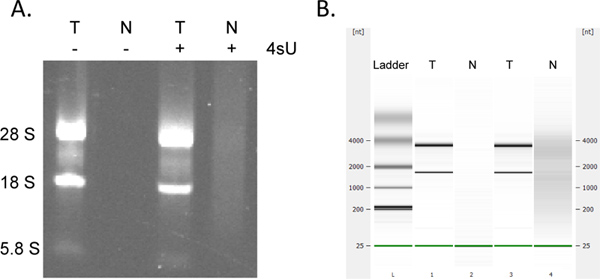
Abbildung 4. Elektrophoretische Analyse der neu transkribiert und RNA. Gesamt-RNA (T) und neu transkribierten RNA (N) aus murinen NIH-3T3-Fibroblasten sowohl in Gegenwart und Abwesenheit von 500 uM 4SU für 1 h wurde durch Agarose-Gelelektrophorese analysiert kultiviert (A) hergestellt und (in der Reihenfolge) unter Verwendung des Agilent Bioanalyzer (B). Kein RNA wurde ohne 4SU Behandlung von Zellen gewonnen. Gereinigt neu transkribiert RNA enthält größere Mengen an hochmolekularen mRNAs und deutlich weniger reifen rRNAs als insgesamtRNA als bemerkenswerte zwischen dem 28S, 18S und 5.8S rRNA-Banden. Klicken Sie hier, um eine größere Abbildung anzuzeigen .
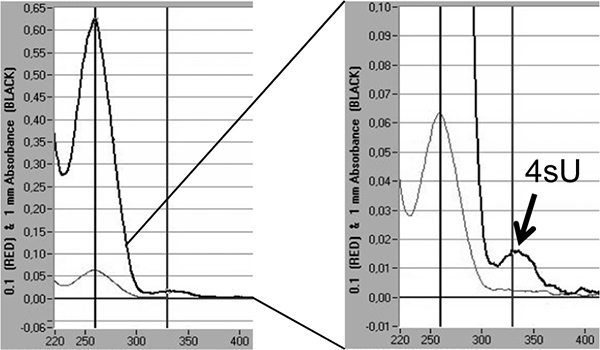
Abbildung 5. Quantifizierung der 4SU Einbau in neu transkribierten RNA durch spektrophotometrische Analyse. Neu transkribierten RNA aus 2 x 100 ug Gesamt-RNA nach 1 h von 200 pM 4SU in murinen NIH-3T3 Fibroblasten gereinigt. Neu transkribierten RNA wurde mit Isopropanol / Ethanol nach Zugabe von 30 ug Nuklease-freies Glykogen gefällt. Spektrophotometrische Untersuchung von neu transkribierten RNA durch eine Nanodrop 1000 Spektralphotometer erhalten wird gezeigt. Die hellgrauen Linien repräsentieren Messungen bei 0,1 mm, während die dickeren, dunkelgrauen Linien repräsentieren Messungen bei 1 mm Flüssigkeitssäule. Auf der rechten Seite, eine Vergrößerung der Spitze des Aussterbens, die ter integriert 4SU-Rückstände gezeigt wird. Basierend auf dem Extinktionskoeffizienten von 18 4SU die Einbauraten 4SU geschätzt werden kann.
| Dauer der Kennzeichnung [min] | Empfohlene 4SU Konzentration [um] |
| 120 | 100-200 |
| 60 | 200 - 500 |
| 15 - 30 | 500 - 1000 |
| <10 | 500 - 2000 |
Tabelle 1. Empfohlene 4SU Konzentrationen.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Metabolische Markierung von neu transkribierten RNA wesentlich verbessert die Leistung von High-Throughput-Technologien wie Microarrays und RNA-seq durch die Bereitstellung geeigneter Vorlagen, um die biologische Frage von Interesse anzusprechen. Das vorliegende Protokoll umfangreich Optimierung. Es erlaubt> 1000-fache Anreicherung von neu transkribierten RNA und bietet hoch reproduzierbare Ergebnisse.
Der experimentelle Aufbau eines 4SU-Tagging Experiment ist von entscheidender Bedeutung, wie neu transkribierten RNA wird die Echtzeit-Transkriptions-Aktivität nur während der Zeit der Exposition von Zellen gegenüber 4SU darzustellen. Wenn die tatsächlichen Änderungen in Transkriptionsraten nach einem Stimulus bereits abgeklungen sind, werden diese verpasste bei der Analyse neu transkribierten RNA, obwohl die Veränderungen in der Gesamt-RNA Ebenen eventuell noch nachweisbar sein. Daher ist ein gutes Verständnis der zugrunde liegenden Biologie wichtig, den Versuchsaufbau sowie die optimale Perioden o definierenf Zeit für 4SU Exposition. Nachfolgend geben wir Empfehlungen und Möglichkeiten zur Vermeidung häufigsten Fallstricke für die wichtigsten Schritte.
Vorbereitung der Stammlösungen und Plastmetall
Alle Stammlösungen müssen bereit sein, mit Nuklease-freiem Wasser werden. Mit in-house gereinigt deionisiertem Wasser kann zu Problemen führen, wenn das Wasser enthält Reduktionsmittel. In einem Fall führte dies zu einem vollständigen Verlust aller markierten RNA. Daher empfehlen wir den Kauf vorgefertigte nukleasefreiem NaCl, Tris-Cl, EDTA, Natriumcitrat und Wasser. Stellen Sie sicher, Nuklease-freien Bedingungen zu allen Zeiten. Dimethylformamid (DMF) löst einige Kunststoffe. Wir fanden, dass unter Verwendung von 25 ml Zellkultur Kunststoffpipetten zu DMF aus dem Bestand Glasflasche übertragen, um 50 ml Falcon-Röhrchen, die Biotin-HPDP Stammlösung war ausreichend, um deutlich reduzieren Ausbeuten der neu transkribierten RNA aus dem gesamten Test. Interessant ist, dass dies nicht negativ auf die biotinylation Effizienz (wie durch Dot-Blot getestet), sondern führte zu einer 75 bis> 90% Verlust der neu transkribierten RNA, die aus den Perlen zurückgewonnen werden konnte. Der Verlust war am stärksten ausgeprägt, wenn die Dauer der Kennzeichnung von 60 auf 30 min oder weniger reduziert wurde. Höchstwahrscheinlich eine Substanz aus der Kunststoff-Pipetten von der DMF eluiert teilweise zerstört die Beschichtung der Streptavidinbeads. Daher sollte die Verwendung von Kunststoff-Materialien nicht bekannt zu sein, mit DMF mit allen Mitteln vermieden werden. Aus den gleichen Gründen sollten Zellschaber nicht zur Gewinnung von Proben aus Trizol Zellkultur-Platten zu verbessern. Es ist interessant festzustellen, dass die mutmaßlichen Substanzen aus den Kunststoffen nach dem DMF oder Trizol eluiert wurden offensichtlich weder durch Chloroform-Extraktion oder Isopropanol / Ethanol-Fällung entfernt.
Zellkultur
Zelldichte auf den Platten ist von entscheidender Bedeutung. In einem Experiment, bei dem die Zellen erwies sich als leicht zu konfluenten (90 -100%), behandelten wir NIH-3T3-Fibroblasten für 30 min mit 100 U / ml Interferon (IFN) α oder γ. In weniger konfluenten Zellen noch 15 min von IFN-Behandlung führte bereits in einem 5 - bis 8-fache Induktion von Genen wie IRF1 oder SOCS3 5. Mit Zellen geringfügig zu konfluenten Microarray-Analyse zeigte keine Induktion von IFN-induzierbaren Genen selbst für die rasch induzierbare Gene wie IRF1 oder SOCS3. Daher ist Zelldichte ein entscheidender Faktor für 4SU-Kennzeichnung Experimente und alle Zellkultur-Platten sollten sorgfältig, bevor Sie die Kennzeichnung untersucht werden.
4SU ist ein photoaktivierbares Ribonukleosid und 4SU-haltige RNA effizient, um Proteine nach der Exposition gegenüber 365 nm Lichtquelle vernetzt. 4SU-behandelten Zellen sollten im Dunkeln und der Exposition gegenüber hellem Licht kultiviert werden sollten vermieden werden. Nach der Entfernung der zellulären Proteine durch Trizol RNA Isolierung dieses Risiko wesentlich reduziert wird.
<p class = "jove_content"> 4SU nicht in zelluläre DNA eingebaut. Es sollte jedoch angemerkt werden, dass die gesamten RNA werden noch geringe Mengen der zellulären DNA werden. Bei Verwendung 4SU-Tagging und q-RT-PCR-Analyse, um virale Genexpression in Cytomegalovirusinfektion studieren fanden wir es notwendig, einen DNaseI Digest Schritt in dem Protokoll zu entfernen konkatemeren der viralen Genomen 19 umfassen. Dies ist wahrscheinlich nicht notwendig, wenn stromabwärts Protokolle, die nicht empfindlich gegenüber der Anwesenheit von DNA.4SU Einbauraten und optimale Konzentration 4SU
4SU wird leicht von Zellen mit intra-und extrazellulären Ebenen wahrscheinlichste Gleichgewicht innerhalb weniger als einer Minute 9,16 gemacht. Die Aufnahme und Eingliederung von 4SU Preise sind konzentrationsabhängig. Daher kann 4SU Konzentration bequem nach der Dauer der Kennzeichnung eingesetzt eingestellt werden. Tabelle 1 berät 4SU Konzentrationen in relation auf die Dauer der Kennzeichnung auf der Grundlage unserer besten persönliche Erfahrung. Für 1 h 4SU Kennzeichnung in Säugerzellen wird 200 uM 4SU ausreichend für die meisten Anwendungen, die sich in etwa ein 4SU Rückstand pro 50 bis 100 Nukleotiden in neu transkribierten RNA in Fibroblasten.
In den letzten paar Jahren haben wir 4SU-Tagging, um ein breites Spektrum von Zelltypen des Menschen und der Maus Ursprungs, einschließlich Fibroblasten, Endothelzellen, Epithelzellen, Knochenmarkstroma Zellen, Makrophagen und T-Zellen aufgetragen. Zusätzlich wurden Zellen von Drosophila und Xenopus erfolgreich eingesetzt. In all diesen Experimenten wurde 4SU Aufnahme sich als sehr effizient bei minimaler Anpassungen 4SU Konzentration für die unterschiedlichen Zelltypen. Beim Einrichten der Verfahren für neue Zelltypen, würden wir zu beschriften Zellen mit steigenden Konzentrationen 4SU-(zB von 50 bis 1600 um) empfehlen und zu analysieren, die die Beziehung von gereinigtem neu transkribiert RNA auf die angelegten 4SU-Konzentrationen (siehe Abbildung 2A / B). Die 4SU-Konzentration bei der die Menge an gereinigtem neu transkribierten RNA tritt ein Plateau zu wählen.
In den Fällen, in denen sich leicht konfluent, Kontakt gehemmt Zellen verwendet werden, würden wir empfehlen, etwas höher 4SU Konzentrationen (zB 500 statt 200 pM) verwenden, um eine effiziente 4SU Einarbeitung zu gewährleisten. Zusätzlich kann in Fällen, in denen Erfassung von sehr kurzen neu transkribierten Transkripte (<200 nt) ist von besonderem Interesse, kann die Konzentration 4SU müssen auch erhöht werden. Dies sollte nicht mit längerer Kennzeichnung Zeiten (z. B.> 1 h) kombiniert werden, um ektopische Wirkungen oder Toxizität zu vermeiden. Schließlich fanden wir, dass die Verwendung zu kleinen Volumen von Zellkulturmedien kann 4SU Einbaueffizienz reduzieren. Wir empfehlen daher, unter Verwendung von 5 ml oder 10 ml Medium pro 10 cm oder 15 cm Schüssel, beziehungsweise.
Vorbereitung der gesamten zellulären RNA
Für den Erfolg dieses Protokolls ist es entscheidend, saubere, RNase-frei gesamte zelluläre RNA zu erhalten. Mit 5 ml Trizol pro 15 cm Schale erzeugt saubere RNA frei von Nukleasen. Wir empfehlen die Verwendung der modifizierten Trizol Protokoll Chomczynski et al. 20. Erstens ist es besser geeignet, um große Mengen von RNA (> 100 ug) als erweiterte Zentrifugalkraft Ergebnisse in fester Pellets, die einfacher zu handhaben während der Waschschritte zu isolieren. Dies erfordert jedoch den Einsatz spezieller Polypropylen-Röhrchen und Adapter als die regulären 15 ml Falcon-Röhrchen Labor nicht überleben mehr als 6.000 × g zentrifugiert. Zweitens verbessert es die Entfernung von DNA und Glykoproteine. Dies wird besonders deutlich, wenn die Vorbereitung RNA von Organen oder Geweben. Drittens ist es nicht die maximale Menge an Gesamt-RNA, die isoliert werden können. Obwohl wir fanden auch spaltenbasierte RNA-Isolierung Methoden (z. B. RNeasy), um RNA von geeigneter Qualität, Standard Spalten are nur in der Lage zu erfassen bis zu 100 ug Gesamt-RNA wodurch die Menge des Ausgangsmaterials. Schließlich wird durch das Entfernen der restliche Ethanol zweimal mit einer Pipette, das Trocknen der RNA, um restliches Ethanol zu entfernen nicht mehr erforderlich. Dadurch wird die Gefahr von Über-Trocknen der RNA, die nur schwer zu lösen kann danach wieder. Grundsätzlich ist 4SU-Kennzeichnung für in vivo, zB durch intravenöse Injektion von Mäusen. Allerdings haben wir festgestellt, dass RNA Reinheit ein großes Problem erfordert die Reinigung von poly-Transkripte vor der Reinigung der neu transkribierten RNA (unveröffentlichte Daten) darstellt.
Biotinylierung und Entfernung von ungebundenen Biotin
Biotin-HPDP ist 100% thiol-specific und bildet eine Disulfidbindung zwischen dem Biotin-Rest und Thiol-markierten RNA-Moleküle transkribiert neu. Biotinylierung Effizienz 4SU-markierten RNA beträgt etwa 30%, wie durch Dot-Blot-Analyse 5 bestimmt. Als Biotin-HPDP ist nicht wasserlösliches kann wirksam durch Chloroform-Extraktion entfernt werden. Während ein einzelner Chloroformextraktion Schritt ausreicht, um die überwiegende Mehrheit der ungebundenen Biotin entfernen wir regelmäßig wiederholen Sie diesen Schritt, um eine vollständige Entfernung zu gewährleisten. Um RNA Verlust während der Chloroform-Extraktion Schritt 2 ml Phase Lock Gel Schwere Rohre (Eppendorf) reduzieren kann nach den Anweisungen des Herstellers verwendet werden. Normalerweise verwenden wir die Phase-Lock-Röhren nur für die zweite Chloroformextraktion Schritt als Vorlage Volumen der ersten Stufe sind oft zu hoch, um direkt mit diesen Rohren. Nach Entfernung des ungebundenen Biotin-HPDP wird RNA von Isopropanol / Ethanolfällung gewonnen. Es ist wichtig, dass die kommerziellen spaltenbasierte Kits beachten, um die biotinylierten RNA (z. B. RNeasy von QIAGEN) erholen sollte nicht verwendet werden, da sie Reduktionsmittel in den Puffer vorgesehen, die Spaltung der Disulfidbrücke und enthalten entfernen Sie das Biotin aus dem neu transkribierten RNA werden .
Die Reinigung des neuenly transkribierten RNA
Fügen Sie nicht mehr als 100 ul biotinylierten RNA zu 100 ul Streptavidinbeads. Hinzufügen weniger Volumen wird bevorzugt. Allerdings sollte das gleiche Volumen an RNA für alle Proben zugesetzt werden. Stellen Sie die RNA-Eingang Lautstärke (zwischen Proben), die Sie auf die Streptavidinbeads hinzufügen, indem Sie einfach das gewünschte Volumen 1x TE zu den Perlen. Ein einfacher Weg, um frische Nuklease-freies 100 mM DTT zu machen ist, um eine ausreichende Menge an DTT Pulver in ein Falcon-Röhrchen auf einem ultra-empfindliche Waage gelegt umfüllen und fügen Sie dann die erforderliche Menge an Nuklease-freiem H 2 O auf 100 mM DTT erzeugen (64,8 ul Wasser pro 1 mg DTT).
Während der Entwicklung von 4SU-Tagging wir getestet Streptavidinbeads von verschiedenen Lieferanten. Eine Reihe von ihnen erzeugt große Mengen Hintergrund. Daher empfehlen wir Ihnen, die Miltenyi Streptavidinbeads als bisher haben wir noch nie irgendwelche Probleme mit Verschleppung von unmarkierten RNA aus Gewebe c erlebtulture abgeleiteten RNA-Proben. Auf diese Weise kann so wenig wie 150 ng der markierten RNA speziell von 150 ug biotinylierten RNA (in 100 l Wasser) gereinigt werden mit 100 ul Streptavidinbeads. Äquilibrierung der Beads mit dem Equilibrierungspuffer mit den Kügelchen geliefert durchgeführt werden und kann leicht verbessern Erfassungsraten 13.
Qualitätskontrollen
Wir empfehlen die Durchführung q-RT-PCR-Kontrolle neu transkribierten RNA lassen, bevor er für das Hochdurchsatz-Analysen. Dies kann auch die Quantifizierung von mehreren Referenz-Gene bekannt, unterschiedlich in der gegebenen experimentellen Einstellung geregelt werden. In Fällen, in denen 4SU-Tagging verwendet, um RNA Zerfallsraten Studie ist, empfehlen wir, eine kurzlebige Transkript (zB myc, fos) und einen langlebigen ein (zB GAPDH) in beide total und neu transkribierten RNA quantifizieren. Das Verhältnis von neu transkribierten / Gesamt-RNA sollte erheblich höher (ca. 5 - bis 10-fach)für die kurzlebigen Transkripte. Basierend auf dem RNA-Halbwertszeit eines Referenz-Gen kann RNA Halbwertszeiten bestimmt werden. Wenn alle drei RNA-Fraktionen (Gesamt-RNA, neu transkribierten RNA und unmarkierten bereits vorhandenen RNA) für vier oder mehr Gene, die Normalisierung der verschiedenen RNA Teilmengen analysiert durch lineare Regressionsanalyse und Qualitätsbewertungen durchgeführt werden kann wie die 7 beschrieben , 21.
Für q-RT-PCR-Analyse, empfehlen wir die Verwendung von 2,5 ul markierte RNA in 20 ul cDNA-Synthese-Mix. Zur optimalen Vergleich der q-RT-PCR-Ergebnisse frieren die cDNA in Aliquots von 5 ul vor der ersten Nutzung. Thaw Rohre kurz vor der Verwendung, fügen Sie 45 ul H 2 O und vorbehaltlich 5 ul der Verdünnungen q-RT-PCR-Analysen. Dies erhöht die Vergleichbarkeit zwischen verschiedenen PCR Läufe.
Neu transkribierten RNA-Proben sollten auf Anzeichen von RNA-Abbau unter Verwendung des Agilent Bioanalyzer bevor sie einer überprüft werdenAnalyse mit hohem Durchsatz (Microarrays oder RNA-seq). Es sei jedoch darauf hingewiesen, dass zusätzliche Banden manchmal von der Agilent Bioanalyzer beachten. Die biologische Bedeutung dieser bleibt unklar. Wie neu transkribiert RNA enthält deutlich weniger ribosomalen RNA, diese Proben gelegentlich scheitern die Agilent Bioanalyzer Qualitätskontrollen. Ist dies nicht durch sichtbare RNA-Abbau Proben von akzeptabler Qualität sind in der Regel gut zu Analyse mit hohem Durchsatz unterworfen werden.
Kompatibilität von neu transkribierten RNA mit Down-Stream-Analysen
Neu transkribiert RNA enthält wesentlich mehr mRNA als Gesamt-RNA. Dies ist vor allem auf die größeren Mengen an Intronsequenzen in neu transkribierten RNA, wenn die Dauer der 4SU-Tagging wird verkürzt zu erhöhen. Deshalb können wir nicht regelmäßig verpflichten sich die Erschöpfung der rRNAs aus neu transkribierten RNA-Proben, da dies erfordert größere Mengen an Ausgangsmaterial während providing eher wenig (~ zweifach) Verstärkung in nicht-rRNA liest. Schließlich ist noch anzumerken, dass die größeren Prozentsatz ungespleißte mit hohem Molekulargewicht Transkripte in neu transkribierten RNA können zusätzliche Fragmentierung benötigen bei der Herstellung von cDNA-Bibliotheken für die nächste Generation Sequenzierung werden. Ergebnisse von der Größe Fragmentierung Schritt sollte daher sorgfältig qualitätsgeprüft.
Datennormalisierung für RNA Halbwertszeit Messungen
Der Standard-Ansatz zur experimentellen Daten für die RNA-Halbwertszeit Messungen normalisieren ist, um alle Daten zu dem RNA-Halbwertszeit von einem gut charakterisierten house-keeping-Gens oder des Median RNA Halbwertszeit in einem bestimmten Zelltyp in früheren Experimenten bestimmt normalisieren. In Säugetierzellen, liegt diese im Bereich von 5 bis 10 h 6,7. Während dieser Ansatz funktioniert auch ganz gut für 4SU-basierten Messungen, sind andere Mittel zur Normalisierung erforderlich, wenn der Median RNA Halbwertszeit nicht bekannt ist oder wenn es may auch durch Veränderungen in der zellulären untersuchten Systems beeinflusst werden, z. B. durch den Knock-out eines RNA Zerfallsweg. 4SU-Tagging bietet eine einzigartige Möglichkeit zur Abschätzung der Median RNA Halbwertszeit auf der Analyse aller drei RNA-Fraktionen, dh gesamten zellulären RNA, neu transkribierten RNA und unmarkiertem bereits vorhandene RNA basiert. Als zelluläre Gesamt-RNA wird in den beiden letztgenannten RNA-Fraktionen ein einfaches lineares Regressionsmodell verwendet, um die drei RNA-Fraktionen zueinander normalisieren und bestimmen den Mittelwert RNA Halbwertszeit 7,16 werden getrennt. Ein Software-Paket ist online verfügbar, um diese Analysen durchzuführen 22.
Ineffiziente Erfassung von Abschriften mit niedrigen Uridingehalt beeinträchtigen RNA Halbwertszeit Messungen was künstlich niedrig neu transkribiert / Gesamt-RNA-Verhältnisse und längere RNA-Halbwertszeiten. Das Ausmaß dieses Problem kann durch Auftragen RNA Halbwertszeiten oder log (neu transkribierten / Gesamt-RNA-Verhältnisse) gegen die uridi beurteilt werdenne Inhalten aller Transkripte 7,15. Dies stellt auch eine gute Qualitätskontrolle, um Unterschiede in 4SU-Einbauraten zwischen unterschiedlichen Proben oder Bedingungen zu beurteilen. In Fällen, in denen eine erhebliche Korrelation zu Uridingehalt beobachtet dies kann für durch bioinformatische bedeutet 15 korrigiert werden. Es sollte jedoch beachtet werden, dass der Beitrag der reifen Transkripte in neu transkribierten RNA nicht leicht von dem viel größeren und damit wesentlich Uridin-reiche Vorstufen unterscheiden werden. Sofern die Verarbeitung Kinetik einer bestimmten Transkript bekannt sind (die sie in der Regel nicht) einfach Korrigieren niedrig Uridingehalt (ineffizient capture) kann grob verzerren RNA Halbwertszeiten. Als solche haben wir vor kurzem gefunden Verarbeitung der meisten menschlichen snoRNAs als höchst ineffizient 9. Wenn wir die neu transkribiert / Gesamt-RNA-Verhältnisse für die geringe Uridingehalt der eher klein (70 bis 300 nt) korrigiert hatte snoRNAs, würde dies in extrem kurzer snoRNA halb liv geführt habenes (<5 min) mit zahlreichen neu transkribiert / Gesamt-RNA-Verhältnisse von mehr als 100%. Deshalb haben wir in der Regel nicht empfohlen Korrektur für niedrige Uridingehalt bei der Messung von RNA-Halbwertszeiten.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Die Autoren erklären, dass sie keine finanziellen Interessen haben.
Acknowledgments
Wir möchten Amie Regan für die sorgfältige Durchsicht des Manuskripts danken. Diese Arbeit wurde von NGFN Plus-Grant # 01GS0801, MRC Stipendium Zuschuss G1002523 und NHSBT Zuschuss WP11-05 LD und DFG FR2938/1-1 zu CCF unterstützt
Materials
| Name | Company | Catalog Number | Comments |
| 4-thiouridine | Carbosynth | T4509 | Prepare 50 mM stock in sterile H2O, store at -20 °C in aliquots of 50-500 μl, discard unused reagent, do not refreeze. |
| Trizol | Invitrogen | 15596026 (100 ml), 15596018 (200 ml) | WARNING - CORROSIVE and HAZARDOUS TO HEALTH! Ensure immediate access to Phenol antidote (PEG-Methanol); Store at 4 °C. |
| Chloroform | Sigma | 372978 | WARNING - HAZARDOUS TO HEALTH |
| Isopropanol | Sigma | 650447 | |
| Sodium citrate, nuclease-free | Sigma | C8532 | Prepare 1.6 M stock solution using nuclease-free water. |
| 5M nuclease-free NaCl | Sigma | 71386 | Stock solution |
| Nuclease-free H2O | Sigma | W4502 | Make 1 ml aliquots in nuclease-free tubes. |
| RNA precipitation buffer | 1.2 M NaCl, 0.8 M sodium citrate in nuclease free water. Prepare in advance under strictly nuclease-free conditions. Store at room temperature in 50 ml falcon tubes. | ||
| Ethanol | Sigma | 459844 | Use with nuclease-free water to prepare 80% ethanol, store at -20 °C. |
| 1 M nuclease-free Tris Cl, pH 7.5 | Lonza | 51237 | Stock solution |
| 500 mM nuclease-free EDTA, pH 8.0 | Invitrogen | 15575-020 | Stock solution |
| 10x Biotinylation Buffer (BB) | 100 mM Tris pH 7.4, 10 mM EDTA in nuclease-free water, make aliquots of 1 ml. | ||
| Dimethylformamide (DMF) | Sigma | D4551 | |
| EZ-Link biotin-HPDP | Pierce | 21341 | Prepare 1 mg/ml stock solution by dissolving 50 mg biotin-HPDP in 50 ml DMF. Gentle warming enhances solubilisation. Store at 4 °C in aliquots of 1 ml. |
| Phase Lock Gel Heavy tubes 2.0 ml | Eppendorf | 0032 005.152 | Optional for the chloroform extraction step. |
| Zeta membrane | BIORAD | 162-0153 | |
| 10x Dot blot binding buffer | 100 mM NaOH, 10 mM EDTA | ||
| Biotin-oligo | 5'-biotin, 25 nucleotides, any sequence | ||
| Sodium dodecyl sulphate | Fisher | BPE9738 | For 100 ml 20% stock solution, add 20 g SDS to 80 ml PBS pH 7-8 and adjust volume to 100 ml. Keep all high-percentage SDS solutions above 20 °C. Warm the solutions slightly should SDS precipitate. |
| EZ-Link Iodoacetyl-LC-Biotin | Pierce | 21333 | Prepare 1 mg/ml stock solution by dissolving 50 mg iodoacetyl-biotin in 50 ml DMF. Gentle warming enhances solubilisation. Store at 4 °C in aliquots of 1 ml. Generates irreversible, thiol-specific biotinylation. |
| Phosphate buffer saline | Gibco | 10010-015 | |
| Dot blot blocking buffer | Mix 20 ml 20% SDS with 20 ml 1 x PBS pH 7-8 and add EDTA to the final concentration of 1 mM. | ||
| Streptavidin-horseradish peroxidase | Vector Laboratories | SA5004 | Store at -20 °C. Mix 10 ml 20% SDS with 10 ml 1 x PBS. Add 20 μl Streptavidin-HRP before use. |
| ECL reagent | GE Healthcare | RNP2109 | Use following the manufacturer's instructions. |
| Super RX, X-RA Film, 18x24 cm | Fujifilm | 47410 19236 | |
| μMacs Streptavidin Kit | Miltenyi | 130-074-101 | Store the beads at 4 °C. |
| Tween 20 | Sigma | P1379 | |
| Washing buffer | 100 mM Tris pH 7.4, 10 mM EDTA, 1 M NaCl, 0.1% Tween 20 in nuclease-free H2O. | ||
| Dithiothreitol (DTT) | Sigma | 43817 | Prepare as 100 mM DTT in nuclease-free H2O, always prepare fresh before use. |
| RNeasy MinElute Kit | Qiagen | 74204 | Store columns at 4 °C, remaining components of the kit at room temperature. |
| 1.5 ml screw-top polypropylene tubes | Sarstedt | 72.692.005 | Compatible with Dimethylformamide |
| 2.0 ml screw-top polypropylene tubes | Sarstedt | 72.694.005 | Compatible with Dimethylformamide |
| 15 ml tubes | BD Falcon | 352096 | Compatible with Dimethylformamide |
| 50 ml tubes | BD Falcon | 352070 | Compatible with Dimethylformamide |
| All solutions/reagents should be stored at room temperature unless otherwise specified. | |||
| Equipment | |||
| UV/VIS spectrophotometer | Thermo Scientific | NanoDrop 1000 | Or equivalent. Use low volume (1-2 μl) for measurements of low RNA concentrations to avoid excessive sample loss. |
| Polypropylene 15 ml centrifuge tubes | VWR International | 525-0153 | In contrast to standard 15 ml tubes, these tolerate up to 15,000 × g |
| High-speed centrifuge | Beckman Coulter | Avanti J-25 | Or equivalent equipment capable of reaching 13,000×g |
| High-speed rotor | Beckman Coulter | JLA-16250 | Or equivalent equipment capable of reaching 13,000×g |
| Adaptors for 15 ml tubes | Laborgeräte Beranek | 356964 | Or equivalent equipment capable of reaching 13,000×g |
| Refrigerated table-top centrifuge | Eppendorf | 5430 R | Or equivalent. |
| Thermomixer | Eppendorf | Thermomixer compact | Or equivalent. |
| Magnetic stand | Miltenyi Biotec | 130-042-109 | One stand holds 8 μMacs columns. |
| Waterbath | Grant | SUB Aqua 5 | Or equivalent. |
| Ultra-fine scale | A&D | GR-202 | Or equivalent. |
| E-Gel iBase Power System | Invitrogen | G6400UK | For RNA gels; or equivalent. |
| E-Gel EX 1% agarose precast gels | Invitrogen | G4020-01 | For RNA gels; or equivalent. |
References
- Brown, P. O., Botstein, D. Exploring the new world of the genome with DNA microarrays. Nature Genetics. 21, 33-37 (1999).
- Mortazavi, A., Williams, B. A., Mccue, K., Schaeffer, L., Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nature Methods. 5, 621-628 (2008).
- Nagalakshmi, U., Wang, Z., et al. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science. 320, 1344-1349 (2008).
- Wilhelm, B. T., Marguerat, S., et al. Dynamic repertoire of a eukaryotic transcriptome surveyed at single-nucleotide resolution. Nature. 453, 1239-U1239 (2008).
- Dölken, L., Ruzsics, Z., et al. High-resolution gene expression profiling for simultaneous kinetic parameter analysis of RNA synthesis and decay. RNA. 14, 1959-1972 (2008).
- Yang, E., van Nimwegen, E., et al. Decay rates of human mRNAs: correlation with functional characteristics and sequence attributes. Genome Res. 13, 1863-1872 (2003).
- Friedel, C. C., Dölken, L., Ruzsics, Z., Koszinowski, U. H., Zimmer, R. Conserved principles of mammalian transcriptional regulation revealed by RNA half-life. Nucleic Acids Res. 37, e115 (2009).
- Nilsen, T. W., Graveley, B. R. Expansion of the eukaryotic proteome by alternative splicing. Nature. 463, 457-463 (2010).
- Windhager, L., Bonfert, T., et al. Ultra short and progressive 4sU-tagging reveals key characteristics of RNA processing at nucleotide resolution. Genome Res. , (2012).
- Melvin, W. T., Milne, H. B., Slater, A. A., Allen, H. J., Keir, H. M. Incorporation of 6-thioguanosine and 4-thiouridine into RNA. Application to isolation of newly synthesised RNA by affinity chromatography. Eur. J Biochem. 92, 373-379 (1978).
- Cleary, M. D., Meiering, C. D., Jan, E., Guymon, R., Boothroyd, J. C. Biosynthetic labeling of RNA with uracil phosphoribosyltransferase allows cell-specific microarray analysis of mRNA synthesis and decay. Nature Biotechnology. 23, 232-237 (2005).
- Kenzelmann, M., Maertens, S., et al. Microarray analysis of newly synthesized RNA in cells and animals. Proc. Natl. Acad. Sci. U.S.A. 104, 6164-6169 (2007).
- Schwanhäusser, B., Busse, D., et al. Global quantification of mammalian gene expression control. Nature. 473, 337-342 (2011).
- Rabani, M., Levin, J. Z., et al. Metabolic labeling of RNA uncovers principles of RNA production and degradation dynamics in mammalian cells. Nat. Biotechnol. , (2011).
- Miller, M. R., Robinson, K. J., Cleary, M. D., Doe, C. Q. TU-tagging: cell type-specific RNA isolation from intact complex tissues. Nat. Methods. 6, 439-441 (2009).
- Miller, C., Schwalb, B., et al. Dynamic transcriptome analysis measures rates of mRNA synthesis and decay in yeast. Mol. Syst. Biol. 7, 458 (2011).
- Weintz, G., Olsen, J. V., et al. The phosphoproteome of toll-like receptor-activated macrophages. Mol. Syst. Biol. 6, 371 (2010).
- Lipsett, M. N. The isolation of 4-thiouridylic acid from the soluble ribonucleic acid of Escherichia coli. Journal of Biological Chemistry. 240, 3975-3978 (1965).
- Marcinowski, L., Liedschreiber, M., et al. Real-time Transcriptional Profiling of Cellular and Viral Gene Expression during Lytic Cytomegalovirus Infection. PLoS Pathog. 8, e1002908 (2012).
- Chomczynski, P., Mackey, K. Short technical reports. Modification of the TRI reagent procedure for isolation of RNA from polysaccharide- and proteoglycan-rich sources. Biotechniques. 19, 942-945 (1995).
- Friedel, C. C., Dölken, L. Metabolic tagging and purification of nascent RNA: implications for transcriptomics. Mol. Biosyst. 5, 1271-1278 (2009).
- Friedel, C. C., Kaufmann, S., Dölken, L., Zimmer, R. HALO - A Java framework for precise transcript half-life determination. Bioinformatics. , (2010).
