

Summary
RNA celular total fornece um modelo pobre para estudar mudanças de curto prazo na síntese de RNA e decadência, bem como a cinética de processamento de RNA. Aqui, descrevemos a marcação metabólica de RNA recentemente transcrito com 4-tiouridina seguido por biotinilação específica do tiol e a purificação do ARN recém transcritos que permitam ultrapassar estas limitações.
Abstract
O desenvolvimento de microarrays de todo o transcriptoma e sequenciamento de próxima geração que revolucionou a nossa compreensão da complexidade da expressão gênica celular. Juntamente com uma melhor compreensão dos mecanismos moleculares envolvidos, medições precisas da cinética subjacentes tornaram-se cada vez mais importante. Aqui, essas metodologias poderosas enfrentam grandes limitações devido às propriedades intrínsecas das amostras de modelos que estudam, ou seja, total de RNA celular. Em muitos casos, mudanças de RNA celular total de ocorrer ou muito devagar ou rápido demais para representar os eventos moleculares subjacentes e sua cinética com resolução suficiente. Além disso, a contribuição de alterações na síntese de ARN, o processamento, e da deterioração não são facilmente diferenciados.
Recentemente, desenvolveu perfis de alta resolução expressão gênica para superar essas limitações. A nossa abordagem baseia-se na marcação metabólica de RNA recentemente transcrito com 4-thiourijante (portanto também designado por 4SU-tagging) seguido por purificação rigorosa do ARN recém transcritos usando biotinilação específica do tiol e as esferas magnéticas revestidas com estreptavidina. A técnica é aplicável a uma ampla gama de organismos, incluindo vertebrados, Drosophila e levedura. Foram aplicados com sucesso 4SU codificação para estudar a cinética em tempo real das actividades de factor de transcrição, proporcionar medições precisas da RNA meia-vida, e obter novos conhecimentos sobre a cinética de processamento do ARN. Finalmente, a modelagem computacional pode ser empregue para gerar uma análise global integrado dos mecanismos moleculares subjacentes.
Introduction
Perfil de expressão gênica é uma ferramenta chave usada para estudar os processos celulares e da rede complexa interação associado. Estudos sobre a abundância de ARNm foram tipicamente o método de escolha para obter conhecimentos básicos sobre os mecanismos moleculares subjacentes. O desenvolvimento de microarrays de todo o transcriptoma 1 e, mais recentemente, o sequenciamento de próxima geração de RNA (RNA-seq) 2-4 alimentou esta abordagem. Embora estas tecnologias revolucionaram nossa compreensão da complexidade da expressão gênica celular, eles enfrentam grandes limitações devido às propriedades intrínsecas de sua amostra do modelo, ou seja, de RNA celular total. Primeiro, as variações a curto prazo nos níveis de ARN total não coincidem alterações nas taxas de transcrição, mas é inerentemente dependente do ARN de semi-vida dos respectivos transcritos. Enquanto uma indução quíntupla de uma transcrição de curta duração, por exemplo, que codifica para um factor de transcrição, serão facilmente detectável no ARN totaldentro de uma hora, a mesma indução de uma transcrição de longa duração, por exemplo, codificação de uma enzima metabólica, ficará praticamente invisível. Além disso, mesmo uma paragem completa (> 1000 vezes down-regulation) na taxa de transcrição de um gene da média com um ARN de semi-vida de cinco horas simplesmente demorar cinco horas para os seus níveis de RNA total de apenas diminuirá duplo . Assim, a análise do RNA total de favorece a detecção de sobre-regulação de transcritos de vida curta, muitos dos quais codificam para factores de transcrição e de genes com funções reguladoras 5. Além disso, a cascata está obscurecido cinética verdadeira de regulação e os eventos de sinalização primários não pode ser diferenciado do secundário. Ambos, por sua vez, pode resultar em viés substancial nas análises a jusante da bioinformática. Em segundo lugar, as alterações nos níveis de ARN total não pode ser atribuída a alterações na síntese de RNA ou deterioração. Medidas deste último requer abordagens invasivos de células, por exemplo, bloqueadores transcriptisobre o uso de actinomicina D 6, e monitorização da deterioração contínua estendida ao longo do tempo de RNA. Com um ARNm de semi-vida média das células de mamífero de 5 - 10 horas 5,7, níveis de ARNm da maioria dos genes somente terão diminuído de menos de duas vezes após várias horas de detenção transcripcional. Estas diferenças relativamente pequenas resultar em medições grosseiramente imprecisas de ARNm de semi-vida para a maioria dos genes celulares, devido à natureza exponencial das equações matemáticas subjacentes. Finalmente, enquanto que o RNA-seq de ARN celular total mostrou que aproximadamente metade dos nossos genes estão sujeitos a eventos de splicing alternativos 8, a cinética subjacentes, bem como os mecanismos dinâmicos orientadores de tecidos e específicos de regulação do contexto de processamento de ARN permanecem pouco compreendidos. Além disso, a contribuição de processamento do RNA para a expressão diferencial de genes, em particular para RNAs não-codificantes, permanece a ser determinada. Ao todo, estas limitações representam grandes obstáculos paramodelagem cinética bioinformática dos mecanismos moleculares subjacentes.
Recentemente, desenvolveu uma abordagem, o perfil de expressão do gene denominado de alta resolução, para ultrapassar estes problemas 5,7,9. Baseia-se na marcação metabólica de RNA recém transcrito usando 4 tiouridina (4SU-tagging), um derivado uridina que ocorre naturalmente, e oferece acesso directo às transcrições recém-transcritos com o mínimo de interferência no crescimento celular e expressão gênica (ver Figura 1) 5, 10-12. A exposição das células eucarióticas a resultados 4SU na sua absorção rápida, a fosforilação de 4SU-trifosfato, e incorporação de RNA recentemente transcrito. Após isolamento do RNA celular total, a fracção de ARN marcado é 4SU-tiol especificamente biotinilados gerar uma ligação de dissulfureto entre a biotina e os transcritos de RNA recentemente. "Total RNA celular", então pode ser quantitativamente separados em rotulada ('recentemente transcrito') e não marcado ("pré-existing ') ARN com elevado grau de pureza usando esferas magnéticas revestidas com estreptavidina. Finalmente, o ARN marcado é recuperado a partir das pérolas por simples adição de um agente redutor (por exemplo ditiotreitol) a clivagem da ligação dissulfureto e libertação do ARN recém transcritos a partir das pérolas.
RNA recém transcrito retrata a atividade transcricional de cada gene, durante o período de exposição 4SU. 4SU-tagging na escala de tempo de minutos, portanto, fornece um retrato instantâneo da expressão gênica eucariótica e um modelo ideal para a jusante análises de bioinformática (por exemplo, análise de promotor). Nos casos em que as condições de estado estacionário pode ser assumidas, os índices de recém-transcrita / total, o recém-transcrito / sem rótulo e sem rótulo / RNA total de proporcionar o acesso não-invasivo para RNA precisos meia-vida 7,13. Além disso, é importante notar que os transcritos de RNA recentemente purificados após tão pouco como 5 minutos de 4SU-marcação (5 min 4SU-RNA) é menor que 15 e 60 min 4SU-RNA.Ao realizar tanto ultra-curtos e progressivamente mais 4SU-tagging em uma única configuração experimental combinado com RNA-seq, a cinética de processamento de RNA são revelados na resolução de nucleotídeos 9. Finalmente, as análises de curso de tempo de RNA recém transcrito e total combinado com modelagem computacional permitem uma análise integrativa da síntese de RNA e decadência 14.
Em conclusão, esta abordagem permite a análise direta da dinâmica da síntese de RNA, processamento e degradação em células eucarióticas. É aplicável em todos os principais organismos modelo, incluindo mamíferos, insetos (Drosophila), anfíbios (Xenopus) e levedura 5,15,16. Ele é diretamente compatível com a análise de microarray 5,17, RNA-seq 9,13,14, e é aplicável in vivo 12,15. Aqui, vamos detalhar a metodologia de rotular, isolar e purificar RNA recém transcrito em células de mamíferos cultivadas. Além disso, potenciomêtroproblemas ai e armadilhas são discutidos.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. Marcação metabólica com 4-tiouridina
Faça um plano detalhado da instalação experimental / programação, por exemplo, quando adicionar o 4SU para cultura de células e quando a colheita das amostras. Plano de pelo menos 5 minutos entre cada condição. Apenas o tratamento de células de uma condição de cada vez. Lidar com máx. 3-5 pratos em um determinado momento. Pega células tão rapidamente quanto possível, para minimizar as alterações nos níveis de temperatura e de CO 2. Evite expor as células à luz brilhante após 4SU é adicionado como isso pode resultar em crosslinking de RNA 4SU-rotulados para proteínas celulares.
Início da rotulagem
- Thaw 4 tiouridina (4SU) imediatamente antes da utilização e pipeta quantidade necessária de 4SU para cada condição em um tubo Falcon estéril.
- Tome a quantidade necessária de meio de cultura celular (5 ml por 10 centímetros prato) fora os pratos e adicionar ao tubo Falcon 4SU contendo e misture bem. Remova e descarte o meio restante dos pratos. <li> Aplicar 4SU contendo meio de volta para os pratos.
Fim de etiquetagem
- Remova o meio de cultura de células a partir de células. Adicionam-se 5 ml de reagente Trizol para cada placa. Para as experiências de complexos, incluindo vários pontos de tempo ou condições, este passo é melhor realizado por duas pessoas, uma remoção do meio, a adição de outro reagente Trizol e colheita do ligado.
- Incubar durante 5 min a temperatura ambiente durante a lise celular completa.
- Usar uma pipeta de 10 ml para enxaguar a placa cuidadosamente com o Trizol adicionado. Isto ajuda a lise celular completa e a recuperação da amostra. Manusear com cuidado como Trizol é extremamente perigoso quando entrar em contato com a pele ou com os olhos! Tem antídoto para queimaduras de fenol na mão (por exemplo, polietileno glicol 300 ou 400 em álcool etílico industrial (70:30)). Transferir as amostras para tubos de polipropileno. Por favor, note que os tubos Falcon padrão não resistir a essas forças de G). As amostras podem ser armazenadas a -20 ° C durante pelo menos um mês até RNA total is preparados.
2. Preparação de RNA Utilizando Modificado Trizol Protocolo
- Adicione 1 ml de clorofórmio (0,2 ml por ml Trizol) e agitar vigorosamente por 15 segundos. Incubar à temperatura ambiente durante 2-3 minutos.
- Centrifugar a 13.000 × g durante 15 min a 4 ° C.
- Transferir fase superior aquosa (contendo o ARN) para um novo tubo de 15 ml de polipropileno.
- Adicionar ½ volume de reacção de ambos os ARN precipitação tampão e isopropanol (por exemplo, a 3 ml de sobrenadante adicionar 1,5 ml de tampão de precipitação de ARN e 1,5 ml de isopropanol).
- Misturar bem. Incubar à temperatura ambiente durante 10 min.
- Centrifugar a 13.000 × g durante 10 min a 4 ° C. Elimine o sobrenadante.
- Girar brevemente (5.000 × g por 30 segundos) e remova isopropanol residual com uma pipeta de 200 mL.
- Adicionar um volume igual de etanol a 75% e agitar até que o tubo destaca pelete. Evite dividi-lo em pequenos pedaços, pois isso pode fazer a remoção de residual etanol difícil.
- Centrifugar a 13.000 × g durante 10 min a 4 ° C. Elimine o sobrenadante.
- Girar RNA brevemente e remover o etanol restante com uma pipeta de 200 mL. Repita o passo e remover o etanol restante com uma pipeta de 20 mL. Depois destas duas etapas, sem posterior secagem do sedimento deverá ser executado.
- Adicionar 100 mL de H 2 O para 100 ug de RNA rendimento esperado e misturar bem por pipetagem para cima e para baixo 5-6 vezes para ajudar na dissolução do RNA.
- Dissolver e desnaturar ARN por aquecimento a 65 ° C durante 10 minutos (agitador) e colocar imediatamente em gelo.
- Medir a concentração de RNA a 260 nm usando um espectrofotómetro NanoDrop, seguindo as instruções do fabricante. Este ARN pode ser armazenada a -80 ° C durante pelo menos um mês.
3. A biotinilação específica do tiol de RNA recentemente transcrito
- Comece com 60-80 ug de RNA celular total.
- Constituem etiquetagem reacção. Pipeta no seguinteordem (RNA por ug):
- 1 ul de tampão 10x Biotinilação
- 7 ul do RNA (contendo 1 ug de RNA diluídas sem nuclease H2O)
- 2 ul biotina-HPDP (1 mg / mL de DMF)
Sempre adicione a biotina-HPDP último e misture imediatamente por pipetagem. No caso da biotina precipita, o conteúdo de DMF pode ser aumentada para uma concentração final de 40%.
- Incubar à temperatura ambiente durante 1,5 horas, com rotação.
- Adicionar um volume igual de clorofórmio. Misture vigorosamente. Incubar durante 2 - 3 minutos, até as fases começam a separar-se e as bolhas começam a desaparecer.
- Centrifugar a 20.000 × g durante 5 min a 4 ° C. Transferir cuidadosamente a fase aquosa superior num novo tubo.
- Repita os passos 3.4 e 3.5 uma vez. Você pode querer realizar esta etapa em 2 mL da fase de bloqueio Gel tubos pesados para reduzir a perda de RNA.
- Precipitação do RNA: adicionar 1/10 do volume de NaCl 5 M e um volume igual deisopropanol à fase aquosa.
- Centrifugar a 20.000 × g durante 20 min a 4 ° C. Elimine o sobrenadante.
- Adicionar um volume igual de 75% de etanol, de centrifugação a 20.000 x g durante 10 min a 4 ° C, desprezar o sobrenadante.
- Girar brevemente e remover o etanol residual, com uma pipeta de 200 uL.
- Girar brevemente e remover o etanol residual com pipeta 20 ul.
- Não permitir que o RNA para secar. Re-suspende-lo em 50-100 ul de H2O (~ 1 mL por 1 jig de RNA). Misture bem por pipetagem cima e para baixo 5-6 vezes.
- Verifique a qualidade da RNA por análise eletroforética para excluir a degradação do RNA.
4. Dot Blot Análise de 4SU-incorporação (Opcional)
4SU incorporação pode ser facilmente determinada por meio de análise de ARN biotinilados dot blot. Este é um passo opcional que permite a resolução de problemas ea estimativa das taxas de incorporação 4SU relação a um controle oligo biotinilado DNA. Para este ensaio nós recommend usando iodoacetil-biotina em vez de biotina-HPDP para biotinylation de RNA 4SU-rotulados no passo 3.2. Isso resulta em uma biotinilação irreversível de 4SU-RNA. Assim, os métodos baseados em colunas (por exemplo RNeasy) pode ser usado para a recuperação de quantidades muito pequenas de RNA biotinilado (por exemplo, 5 mg). Enquanto o RNA biotinilado utilizando biotina-HPDP também é adequado para este ensaio, o sinal resultante é mais fraca e a razão sinal-ruído menos favorável (Figura 3).
- Siga o protocolo para 4SU a rotulagem e isolamento de RNA total de celulares, conforme descrito nas seções 1 e 2.
- Biotinylate 4SU marcado RNA, conforme descrito na seção 3 substituindo biotina-HPDP com iodoacetil-biotina e fazer duas extrações clorofórmio para remover completamente o excesso de resíduos iodoacetil-biotina.
- Recuperar ARN biotinilados por precipitação de isopropanol / etanol, como descrito, ou usando uma abordagem baseada em coluna (por exemplo, RNeasy) no caso de pequenas quantidades de RNA (<10 ug) São utilizados.
- Incubar a membrana Zeta em água livre de nuclease, com balanço, durante 10 min.
- Aqui a membrana para fora da água sem nuclease e remover fluidos excessivos pela colocação da membrana entre duas toalhas de papel limpas e pressionando firmemente. Secagem ao ar da membrana durante 5 min irá resultar em pontos mais agradáveis.
- Para cada amostra, preparar 20 ul de RNA de 200 ng / mL, utilizando tampão de ligação de ponto de gelo mancha fria (NaOH 10 mM, EDTA 1 mM). Aplicar 5 ul desta diluição (isto é, 1 mg de RNA), assim como três diluições de 10 vezes subsequentes (isto é, 100, 10 e 1 ng de ARN, respectivamente) para a membrana Zeta por pipetagem. Pipetagem através de uma prateleira vazia de ponteiras podem ser empregues para proporcionar o espaçamento uniformemente distribuída. Alternativamente, o uso de um aparelho de dot blot de acordo com as instruções do fabricante.
- Aplicar 5 ul do DNA marcado com biotina-oligo em concentrações variando entre 20 ng / mL e 20 pg / mL (isto é, 100-0,1 ng oligo) cont como positivarol à membrana por pipetagem. Use uma amostra 4SU ingênuo biotinilado como controle negativo.
- Ar seco a membrana por 5 min.
- Incubar a membrana durante 30 minutos em 40 ml de tampão de bloqueio com agitação.
- Incubar a membrana com 10 ml de 1:1000 de estreptavidina-peroxidase de rábano silvestre durante 15 min (5 ml de PBS + 5 ml de SDS a 20% + 10 ul de estreptavidina-peroxidase de rábano silvestre)
- Lavar membrana duas vezes em 40 ml de PBS + 10% de SDS (20 ml de PBS + 20 ml de SDS a 20%) durante 5 min.
- Lavar membrana duas vezes em 40 ml de PBS + 1% de SDS (38 ml de PBS + 2 ml SDS a 20%) durante 5 min.
- Lavar membrana duas vezes em 40 ml de PBS + 0,1% de SDS (40 ml de PBS + 200 ul de 20% SDS) durante 5 min.
- Remover o líquido excessivo, colocando membrana entre duas toalhas de papel limpo e pressionando-os firmemente.
- Visualize ligada à membrana HRP utilizando ECL acordo com as instruções do fabricante.
- Colocar a membrana em folha de plástico / saco, remover as bolhas de ar e incubar durante 2 min no escuro.
- Expor membranafilme por 1-5 min.
5. Separação de RNA etiquetados e não etiquetados Usando revestidas de estreptavidina Magnetic Beads
- Calor tampão de lavagem (3 ml por amostra) a 65 ° C num banho de água.
- Prepare ditiotreitol fresco 100 mm (TDT) em nuclease livre de H 2 O. Fazê-lo por decantação 15 - 30 mg de pó de DTT em um ambiente limpo 50 ml tubo Falcon colocado na escala ultra-fino. Pesar e adicionar a quantidade necessária de livre de nuclease de H 2 O.
- Calor biotinilado amostras de RNA a 65 ° C durante 10 minutos para desnaturar e colocar imediatamente em gelo.
- Lugar μMacs colunas no suporte magnético. Recomenda-se não processar mais de 12 amostras de cada vez (6-8 amostras são ideais).
- Colunas Miltenyi pré-equilibrar com 1 ml a temperatura ambiente tampão de lavagem. Isso levará cerca de 15 min.
- Enquanto isso, adicionar 100 ul de pérolas de estreptavidina a 50-100 ul de ARN biotinilados. Incubar à temperatura ambiente durante 15 minutos com rotação. < li> Se qualquer uma das colunas não iniciou o escoamento até agora isto pode ser facilitado por pressionando suavemente sobre o topo da coluna com um dedo de luva. Uma vez que o fluxo começou as colunas drenar rapidamente.
- Aplicar o ARN / contas às colunas. Descarte o flow-through, a menos que você deseja recuperar a fração de RNA não marcado (ver secção 7).
- Lavar três vezes com 0,9 ml de 65 ° C, tampão de lavagem (1 ml ponteiras encolher quando pipetagem tampões a 65 ° C).
- Lavar três vezes com 0,9 ml de tampão de lavagem à temperatura ambiente.
- Pipetar 700 ul tampão RLT (RNeasy kit MinElute limpeza, Qiagen) para novos tubos de 2 ml e colocá-los sob as colunas.
- Eluir os transcritos de RNA recentemente no buffer RLT pela adição de 100 ul de 100 mM de DTT para as colunas.
- Realizar uma segunda ronda de eluição de 3 min mais tarde no mesmo tubo, adicionando mais 100 ul de 100 mM de DTT.
6. A recuperação de ARN recentemente transcrito
onte ú "> Continuar com o Cleanup MinElute RNeasy (Qiagen) protocolo seguindo as instruções do fabricante. Elute em 25 ul de duas concentrações livre de nuclease H O. Medida RNA usando um espectrofotômetro Nanodrop. Para evitar a necessidade de descongelar e voltar a congelar RNA antes da apresentação para um ensaio de elevado débito, recomendamos preparar ADNc imediatamente após o RNA recentemente transcrito é purificado. Use 2,5 ul de RNA recentemente transcrito em 20 ul da mistura de síntese de cDNA para a síntese de cDNA, seguindo as instruções do fabricante. realizar os controlos de qRT-PCR utilizando um : 10 diluições da mistura de cDNA de RNA Armazenar a -80 ° C..7. Recuperação de, consolidado ARN não marcado (Opcional)
Caso o RNA não ligado tem de ser recuperado, recolher e combinar o fluxo através de (após a adição da solução de pérolas de estreptavidina-RNA às colunas) e a primeira lavagem para a precipitação subsequente. Normalmente, é suficiente para precipitar apenas 50% do ARN não ligado como this vai conter> 80% do material de partida.
- Adicionar um volume igual de isopropanol (sem sal deve ser adicionado como o tampão de lavagem já contiver um M de NaCl).
- Centrifugar a 20.000 × g durante 20 min a 4 ° C. Elimine o sobrenadante.
- Adicionar um volume igual de 75% de etanol, de centrifugação a 20.000 x g durante 10 min a 4 ° C, desprezar o sobrenadante.
- Girar brevemente e remover o etanol residual, com uma pipeta de 200 uL.
- Girar brevemente e remover o etanol residual com pipeta 20 ul.
- Não permitir que o RNA para secar. Ressuspender em 100 ul de H2O Misture bem por pipetagem cima e para baixo 5-6 vezes. Incubar a 65 ° C durante 10 min com agitação e transferir directamente em gelo.
- Verifique a qualidade da RNA por análise eletroforética para excluir a degradação do RNA.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
1. Material de Partida e os rendimentos esperados
Após 1 hora (hr) de 4SU-exposição de RNA recentemente transcrito representa cerca de 1-4% do RNA celular total. Esta será mais baixa em células de crescimento a ser preso em que já não conta para sintetizar o RNA para o crescimento celular / replicação. Quando a rotulagem por 1 hr, recomendamos iniciar o ensaio com 60-80 mg de RNA total. Começando com menos de 30 ug de RNA total de resultados em peletes pequenas de RNA que são difíceis de ver, após a etapa de biotinilação e, portanto, pode ser facilmente perdido. Os níveis de ARN de entrada pode ser aumentado para tanto como 150 ug por períodos muito curtos de marcação (por exemplo, 5 - 10 min.) Quando a duração da RNA rotulagem é reduzido de 1 hora e 5 min a contribuição de seqüências intrônicas de curta duração, nos aumentos de RNA recém-transcrita a partir de ~ 60% para ~ 80% 9. Como intrões são substancialmente mais longo em comparação com as sequências de codificação, bem como 5'-e 3'-UTRs, a quantidade de recém transcritosRNA, que pode ser purificado seguindo a curto ou mesmo ultra-curto 4SU-tagging, não cair linearmente. Como tal, obtivemos> 0,5% de ARN total após 5 min de 4SU codificação em linhas não aderentes de células B humanas 9. Deve, contudo, notar-se que a maior concentração de durações 4SU e ligeiramente mais longo do rótulo pode ser necessária para atingir taxas de incorporação 4SU semelhantes em células aderentes. Enquanto ainda uma baixa taxa 4SU-incorporação permitirá a captura eficiente e purificação de grandes transcrições, uridine ricos, muito curto transcrição com baixo teor de uridina (por exemplo, miRNAs) são susceptíveis de escapar de purificação mesmo quando se utiliza altas concentrações 4SU (> 1 mm). Em fibroblastos murinos NIH-3T3, 1 hr de 200 mM exposição 4SU rotulados RNA recém transcrito com cerca de um 4SU resíduo por 50-100 nucleotídeos (nt) 5. Isso deve permitir a recuperação altamente eficiente de transcritos> 500 - 1000 nt de comprimento. Assim, observou-se apenas um pequeno tamanho do transcritopreconceito quando rotulagem para uma hr usando 200 mM 4SU em ambos os fibroblastos murinos e células B humanas 7. Enquanto 1 hr de 200 uM 4SU não resultou em qualquer alteração significativa nos níveis de transcritos celulares em fibroblastos de murino, a exposição prolongada das células a ≥ 200 uM 4SU tem como resultado um aumento mensurável défice em 24 horas (dados não publicados). Assim, tanto a duração da rotulagem e da 4SU-concentração empregada deve ser minimizado para evitar efeitos ectópica ou tóxicos. Uma maneira fácil de determinar o mínimo 4SU concentração necessário para a recuperação eficiente de RNA recém-transcrita é purificar RNA recém transcrito a seguir 4SU a rotulagem com concentrações crescentes de 4SU (por exemplo 50-1600 M). Como mostrado nas Figuras 2A e 2B, a recuperação do ARN recém transcritos marcados durante 1 hora em fibroblastos humanos primários aumentaram substancialmente entre 50 e 200 fiM 4SU mas depois começou a estagnar.
2. PontoBlot Quantificação da Incorporação 4SU (opcional)
Em alguns casos, pode ser de interesse para medir a quantidade de incorporação 4SU em ARN total. Isto é melhor realizado por análise em dot blot do ARN biotinilados utilizando um conjugado de estreptavidina. Devido à sua natureza química iodoacetil-biotina é mais reactiva ao tiol-grupos que biotina-HPDP resultando na biotinilação de praticamente todos os resíduos 4SU recentemente transcrito em RNA. É importante notar que, tal como biotina-HDPD, iodoacetil-biotina não é solúvel em água e é, portanto, eficazmente removidas por extracção com clorofórmio, como realizado para a biotina-HPDP. Portanto, as condições de reacção idênticas e as concentrações podem ser empregues como quando se utiliza biotina-HPDP. No entanto, iodoacetil-biotina não é reversível. Não pode portanto ser utilizado para a purificação de RNA recentemente transcrito nas abordagens baseadas em coluna. Enquanto o uso de iodoacetil-biotina permite quantificar 4SU-incorporação, as medições baseadas em biotina-HPDP considerar tanto4SU-incorporação e eficiência biotinylation. Empregando os dois reagentes de biotinilação para a mesma amostra, permite a medição da eficiência de biotinilação de ARN-4SU incorporado. A eficiência de biotinilação biotina-HPDP para 4SU marcado ARN parece ser cerca de três vezes menos do que a de iodoacetil-biotina, o que indica que apenas cerca de uma em cada três 4SU resíduos recentemente transcrito em RNA é, na verdade, por biotinilado biotina-HPDP (Figura 3). Comparando as intensidades de sinal de amostras com o controlo do DNA oligo biotinilado, as densidades de biotinilação pode ser medido. Para a maioria das linhas de células de mamíferos um sinal positivo ainda deveriam ser detectáveis em 10 ng de ARN biotinilados após 1 h de 200 uM 4SU rotulagem. Uma fraco sinal de fundo é geralmente detectável para a concentração mais elevada (1 ug) de ARN não marcados.
3. Purificação de RNA recentemente transcrito
A recuperação de ARN recentemente transcrito é altamente quanquantitativa. Se você começou com a mesma concentração de RNA que você pode esperar para obter a mesma quantidade de RNA recém-transcritos para todas as amostras. Como muitos ensaios baseados em colunas, a recolha de RNA recentemente transcrito utilizando o kit MinElute RNeasy pode resultar na absorção adicional a 230-260 nm (presença de detergentes derivados dos tampões de lavagem), o que pode interferir com as medições de OD 260. Isto é visto em menor grau quando se utiliza um tubo de recolha de 2 ml de fresco para cada passo de centrifugação. No entanto, quaisquer medições excessivamente elevados de OD (maior> 2 vezes do que as outras amostras) devem ser consideradas com cautela, particularmente se de DO 260/280 rácios são <1,7. Para as análises a jusante é, portanto, muitas vezes, melhor usar a mesma quantidade de volume de RNA molde para todas as amostras. Nos casos em que os rendimentos de RNA marcadas são mais baixos do que o esperado para a verificação de sinais de degradação do ARN por análise de eletroforese. RNA recém transcrito contém quantidades significativamente maiores de grandes transcrições, unspliced com as bandas rRNA típicos sendo muito menos proeminente (Figura 4).
4. Quantificação de RNA recentemente transcrito
Finalmente, a taxa de incorporação de 4SU recém transcritos do ARN pode ser quantificada por análise espectrofotométrica directamente com base no máximo de absorção de 4SU a 330 nm e o OD 330/260 rácio 5,18. Isto requer> 3 ug de ARN marcado concentrados num pequeno volume (10-20 mL) de isopropanol / precipitação com etanol. Para evitar a perda do pequeno RNA pellet co-precipitação com 30 mg de glicogênio nuclease-free (Fermentas, # R0551) deve ser realizada. Um pico adicional é visível a 330 nm reflectindo a taxa de incorporação de 4SU recentemente transcrito em RNA (Figura 5).
/ Files/ftp_upload/50195/50195fig1highres.jpg "/>
Figura 1. . Princípio da marcação metabólica com 4-tiouridina (4SU) 4SU é adicionada às células para a (5 - 120 minutos), o tempo necessário seguido de preparação de RNA celular total. Após a biotinilação específica do tiol, o RNA celular total é separado em 4SU-rotulados, RNA recentemente transcrito, e não marcados, RNA pré-existentes, utilizando pérolas magnéticas revestidas com estreptavidina. RNA recém transcrito está recuperado das contas usando um agente redutor que corta as pontes dissulfeto que ligam as RNA recém transcrito para as contas. Clique aqui para ver a figura maior .
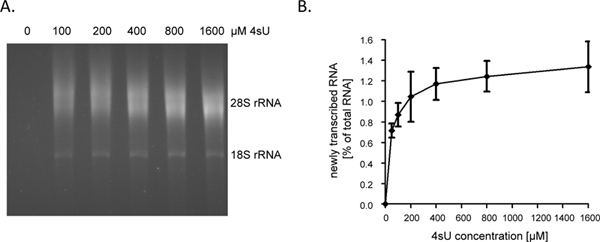
Figura 2. A recuperação de ARN recentemente transcrito sequência concentrações crescentes. 4SU (A) fibroblastos primários do prepúcio humano (HFF) foram incubadas com 100, 200, 400, 800 ou 1600 uM de 4SU. Novos transcritos de ARN foi purificada a partir de 50 ug de RNA celular total e sujeita a análise eletroforética. Como esperado, um aumento dependente da concentração recuperados RNA recém transcrito foi observado que começou a estabilizar em concentrações mais elevadas. (B) Valores de purificada RNA recém transcrito foram quantificadas utilizando o software ImageJ 1.45s. Os dados combinados de quatro experiências independentes sobre as quantidades de RNA recentemente transcrito recuperado seguindo diferentes concentrações de 4SU de rotulagem que variam a partir de qualquer 50-800 4SU fiM (n = 2) ou 100 -. 4SU 1,600 fiM (n = 2) são mostrados clique aqui para ver maior figura .
upload/50195/50195fig3.jpg "alt =" Figura 3 "fo: content-width =" 4.5in "fo: src =" / files/ftp_upload/50195/50195fig3highres.jpg "/>
Figura 3. Estimativa da incorporação 4SU em RNA total 4SU-rotulados usando a análise dot blot. RNA total foi isolado a partir de fibroblastos NIH-3T3 de murino ou de fibroblastos de prepúcio humano (HFF) foram incubadas com 200 uM 4SU durante uma hora. Sem 4SU foi adicionada a um prato como controlo negativo. Para tanto HFF contacto inibido (n = células não-crescimento) e de células de cultura (y) foram incluídos. O ARN foi isolado utilizando o reagente Trizol e subsequentemente conjugado com biotina-HPDP ou iodoacetilo-biotina. A concentração de cada uma das amostras foi ajustada a 200 ng / jil e 5 ul desta diluição (isto é, 1 mg de RNA), assim como três subsequentes diluições de 10 vezes (isto é, 100, 10 e 1 ng de ARN, respectivamente), foram todos visto em um pedaço de membrana Zeta. 5 ul de diluições de DNA marcado com biotina-oligo foram colocados sobre a membrana, como controlos positivos em concentratiões variando de 20 ng / mL até 20 pg / mL (isto é, 100-0,1 ng, respectivamente). Densidade Biotina foi sondada utilizando um conjugado de estreptavidina-peroxidase de rábano.
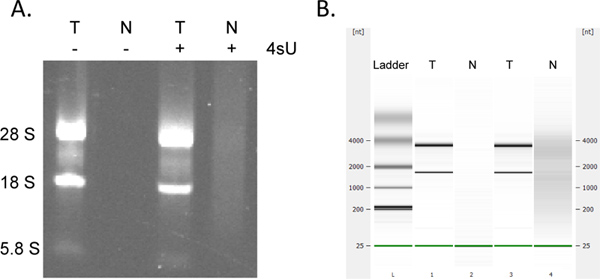
Figura 4. Análise eletroforética de RNA recentemente transcrito e total. ARN total (T) e RNA recentemente transcrito (N), preparada a partir de fibroblastos murinos NIH-3T3 cultivadas tanto na presença e na ausência de 500 uM 4SU durante 1 hora foi analisado por electroforese em gel de agarose (A) e (na mesma ordem), utilizando o Agilent Bioanalyzer (B). Nenhum ARN foi recuperado sem tratamento 4SU de células. Purificada ARN recém transcritos contém maiores quantidades de ARNm de elevado peso molecular e de rRNAs significativamente menos do que o total madurosRNA como notável entre o 28S, 18S e 5.8S rRNA bandas. Clique aqui para ver a figura maior .
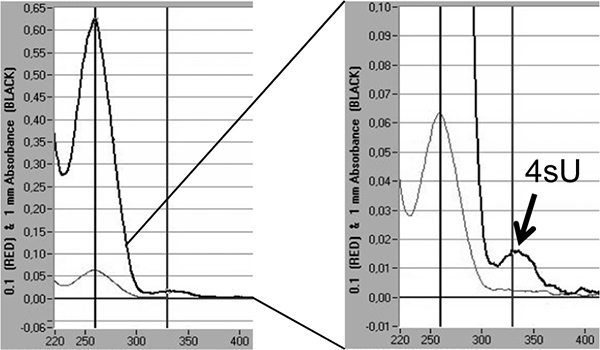
Figura 5. Quantificação da incorporação 4SU em RNA recém transcrito pela análise espectrofotométrica. RNA recém transcrito purificados a partir de 2 x 100 mg RNA total após 1 hora de 200 mM 4SU em murinos fibroblastos NIH-3T3. Novos transcritos de ARN foi precipitado com isopropanol / etanol após adição de 30 ug de glicogénio livre de nuclease. A análise espectrofotométrica do RNA recém transcrito obtidos por um Nanodrop 1000 espectrofotômetro é mostrado. As linhas cinza claro representam medições de 0,1 mm enquanto os mais grossos, linhas cinzentas escuras representam medições em coluna de fluido de 1 mm. À direita, uma ampliação do pico de extinção representando tele incorporou 4SU-resíduos é mostrado. Com base no coeficiente de extinção de 18 4SU as taxas de incorporação de 4SU pode ser estimada.
| Duração de rotulagem [min] | Concentração 4SU recomendado [mM] |
| 120 | 100-200 |
| 60 | 200-500 |
| 15-30 | 500 - 1000 |
| <10 | 500 - 2000 |
Tabela 1. Concentrações 4SU recomendado.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Marcação metabólica de RNA recém-transcrita aumenta substancialmente o poder das tecnologias de alto rendimento, como microarrays e RNA-seq, fornecendo modelos mais adequados para lidar com a questão biológica de interesse. O presente protocolo foi extensivamente otimização. Ele permite o enriquecimento> 1.000 vezes maior de RNA recentemente transcrito e proporciona resultados altamente reprodutíveis.
O delineamento experimental do experimento 4SU-tagging é de crucial importância como RNA recém transcrito vai retratar a atividade transcricional em tempo real apenas durante o tempo de exposição das células à 4SU. Se as mudanças reais nas taxas de transcrição a seguir um estímulo já diminuído, estas serão perdidas quando se analisa RNA recém-transcritas, embora as mudanças nos níveis de RNA total pode ainda ser detectável. Portanto, uma boa compreensão da biologia subjacente é importante definir o arranjo experimental bem como a óptima períodos of tempo para a exposição 4SU. Abaixo, fornecemos recomendações e formas para evitar armadilhas comuns para os passos mais decisivos.
Preparação de soluções de reserva e utensílios de plástico
Todas as soluções de armazém devem ser preparadas utilizando água livre de nuclease. Usando em casa água purificada desionizada pode resultar em problemas quando a água contém agentes redutores. Em um caso, o que resultou na perda completa de todos os ARN marcado. Por isso, recomendamos a compra de pré-fabricados nuclease livre de NaCl, Tris-Cl, EDTA, citrato de sódio e água. Assegurar condições livre de nuclease em todos os momentos. Dimetilformamida (DMF), dissolve-se alguns materiais plásticos. Nós descobrimos que a utilização de 25 ml, pipetas de plástico de cultura de células para transferir DMF a partir do seu frasco de vidro de estoque para tubos Falcon de 50 ml, para se preparar a solução de biotina-HPDP foi suficiente para reduzir substancialmente a produção de RNA recentemente transcrito de todo o ensaio. Curiosamente, isso não afetou negativamente o biotinileficiência ção (como testado por dot blot), mas resultou numa 75 a> 90% de perda de RNA recentemente transcrito que poderiam ser recuperados a partir das esferas. A perda foi mais pronunciada quando a duração da marcação foi reduzida de 60 ou inferior a 30 min. Muito provavelmente, uma substância eluído das pipetas de plástico pelo DMF parcialmente destruído o revestimento das esferas de estreptavidina. Portanto, a utilização de materiais de plástico não são conhecidos por serem compatíveis com DMF deve ser evitado por todos os meios. Pelas mesmas razões, raspadores de células não deve ser utilizado para melhorar a recuperação de amostras de Trizol placas de cultura de células. É interessante notar que as substâncias putativos eluídas dos plásticos pela DMF ou Trizol aparentemente não foram removidas por extracção nem clorofórmio isopropanol / precipitação com etanol.
A cultura celular
A densidade celular nas placas é de crucial importância. Numa experiência onde as células pareceram ser ligeiramente demasiado confluente (90 -100%), foi tratada fibroblastos murinos NIH-3T3 durante 30 min com 100 U / ml de interferão (IFN) ou α γ. Em células confluentes mesmo menos de 15 min de tratamento com IFN já resultou em um de 5 - a 8 vezes a indução de genes ou como IRF1 SOCS3 5. Com células sendo ligeiramente análise microarray também confluente não mostrou qualquer indução de genes IFN-induzidas, mesmo para o induzível mais rapidamente genes como IRF1 ou SOCS3. Portanto, a densidade de células é um fator crucial para experimentos 4SU de rotulagem e todas as placas de cultura de células deve ser cuidadosamente examinada antes de iniciar a marcação.
4SU fotoactivável é um ribonucleósido e 4SU contendo ARN é reticulado de forma eficiente a proteínas após a exposição à fonte de luz de 365 nm. 4SU células tratadas devem ser cultivadas no escuro, e a exposição a luz intensa deve ser evitada. Após a remoção das proteínas celulares por isolamento de RNA de Trizol este risco é reduzido substancialmente.
<classe p = "jove_content"> 4SU não é incorporada no ADN celular. Deve, contudo, notar-se que o RNA total ainda conter pequenas quantidades de ADN celular. Ao utilizar 4SU-tagging e análise q-RT-PCR para estudar a expressão gênica viral na infecção por citomegalovírus achamos necessário incluir um resumo passo ADNasel no protocolo para remover os genomas virais concatemeric 19. Esta não é, provavelmente, quando necessário, utilizando protocolos a jusante, que não são sensíveis à presença de DNA.Taxas de incorporação 4SU e concentração 4SU ideal
4SU é facilmente absorvido pelas células com níveis intra e extra-celular mais provável de equilíbrio dentro de menos de um minuto 9,16. Taxas de 4SU absorção e incorporação são dependente da concentração. Portanto, a concentração 4SU pode ser convenientemente ajustado de acordo com a duração empregada de rotulagem. Tabela 1 fornece pareceres sobre as concentrações 4SU em relção com a duração de rotulagem com base no nosso melhor experiência pessoal. Para uma HR de rotulagem 4SU em células de mamíferos, a 200 uM 4SU será suficiente para a maioria das aplicações, resultando em cerca de um resíduo 4SU por 50 a 100 nucleótidos de RNA recentemente transcrito em fibroblastos.
Nos últimos anos, foi aplicado 4SU codificação para uma ampla gama de tipos de células de origem humana e de murino, incluindo fibroblastos, células endoteliais, células epiteliais, células do estroma da medula óssea, macrófagos e células-T. Além disso, as células de Drosophila e Xenopus foram usadas com êxito. Em todas estas experiências, a incorporação 4SU foi encontrado para ser altamente eficiente exigindo adaptações mínimas na concentração 4SU para os diferentes tipos de células. Ao configurar o método de novos tipos de células, nós recomendamos às células etiqueta com o aumento 4SU-concentração (por exemplo, variando 50-1600 M) e analisar a relação entre purificada recentemente transcrito RNA às 4SU-concentrações aplicadas (ver Figura 2A / B). O 4SU-concentração para a qual a quantidade de transcritos de RNA recentemente purificada entra num planalto devem ser escolhidos.
Nos casos em que muito confluentes, contato células inibidos são utilizados, recomendamos usar concentrações 4SU ligeiramente mais elevadas (por exemplo, 500 em vez de 200 M) para garantir a eficiente incorporação 4SU. Além disso, nos casos em que a captura de transcritos recém-transcritas muito curtos (<200 nt) é de particular interesse, a concentração 4SU também pode precisar de ser aumentada. Isto não deve ser combinado com tempos prolongados de rotulagem (por exemplo> 1 hora) a fim de evitar os efeitos de toxicidade ou ectópicas. Finalmente, verificou-se que o uso de muito pequeno volume de meio de cultura de células pode reduzir a eficiência da incorporação 4SU. Recomendamos, portanto, usando 5 ml ou 10 ml de meio por 10 centímetros ou 15 centímetros de prato, respectivamente.
Preparação de ARN total celular
Para o sucesso deste protocolo é fundamental para obter limpo, livre de RNase total de RNA celular. Usando 5 ml Trizol por 15 centímetros prato produz RNA limpas livres de nucleases. Recomendamos o uso do protocolo Trizol modificado por Chomczynski et al. 20. Em primeiro lugar, é melhor adaptado para isolar grandes quantidades de RNA (> 100 mg) como reforço centrífugas força resulta em aglomerados firmes que são mais fáceis de manipular durante as etapas de lavagem. No entanto, isto requer o uso de tubos de polipropileno especiais e adaptadores como os 15 ml de tubos regulares Falcon laboratoriais não sobrevivem mais do que 6.000 x g. Em segundo lugar, ele melhora a remoção de DNA e glicoproteínas. Isto torna-se particularmente evidente quando se preparava RNA de órgãos ou tecidos. Em terceiro lugar, não se limita a quantidade máxima de ARN total, que podem ser isolados. Embora também encontramos métodos de isolamento de RNA baseados em colunas (por exemplo RNeasy) para fornecer RNA de qualidade adequada, colunas padrão are apenas capaz de capturar até 100 ug de ARN total limitando assim a quantidade de material de partida. Por último, por remoção do restante etanol por duas vezes com uma pipeta, a secagem do ARN para remover o etanol residual, não é mais necessária. Isto elimina o risco de sobre-secagem do RNA, que podem ser difíceis de se dissolver novamente em seguida. Em princípio, 4SU-marcação in vivo é aplicável, por exemplo, por injecção intravenosa de ratos. No entanto, observou-se que a pureza RNA representa um grande problema que requer a purificação de poli-transcrições antes de purificação de RNA recém-transcrita (dados não publicados).
Biotinilação e remoção de biotina não ligado
Biotina-HPDP é 100% específica do tiol e forma uma ligação de dissulfureto entre o resíduo de biotina e moléculas de RNA recentemente transcrito tiol-etiquetados. Biotinilação eficiência do RNA 4SU marcada é de cerca de 30%, conforme determinado por análise de mancha de pontos 5. A biotina-HPDP não é solúvel em águaque pode ser eficientemente removido por extracção com clorofórmio. Enquanto um único passo de extracção de clorofórmio é suficiente para remover a grande maioria de biotina não acoplada repetimos regularmente este passo para assegurar a remoção completa. Para reduzir a perda de RNA durante o clorofórmio extração passo 2 ml Fase de bloqueio de Gel tubos pesados (Eppendorf) pode ser usado seguindo as instruções do fabricante. Normalmente usamos os tubos de bloqueio de fase apenas para o segundo passo de extracção com clorofórmio, como o volume do molde do primeiro degrau são frequentemente demasiado alta para ser directamente compatível com esses tubos. Após a remoção de biotina-HPDP desacoplado, o RNA é recuperado por isopropanol / precipitação com etanol. É importante notar que os kits comerciais baseados em colunas para recuperar o RNA biotinilado (por exemplo RNeasy da Qiagen), não deve ser usado uma vez que contêm agentes redutores nos tampões fornecidos, que clivam a ligação dissulfureto e remover a biotina a partir do RNA recentemente transcrito .
Purificação de novoly RNA transcrito
Não adicione mais de 100 RNA biotinilado ul a 100 ul de contas estreptavidina. Ao aumentar de volume é menos preferido. No entanto, o mesmo volume de ARN deve ser acrescentado em todas as amostras. Ajustar o volume de entrada de RNA (entre amostras) que você adiciona às contas estreptavidina simplesmente adicionando o volume necessário de 1x TE para as contas. Uma maneira fácil de fazer sem nuclease mM DTT fresco 100 é de decantar uma quantidade suficiente de pó DTT para um tubo Falcon colocado numa escala ultra-sensível e, em seguida, adicionar a quantidade necessária de livre de nuclease de H 2 O para gerar 100 mM de DTT (64,8 mL de água por 1 mg DTT).
Durante o desenvolvimento do 4SU codificação testámos esferas de estreptavidina a partir de vários fornecedores. Alguns deles gerado grandes quantidades de fundo. Portanto, recomendamos fortemente o uso do Streptavidin contas Miltenyi como, até agora, nunca tiveram quaisquer problemas com carry-over de RNA não marcados a partir do tecido cAmostras de RNA ultura derivados. Desta forma, tão pouco como 150 ng de ARN marcado pode ser especificamente purificada a partir de 150 ug de ARN biotinilado (em 100 ul de água), utilizando 100 ul de contas de estreptavidina. O equilíbrio das contas com tampão de equilíbrio fornecido com os grânulos podem ser realizados e podem melhorar ligeiramente as taxas de captura 13.
Controles de qualidade
Recomendamos a realização dos controlos q-RT-PCR em RNA recém transcrito antes de submetê-lo para análise de alto rendimento. Isto pode incluir a quantificação de vários genes de referência conhecidos diferencialmente regulados na dada situação experimental. Nos casos em que 4SU-tagging é empregada para estudar as taxas de decaimento de RNA, recomendamos para quantificar a transcrição de curta duração (por exemplo, myc, fos) e de longa duração um (por exemplo, GAPDH) de RNA total e recém-transcritas. A razão do ARN recém transcritos / total deve ser substancialmente maior (~ 5 - a 10 vezes)para as transcrições de curta duração. Com base no ARN de semi-vida de um gene de referência, RNA meia-vida pode ser determinada. Se todas as três fracções de ARN (RNA total, RNA recentemente transcrito de ARN não marcado e pré-existente) são analisados por quatro ou mais genes, a normalização dos diferentes subconjuntos de ARN pode ser realizada por meio de análise de regressão linear, e as contagens de controlo de qualidade podem ser determinados como descrito 7 , 21.
Para a análise q-RT-PCR, recomendamos a utilização de 2,5 mL de RNA marcados em 20 l mix síntese de cDNA. Para fins de comparação óptima dos resultados Q-RT-PCR do cDNA congelar em alíquotas de 5 ul, antes da primeira utilização. Descongelar tubos imediatamente antes da utilização, adicionar 45 ul de H2O e 5 ul sujeito das diluições para análises Q-RT-PCR. Isso aumenta significativamente a comparabilidade entre diferentes execuções de PCR.
Amostras de RNA recém transcrito deve ser verificado para os sinais de degradação do RNA usando o Bioanalyzer Agilent antes de submetê-los aanálise de alto rendimento (microarrays ou RNA-seq). Deve, contudo, notar-se que as bandas adicionais são, por vezes, observada pelo Bioanalyzer da Agilent. O significado biológico deste permanece obscura. Como RNA recém transcrito contém RNA significativamente menos ribossômico, essas amostras ocasionalmente falhar o Agilent controles de qualidade Bioanalyzer. Se isto não é devido a RNA amostras visíveis de degradação de qualidade aceitável são geralmente fino para ser submetido a uma análise de alto rendimento.
Compatibilidade de RNA recém-transcrita com análises a jusante
Novos transcritos de ARN contém substancialmente mais do que o ARNm de RNA total. Isto é principalmente devido às grandes quantidades de seqüências intrônicas em RNA recém transcrito que aumentam quando a duração da 4SU-tagging é encurtado. Portanto, não assumimos regularmente o esgotamento dos rRNAs de amostras de RNA recém transcrito como este requer grandes quantidades de matéria-prima, enquanto providmuito pouco ganho ing (~ duplo) em não-rRNA lê. Por último, resta assinalar que a maior percentagem de unspliced, transcrições de elevado peso molecular presentes no RNA recentemente transcrito pode exigir fragmentação adicional quando da preparação de bibliotecas de cDNA para a sequenciação de última geração. Os resultados da etapa de fragmentação, por conseguinte, o tamanho deve ser de qualidade controlada cuidadosamente.
A normalização de dados para RNA medidas de meia-vivas
O método padrão para normalizar os dados experimentais para RNA de medidas de meia-vida é normalizar todos os dados para o RNA de meia-vida de um gene de manutenção da casa bem caracterizada a mediana ou RNA meia-vida num determinado tipo de célula determinado em experiências anteriores. Em células de mamífero, este último encontra-se na gama de 5 a 10 h 6,7. Embora essa abordagem também funciona muito bem para as medições baseadas em 4SU, outros meios para a normalização são necessários se a mediana RNA meia-vida não é conhecido ou se may ainda ser afetada por alterações no sistema celular em estudo, por exemplo, pelo knock-out de um decaimento via RNA. 4SU-marcação oferece uma maneira única de estimar a mediana do RNA meia-vida com base na análise de todas as três fracções de RNA, ou seja ARN celular total, RNA recentemente transcrito e o RNA não marcadas pré-existentes. Como RNA celular total é dividida em duas últimas fracções de RNA um modelo de regressão linear simples podem ser utilizados para normalizar as três fracções de RNA para o outro e determinar a mediana do RNA meia-vida 7,16. Um pacote de software está disponível on-line para realizar essas análises 22.
Captura ineficiente de transcrições com baixo teor de uridina pode afetar RNA medições de meia-vida, resultando em índices artificialmente RNA recém transcrito / total de baixos e prolongados RNA meias-vidas. A extensão deste problema pode ser avaliado através da representação gráfica de RNA de semi-vida ou o log (rácios de RNA total recentemente transcrito /) contra a uridiconteúdo ne de todas as transcrições 7,15. Isso também fornece um bom controle de qualidade para avaliar as diferenças nas taxas de 4SU-incorporação entre as diferentes amostras ou condições. Nos casos em que uma correlação substancial no conteúdo de uridina é observada esta pode ser corrigida por bioinformática meios 15. No entanto, deve notar-se que a contribuição de transcritos maduros recentemente transcrito em ARN não pode ser facilmente diferenciado do muito maior e, portanto, muito mais precursores de uridina-ricos. A menos que a cinética de transformação de um dado transcrição são conhecidos (o que geralmente não são), simplesmente corrigindo baixo teor de uridina (captura ineficiente) pode distorcer grosseiramente RNA meias-vidas. Como tal, recentemente descobriu processamento da maioria dos humanos snoRNAs ser altamente ineficiente 9. Se tivéssemos corrigido os índices de RNA recém transcrito / total para o baixo teor uridine do pequeno (70-300 nt) snoRNAs, isso teria resultado em curtíssimo snoRNA meia lives (<5 min) com vários índices de RNA recém transcrito / total superior a 100%. Portanto, nós geralmente não recomendo corrigindo baixo teor uridine ao medir RNA meias-vidas.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Os autores declaram que não têm interesses financeiros concorrentes.
Acknowledgments
Gostaríamos de agradecer a Amie Regan para a leitura cuidadosa do manuscrito. Este trabalho foi apoiado por NGFN Além disso concessão # 01GS0801, MRC bolsista G1002523 e NHSBT concessão WP11-05 para LD e DFG concessão FR2938/1-1 para CCF
Materials
| Name | Company | Catalog Number | Comments |
| 4-thiouridine | Carbosynth | T4509 | Prepare 50 mM stock in sterile H2O, store at -20 °C in aliquots of 50-500 μl, discard unused reagent, do not refreeze. |
| Trizol | Invitrogen | 15596026 (100 ml), 15596018 (200 ml) | WARNING - CORROSIVE and HAZARDOUS TO HEALTH! Ensure immediate access to Phenol antidote (PEG-Methanol); Store at 4 °C. |
| Chloroform | Sigma | 372978 | WARNING - HAZARDOUS TO HEALTH |
| Isopropanol | Sigma | 650447 | |
| Sodium citrate, nuclease-free | Sigma | C8532 | Prepare 1.6 M stock solution using nuclease-free water. |
| 5M nuclease-free NaCl | Sigma | 71386 | Stock solution |
| Nuclease-free H2O | Sigma | W4502 | Make 1 ml aliquots in nuclease-free tubes. |
| RNA precipitation buffer | 1.2 M NaCl, 0.8 M sodium citrate in nuclease free water. Prepare in advance under strictly nuclease-free conditions. Store at room temperature in 50 ml falcon tubes. | ||
| Ethanol | Sigma | 459844 | Use with nuclease-free water to prepare 80% ethanol, store at -20 °C. |
| 1 M nuclease-free Tris Cl, pH 7.5 | Lonza | 51237 | Stock solution |
| 500 mM nuclease-free EDTA, pH 8.0 | Invitrogen | 15575-020 | Stock solution |
| 10x Biotinylation Buffer (BB) | 100 mM Tris pH 7.4, 10 mM EDTA in nuclease-free water, make aliquots of 1 ml. | ||
| Dimethylformamide (DMF) | Sigma | D4551 | |
| EZ-Link biotin-HPDP | Pierce | 21341 | Prepare 1 mg/ml stock solution by dissolving 50 mg biotin-HPDP in 50 ml DMF. Gentle warming enhances solubilisation. Store at 4 °C in aliquots of 1 ml. |
| Phase Lock Gel Heavy tubes 2.0 ml | Eppendorf | 0032 005.152 | Optional for the chloroform extraction step. |
| Zeta membrane | BIORAD | 162-0153 | |
| 10x Dot blot binding buffer | 100 mM NaOH, 10 mM EDTA | ||
| Biotin-oligo | 5'-biotin, 25 nucleotides, any sequence | ||
| Sodium dodecyl sulphate | Fisher | BPE9738 | For 100 ml 20% stock solution, add 20 g SDS to 80 ml PBS pH 7-8 and adjust volume to 100 ml. Keep all high-percentage SDS solutions above 20 °C. Warm the solutions slightly should SDS precipitate. |
| EZ-Link Iodoacetyl-LC-Biotin | Pierce | 21333 | Prepare 1 mg/ml stock solution by dissolving 50 mg iodoacetyl-biotin in 50 ml DMF. Gentle warming enhances solubilisation. Store at 4 °C in aliquots of 1 ml. Generates irreversible, thiol-specific biotinylation. |
| Phosphate buffer saline | Gibco | 10010-015 | |
| Dot blot blocking buffer | Mix 20 ml 20% SDS with 20 ml 1 x PBS pH 7-8 and add EDTA to the final concentration of 1 mM. | ||
| Streptavidin-horseradish peroxidase | Vector Laboratories | SA5004 | Store at -20 °C. Mix 10 ml 20% SDS with 10 ml 1 x PBS. Add 20 μl Streptavidin-HRP before use. |
| ECL reagent | GE Healthcare | RNP2109 | Use following the manufacturer's instructions. |
| Super RX, X-RA Film, 18x24 cm | Fujifilm | 47410 19236 | |
| μMacs Streptavidin Kit | Miltenyi | 130-074-101 | Store the beads at 4 °C. |
| Tween 20 | Sigma | P1379 | |
| Washing buffer | 100 mM Tris pH 7.4, 10 mM EDTA, 1 M NaCl, 0.1% Tween 20 in nuclease-free H2O. | ||
| Dithiothreitol (DTT) | Sigma | 43817 | Prepare as 100 mM DTT in nuclease-free H2O, always prepare fresh before use. |
| RNeasy MinElute Kit | Qiagen | 74204 | Store columns at 4 °C, remaining components of the kit at room temperature. |
| 1.5 ml screw-top polypropylene tubes | Sarstedt | 72.692.005 | Compatible with Dimethylformamide |
| 2.0 ml screw-top polypropylene tubes | Sarstedt | 72.694.005 | Compatible with Dimethylformamide |
| 15 ml tubes | BD Falcon | 352096 | Compatible with Dimethylformamide |
| 50 ml tubes | BD Falcon | 352070 | Compatible with Dimethylformamide |
| All solutions/reagents should be stored at room temperature unless otherwise specified. | |||
| Equipment | |||
| UV/VIS spectrophotometer | Thermo Scientific | NanoDrop 1000 | Or equivalent. Use low volume (1-2 μl) for measurements of low RNA concentrations to avoid excessive sample loss. |
| Polypropylene 15 ml centrifuge tubes | VWR International | 525-0153 | In contrast to standard 15 ml tubes, these tolerate up to 15,000 × g |
| High-speed centrifuge | Beckman Coulter | Avanti J-25 | Or equivalent equipment capable of reaching 13,000×g |
| High-speed rotor | Beckman Coulter | JLA-16250 | Or equivalent equipment capable of reaching 13,000×g |
| Adaptors for 15 ml tubes | Laborgeräte Beranek | 356964 | Or equivalent equipment capable of reaching 13,000×g |
| Refrigerated table-top centrifuge | Eppendorf | 5430 R | Or equivalent. |
| Thermomixer | Eppendorf | Thermomixer compact | Or equivalent. |
| Magnetic stand | Miltenyi Biotec | 130-042-109 | One stand holds 8 μMacs columns. |
| Waterbath | Grant | SUB Aqua 5 | Or equivalent. |
| Ultra-fine scale | A&D | GR-202 | Or equivalent. |
| E-Gel iBase Power System | Invitrogen | G6400UK | For RNA gels; or equivalent. |
| E-Gel EX 1% agarose precast gels | Invitrogen | G4020-01 | For RNA gels; or equivalent. |
References
- Brown, P. O., Botstein, D. Exploring the new world of the genome with DNA microarrays. Nature Genetics. 21, 33-37 (1999).
- Mortazavi, A., Williams, B. A., Mccue, K., Schaeffer, L., Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nature Methods. 5, 621-628 (2008).
- Nagalakshmi, U., Wang, Z., et al. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science. 320, 1344-1349 (2008).
- Wilhelm, B. T., Marguerat, S., et al. Dynamic repertoire of a eukaryotic transcriptome surveyed at single-nucleotide resolution. Nature. 453, 1239-U1239 (2008).
- Dölken, L., Ruzsics, Z., et al. High-resolution gene expression profiling for simultaneous kinetic parameter analysis of RNA synthesis and decay. RNA. 14, 1959-1972 (2008).
- Yang, E., van Nimwegen, E., et al. Decay rates of human mRNAs: correlation with functional characteristics and sequence attributes. Genome Res. 13, 1863-1872 (2003).
- Friedel, C. C., Dölken, L., Ruzsics, Z., Koszinowski, U. H., Zimmer, R. Conserved principles of mammalian transcriptional regulation revealed by RNA half-life. Nucleic Acids Res. 37, e115 (2009).
- Nilsen, T. W., Graveley, B. R. Expansion of the eukaryotic proteome by alternative splicing. Nature. 463, 457-463 (2010).
- Windhager, L., Bonfert, T., et al. Ultra short and progressive 4sU-tagging reveals key characteristics of RNA processing at nucleotide resolution. Genome Res. , (2012).
- Melvin, W. T., Milne, H. B., Slater, A. A., Allen, H. J., Keir, H. M. Incorporation of 6-thioguanosine and 4-thiouridine into RNA. Application to isolation of newly synthesised RNA by affinity chromatography. Eur. J Biochem. 92, 373-379 (1978).
- Cleary, M. D., Meiering, C. D., Jan, E., Guymon, R., Boothroyd, J. C. Biosynthetic labeling of RNA with uracil phosphoribosyltransferase allows cell-specific microarray analysis of mRNA synthesis and decay. Nature Biotechnology. 23, 232-237 (2005).
- Kenzelmann, M., Maertens, S., et al. Microarray analysis of newly synthesized RNA in cells and animals. Proc. Natl. Acad. Sci. U.S.A. 104, 6164-6169 (2007).
- Schwanhäusser, B., Busse, D., et al. Global quantification of mammalian gene expression control. Nature. 473, 337-342 (2011).
- Rabani, M., Levin, J. Z., et al. Metabolic labeling of RNA uncovers principles of RNA production and degradation dynamics in mammalian cells. Nat. Biotechnol. , (2011).
- Miller, M. R., Robinson, K. J., Cleary, M. D., Doe, C. Q. TU-tagging: cell type-specific RNA isolation from intact complex tissues. Nat. Methods. 6, 439-441 (2009).
- Miller, C., Schwalb, B., et al. Dynamic transcriptome analysis measures rates of mRNA synthesis and decay in yeast. Mol. Syst. Biol. 7, 458 (2011).
- Weintz, G., Olsen, J. V., et al. The phosphoproteome of toll-like receptor-activated macrophages. Mol. Syst. Biol. 6, 371 (2010).
- Lipsett, M. N. The isolation of 4-thiouridylic acid from the soluble ribonucleic acid of Escherichia coli. Journal of Biological Chemistry. 240, 3975-3978 (1965).
- Marcinowski, L., Liedschreiber, M., et al. Real-time Transcriptional Profiling of Cellular and Viral Gene Expression during Lytic Cytomegalovirus Infection. PLoS Pathog. 8, e1002908 (2012).
- Chomczynski, P., Mackey, K. Short technical reports. Modification of the TRI reagent procedure for isolation of RNA from polysaccharide- and proteoglycan-rich sources. Biotechniques. 19, 942-945 (1995).
- Friedel, C. C., Dölken, L. Metabolic tagging and purification of nascent RNA: implications for transcriptomics. Mol. Biosyst. 5, 1271-1278 (2009).
- Friedel, C. C., Kaufmann, S., Dölken, L., Zimmer, R. HALO - A Java framework for precise transcript half-life determination. Bioinformatics. , (2010).
