

Summary
L'RNA cellulare totale fornisce un modello povero per studiare variazioni a breve termine in sintesi dell'RNA e il decadimento nonché la cinetica di RNA processing. Qui, descriviamo marcatura metabolica di RNA trascritto recente con 4 thiouridine seguito da biotinylation tiolo-specifica e purificazione dell'RNA trascritto appena consentano di superare queste limitazioni.
Abstract
Lo sviluppo di microarrays intero trascrittoma e sequenziamento di nuova generazione ha rivoluzionato la nostra comprensione della complessità di espressione genica cellulare. Insieme a una migliore comprensione dei meccanismi molecolari coinvolti, misure precise della cinetica sottostanti sono diventati sempre più importanti. Qui, questi potenti metodologie incontrano notevoli limitazioni dovute alle proprietà intrinseche dei campioni template studiano, cioè RNA cellulare totale. In molti casi cambiamenti RNA cellulare totale verificano o troppo lentamente o troppo velocemente per rappresentare gli eventi molecolari sottostanti e la loro cinetica con risoluzione sufficiente. Inoltre, il contributo delle alterazioni della sintesi dell'RNA, elaborazione e decadimento non sono facilmente differenziate.
Recentemente abbiamo sviluppato ad alta risoluzione-profili di espressione genica di superare queste limitazioni. Il nostro approccio si basa sulla marcatura metabolica di nuova RNA trascritto con 4 thiouridine (quindi anche denominato 4SU-tagging) seguita da una rigorosa purificazione dell'RNA trascritto recente utilizzando biotinylation tiolo-specifici e biglie magnetiche rivestite di streptavidina. È applicabile ad una vasta gamma di organismi vertebrati, compresi Drosophila e lieviti. Abbiamo applicato con successo 4SU-tagging per studiare la cinetica in tempo reale delle attività di fattore di trascrizione, di fornire misure precise di RNA emivita, e ottenere nuove informazioni sulle cinetiche di trasformazione dell'RNA. Infine, modellazione computazionale può essere impiegato per generare un, analisi completa integrata dei meccanismi molecolari sottostanti.
Introduction
Profili di espressione genica è uno strumento chiave utilizzata per lo studio dei processi cellulari e la rete associata complessa interazione. Studi su mRNA abbondanza sono stati in genere il metodo di scelta per ottenere intuizioni fondamentali sui meccanismi molecolari sottostanti. Lo sviluppo di microarrays intero trascrittoma-1 e, più recentemente, di sequenziamento di nuova generazione di RNA (RNA-Seq) 2-4 alimentato questo approccio. Mentre queste tecnologie hanno rivoluzionato la nostra comprensione della complessità di espressione genica cellulare, si trovano ad affrontare gravi limitazioni dovute alle proprietà intrinseche delle loro campione modello, cioè RNA cellulare totale. In primo luogo, i cambiamenti a breve termine dei livelli totali di RNA non corrispondono a variazioni dei tassi di trascrizione, ma sono intrinsecamente dipendente dalla RNA emivita delle rispettive trascrizioni. Mentre un quintuplo induzione di un trascritto di breve durata, per esempio codifica per un fattore di trascrizione, sarà prontamente rilevabile in RNA totaleentro un'ora, la stessa induzione di una trascrizione vita lunga, ad esempio la codifica per un enzima metabolico, rimane praticamente invisibile. Inoltre, anche un completo spegnimento (> 1.000 volte down-regulation) del tasso di trascrizione di un gene medio con un RNA emivita di cinque ore sarà semplicemente prendere cinque ore per i suoi livelli di RNA totale di diminuire solo del duplice . Pertanto, l'analisi di RNA totale favorisce l'individuazione di up-regolazione di trascritti di breve durata, molti dei quali codificano per fattori di trascrizione e geni con funzioni regolatrici 5. Inoltre, la vera cascata cinetica di regolazione è oscurata e segnalazione di eventi primari non può essere differenziata da secondario. Entrambi, a loro volta, possono generare una distorsione nella valle analisi bioinformatica. In secondo luogo, alterazioni nei livelli totali di RNA non può essere attribuito a cambiamenti nella sintesi di RNA o di decadenza. Misurazioni di quest'ultimo richiedono approcci invasivi cellule, ad esempio bloccanti trascrizionisull'uso actinomicina D 6, e il monitoraggio prolungato di continuo decadimento RNA nel tempo. Con un mRNA emivita media nelle cellule di mammifero di 5-10 ore 5,7, i livelli di mRNA della maggior parte dei geni saranno solo sono diminuiti di meno di due volte a seguito di diverse ore di arresto trascrizionale. Piuttosto queste piccole differenze determinare misurazioni grossolanamente imprecise di mRNA emivite per la maggioranza dei geni cellulari a causa della natura esponenziale delle equazioni matematiche sottostanti. Infine, mentre l'RNA-Seq di RNA cellulare totale ha rivelato che circa la metà dei nostri geni sono soggetti ad eventi alternative splicing 8, la cinetica di fondo, nonché i meccanismi dinamici che guidano tessuto e contesto-specifica regolamentazione del trattamento RNA sono ancora limitate. Inoltre, il contributo di RNA elaborazione per l'espressione genica differenziale, in particolare per RNA non codificanti, rimane da determinare. Complessivamente, queste limitazioni rappresentano i principali ostacoli permodellazione cinetica bioinformatica dei meccanismi molecolari sottostanti.
Recentemente abbiamo sviluppato un approccio, definito ad alta risoluzione profili di espressione genica, per superare questi problemi 5,7,9. Si basa sulla marcatura metabolica di RNA appena trascritto utilizzando 4 thiouridine (4SU-tagging), un derivato uridina naturale, e offre accesso diretto alle trascrizioni appena trascritto con la minima interferenza nella crescita cellulare e l'espressione genica (vedi Figura 1) 5, 10-12. L'esposizione delle cellule eucariotiche ai risultati 4SU nella sua rapida diffusione, la fosforilazione di 4SU-trifosfato, e l'incorporazione in RNA appena trascritto. Dopo aver isolato RNA cellulare totale, la frazione di RNA 4SU marcato è tiolo specificamente biotinilato generando un legame disolfuro tra biotina e l'RNA appena trascritto. 'RNA cellulare totale' può quindi essere quantitativamente separati in etichetta ('appena trascritto') e senza etichetta ('pre-existing '), l'RNA con elevata purezza utilizzando biglie magnetiche rivestite di streptavidina. Infine, etichettato RNA viene recuperato dalle perline semplicemente aggiungendo un agente riducente (ad esempio ditiotreitolo) scindere il legame disolfuro e libera gli RNA appena trascritto dalle perline.
Recentemente trascritto RNA raffigura l'attività trascrizionale di ogni gene durante il periodo di tempo di esposizione 4SU. 4SU-tagging nella scala temporale di minuti quindi fornisce una fotografia istantanea di espressione genica eucariotica e un modello ideale per a valle bioinformatici analisi (ad esempio l'analisi del promotore). Nei casi in cui le condizioni di steady-state possono essere assunte, i rapporti di recente trascritto / totale, appena trascritte / senza etichetta e senza etichetta / RNA totale forniscono l'accesso non invasivo per RNA precise emivita 7,13. Inoltre, è importante notare che l'RNA appena trascritto purificato dopo appena 5 minuti di 4SU-tagging (5 min 4SU-RNA) è più giovane di 15 e 60 min 4SU-RNA.Quando si esegue entrambi ultra-corta e progressivamente più 4SU-tagging in una singola impostazione sperimentale combinato con RNA-Seq, la cinetica di trasformazione dell'RNA vengono rivelati alla risoluzione nucleotide 9. Infine, l'analisi tempo-corso di RNA appena trascritto e totale in combinazione con modelli computazionali consentono un'analisi integrativa di sintesi di RNA e di decadimento 14.
In conclusione, questo approccio permette l'analisi diretta della dinamica di sintesi dell'RNA, trasformazione e degradazione nelle cellule eucariotiche. È applicabile in tutti i principali organismi modello tra cui mammiferi, insetti (Drosophila), anfibi (Xenopus), e lievito 5,15,16. Esso è direttamente compatibile con microarray analisi 5,17, RNA-Seq 9,13,14, ed è applicabile in vivo 12,15. Qui, abbiamo dettaglio la metodologia di etichettare, isolare e purificare l'RNA appena trascritto in cellule di mammifero in coltura. Inoltre, potenziometrosono discussi Al problemi e le insidie.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. Marcatura metabolica con 4 thiouridine
Fare un piano dettagliato del setup sperimentale / programma, ad esempio, quando aggiungere il 4SU di coltura cellulare e quando raccogliere i campioni. Piano per almeno 5 minuti tra ogni condizione. Solo il trattamento di cellule di una condizione alla volta. Maneggiare max. 3-5 piatti in un dato momento. Maneggiare le cellule più rapidamente possibile per ridurre al minimo i cambiamenti di temperatura e livelli di CO 2. Evitare di esporre le cellule alla luce dopo 4SU viene aggiunto in quanto ciò potrebbe causare la reticolazione di RNA 4SU-etichettati per le proteine cellulari.
Avvio di etichettatura
- Scongelare a 4 thiouridine (4SU) subito prima dell'uso e pipetta quantità necessaria di 4SU per ogni condizione in un tubo Falcon sterile.
- Prendete la quantità di mezzo di coltura cellulare (5 ml per 10 cm di piatto) fuori i piatti e aggiungere al 4SU contenenti tubo Falcon e mescolare accuratamente. Rimuovere e gettare la media residua dai piatti. <li> Applicare 4SU contenente schienale medio per i piatti.
Fine di etichettatura
- Rimuovere coltura cellulare da cellule. Aggiungere 5 ml di Trizol per ogni piatto. Per gli esperimenti complessi, tra cui più punti o condizioni di tempo, questo passo è fatto meglio da due persone, una rimozione del mezzo, l'altra aggiungendo Trizol e raccogliendo il lisato.
- Incubare per 5 minuti a temperatura ambiente per lisi cellulare completa.
- Utilizzare una pipetta da 10 ml per lavare il piatto con cura con il Trizol aggiunto. Questo favorisce la lisi cellulare completo e il recupero del campione. Maneggiare con cura, come Trizol è estremamente pericoloso al contatto con la pelle o con gli occhi! Avere antidoto per le ustioni fenolo a portata di mano (per esempio polietilenglicole 300 o 400 in alcool denaturato industriale (70:30)). Campioni di trasferimento a tubi di polipropilene. Si prega di notare che i tubi Falcon standard non resistere a queste forze elevate g). I campioni possono essere conservati a -20 ° C per almeno un mese fino RNA totale is preparato.
2. RNA Preparazione Utilizzo Modificata Trizol protocollo
- Aggiungere 1 ml di cloroformio (0.2 ml per ml Trizol) e agitare vigorosamente per 15 sec. Incubare a temperatura ambiente per 2 - 3 min.
- Centrifugare a 13.000 xg per 15 min a 4 ° C.
- Trasferire fase superiore acquosa (contenente l'RNA) in una nuova provetta di polipropilene 15 ml.
- Aggiungere ½ il volume di reazione di precipitazione sia RNA tampone e isopropanolo (3 ml per es surnatante aggiungere 1,5 ml di tampone RNA precipitazione e 1,5 ml di isopropanolo).
- Mescolare bene. Incubare a temperatura ambiente per 10 min.
- Centrifugare a 13.000 xg per 10 min a 4 ° C. Scartare il surnatante.
- Centrifugare brevemente (5.000 × g per 30 sec) e rimuovere residui di isopropanolo con pipetta 200 microlitri.
- Aggiungere un uguale volume di etanolo 75% e agitare fino a quando il tubo stacca pellet. Evitare di romperlo in tanti piccoli pezzi come questo può rendere la rimozione di residuil difficile etanolo.
- Centrifugare a 13.000 xg per 10 min a 4 ° C. Scartare il surnatante.
- Spin down RNA brevemente ed eliminare l'etanolo residuo con una pipetta 200 microlitri. Ripetere il passaggio ed eliminare l'etanolo residuo con una pipetta 20 ml. Dopo questi due passaggi, senza ulteriori asciugatura del pellet dovrebbero essere eseguite.
- Aggiungere 100 ml di H 2 O a 100 pg di RNA rendimento atteso e mescolare bene pipettando su e giù 5 - 6 volte per aiutare nella dissoluzione del RNA.
- Sciogliere e denaturare RNA mediante riscaldamento a 65 ° C per 10 min (agitatore) ed immediatamente posto sul ghiaccio.
- Misurare la concentrazione di RNA a 260 nm utilizzando uno spettrofotometro NanoDrop, seguendo le istruzioni del produttore. Questo RNA può essere conservato a -80 ° C per almeno un mese.
3. Tiolo-specifica Biotinilazione di recente RNA trascritto
- Inizia con 60 - 80 mg di RNA cellulare totale.
- Costituiscono etichettatura reazione. Pipettare nella seguenteordine (per RNA mcg):
- 1 ml 10x Buffer Biotinilazione
- 7 RNA microlitri (contenente 1 RNA mcg diluito in nucleasi H 2 O)
- 2 microlitri biotina-HPDP (1 mg / ml di DMF)
Aggiungere sempre la biotina-HPDP scorso e mescolare immediatamente da pipettaggio. Nel caso la biotina precipita, contenuti DMF può essere aumentata a una concentrazione finale del 40%.
- Incubare a temperatura ambiente per 1,5 ore con rotazione.
- Aggiungere un uguale volume di cloroformio. Mescolare energicamente. Incubare per 2-3 minuti fino a quando le fasi cominciano a separarsi e bolle iniziano a scomparire.
- Centrifugare a 20.000 xg per 5 minuti a 4 ° C. Trasferire accuratamente la fase superiore acquosa in una nuova provetta.
- Ripetere i passi 3.4 e 3.5 una volta. Si consiglia di eseguire questo passaggio in 2 ml di blocco Gel tubi pesanti fase per ridurre la perdita di RNA.
- RNA precipitazione: add 1/10 del volume di 5 M NaCl e un volume uguale diisopropanolo alla fase acquosa.
- Centrifugare a 20.000 xg per 20 min a 4 ° C. Scartare il surnatante.
- Aggiungere un uguale volume di etanolo 75%, centrifugare a 20.000 xg per 10 min a 4 ° C, scartare il surnatante.
- Spin brevemente ed eliminare l'etanolo residuo con 200 microlitri pipetta.
- Spin brevemente ed eliminare l'etanolo residuo con 20 microlitri pipetta.
- Non permettere che l'RNA si asciughino. Risospendere in 50 - 100 ml H 2 O (~ 1 ml per 1 mg di input RNA). Mescolare bene pipettando su e giù per 5-6 volte.
- Verificare la qualità dell'RNA mediante analisi electrophoretical per escludere la degradazione dell'RNA.
4. Dot Blot Analisi di 4SU-incorporazione (Opzionale)
Incorporazione 4SU può essere facilmente determinata da analisi dot blot di RNA biotinilato. Questo è un passaggio facoltativo che consente la risoluzione dei problemi e la stima dei tassi di incorporazione 4SU relativi a un controllo biotinilato oligo DNA. Per questo test abbiamo recommend utilizzando iodoacetyl-biotina invece di biotina-HPDP per biotinilazione di RNA 4SU-etichettati in fase 3.2. Ciò si traduce in un biotinilazione irreversibile di 4SU-RNA. Pertanto, (ad es RNeasy) possono essere utilizzati metodi basati colonna per il recupero di molto più piccole quantità di RNA biotinilato (ad esempio 5 mg). Mentre RNA biotinilato utilizzando biotina-HPDP è adatto anche per questo saggio, il segnale risultante è più debole e il rapporto segnale-rumore meno favorevole (Figura 3).
- Seguire il protocollo per 4SU-etichettatura e l'isolamento di RNA cellulare totale, come descritto nelle sezioni 1 e 2.
- Biotinylate 4SU marcato RNA come descritto nel capitolo 3 sostituendo biotina-HPDP con iodoacetyl-biotina ed effettuare due estrazioni cloroformio per rimuovere completamente i residui di iodoacetyl-biotina eccessivi.
- Recuperare RNA biotinilati da isopropanolo / etanolo precipitazione come descritto o con un approccio basato su colonne (es RNeasy) nel caso di piccole quantità di RNA (<10 mcg) Sono utilizzati.
- Incubare la membrana Zeta in acqua priva di nucleasi con dondolo per 10 min.
- Prendere la membrana fuori dall'acqua nucleasi e rimuovere i liquidi eccessivi mettendo membrana tra due tovaglioli di carta puliti e premendo con decisione. Aria di essiccazione la membrana per 5 minuti si tradurrà in punti più belli.
- Per ogni campione, preparare 20 ml di RNA 200 ng / ml usando ghiaccio freddo dot blot tampone di legame (10 mM NaOH, 1 mM EDTA). Applicare 5 ml di questa diluizione (cioè 1 mg di RNA) e tre successive diluizioni di 10 volte (cioè 100, 10, e 1 ng di RNA, rispettivamente) alla membrana Zeta pipettando. Pipettaggio attraverso un rack vuoto di puntali possono essere impiegati per fornire spaziatura uniformemente distribuito. In alternativa, utilizzare un apparato di dot blot secondo le istruzioni del produttore.
- Applicare 5 pl della biotina marcato DNA oligo a concentrazioni comprese tra 20 ng / ml a 20 pg / ml (ossia 100-0,1 ng oligo) come cont positivorol alla membrana mediante pipettaggio. Utilizzare un biotinilato, campione 4SU naive come controllo negativo.
- Far asciugare la membrana per 5 min.
- Incubare la membrana per 30 minuti in 40 ml di tampone di bloccaggio con dondolo.
- Incubare la membrana con 10 ml di 1:1.000 streptavidina-perossidasi di rafano per 15 min (5 ml di PBS + 5 ml di 20% SDS + 10 pl streptavidina-perossidasi di rafano)
- Lavare membrana due volte in 40 ml di PBS + 10% SDS (20 ml di PBS + 20 ml di 20% SDS) per 5 min.
- Lavare membrana due volte in 40 ml di PBS + 1% SDS (38 ml PBS + 2 ml di 20% SDS) per 5 min.
- Lavare membrana due volte in 40 ml di PBS + 0,1% SDS (40 ml PBS + 200 microlitri 20% SDS) per 5 min.
- Rimuovere il liquido eccessivo mettendo membrana tra due tovaglioli di carta puliti e premendo su di loro con fermezza.
- Visualizza legata alla membrana HRP utilizzando ECL le istruzioni del produttore.
- Trasferire la membrana in pellicola plastica / sacchetto, rimuovere le bolle d'aria e incubare per 2 minuti al buio.
- Esporre membranapellicola per 1-5 min.
5. Separazione di RNA etichettati e senza etichetta Uso rivestite di streptavidina biglie magnetiche
- Calore tampone di lavaggio (3 ml per campione) a 65 ° C in un bagno d'acqua.
- Preparare fresco 100 mM ditiotreitolo (DTT) in nucleasi H 2 O. Fatelo per decantazione 15-30 mg di polvere DTT in un ambiente pulito tubo da 50 ml Falcon collocato sulla scala ultra-fine. Pesare e aggiungere la quantità necessaria di nucleasi H 2 O.
- Calore biotinilato campioni di RNA a 65 ° C per 10 minuti per denaturare ed immediatamente posto sul ghiaccio.
- Luogo μMacs colonne nel supporto magnetico. Si raccomanda di non elaborare più di 12 campioni per volta (6-8 campioni sono ottimali).
- Pre-equilibrare colonne MILTENYI con 1 ml di tampone di lavaggio temperatura ambiente. Questo richiederà circa 15 min.
- Nel frattempo, aggiungere 100 ml di perline streptavidina a 50 - 100 ml di RNA biotinilato. Incubare a temperatura ambiente per 15 min con rotazione. < li> Se una qualsiasi delle colonne non ha avviato drenante ormai questo può essere facilitato da una leggera pressione sulla parte superiore della colonna con un dito guantato. Una volta che il flusso è avviato colonne drenano facilmente.
- Applicare le RNA / branelli per le colonne. Gettare il flow-through a meno che non si desidera recuperare la frazione di RNA non marcato (vedi sezione 7).
- Lavare tre volte con 0,9 ml di 65 ° C tampone di lavaggio (1 ml puntali si restringono quando pipettaggio buffer a 65 ° C).
- Lavare tre volte con 0,9 ml di tampone di lavaggio temperatura ambiente.
- Pipetta 700 microlitri Buffer RLT (RNeasy MinElute Cleanup Kit, Qiagen) in nuovi ml provette 2 e metterli sotto le colonne.
- Eluire l'RNA appena trascritto nel buffer RLT con l'aggiunta di 100 ml di 100 mM DTT alle colonne.
- Eseguire una eluizione secondo turno 3 minuti più tardi nella stessa provetta con l'aggiunta di altri 100 ml di 100 mM DTT.
6. Recupero di recente RNA trascritto
ontent "> Continua con la Pulitura MinElute RNeasy (Qiagen) protocollo seguendo le istruzioni del produttore. Elute in 25 H 2 O. concentrazioni di RNA Misura nucleasi microlitri utilizzando uno spettrofotometro NanoDrop. Per evitare la necessità di scongelare e ricongelare RNA prima di presentare ad un saggio ad alta prestazione, si consiglia di preparare cDNA subito dopo la nuova trascritto RNA è purificato. Utilizzare 2,5 ml di RNA appena trascritto in 20 microlitri mix sintesi del DNA per la sintesi del DNA seguendo le istruzioni del produttore. Eseguire controlli di RT-PCR quantitativa utilizzando 1 : 10 diluizioni del mix cDNA Conservare RNA a -80 ° C..7. Recupero di Senza etichetta, Unbound RNA (Opzionale)
Nel caso l'RNA non associato devono essere recuperati; raccogliere e combinare il flusso passante (dopo l'aggiunta della soluzione perline RNA-streptavidina alle colonne) ed il primo lavaggio per successiva precipitazione. Di solito è sufficiente per far precipitare solo il 50% del non legato RNA come this conterrà> 80% del materiale di partenza.
- Aggiungere un uguale volume di isopropanolo (senza sale deve essere aggiunto come tampone di lavaggio contiene già 1 M NaCl).
- Centrifugare a 20.000 xg per 20 min a 4 ° C. Scartare il surnatante.
- Aggiungere un uguale volume di etanolo 75%, centrifugare a 20.000 xg per 10 min a 4 ° C, scartare il surnatante.
- Spin brevemente ed eliminare l'etanolo residuo con 200 microlitri pipetta.
- Spin brevemente ed eliminare l'etanolo residuo con 20 microlitri pipetta.
- Non permettere che l'RNA si asciughino. Risospendere in 100 ml H 2 O. Mescolare bene pipettando su e giù per 5-6 volte. Incubare a 65 ° C per 10 minuti con agitazione e trasferire direttamente su ghiaccio.
- Verificare la qualità dell'RNA mediante analisi electrophoretical per escludere la degradazione dell'RNA.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
1. Materiale di partenza e di rendimenti attesi
A seguito di 1 ora (ora) di 4SU-esposizione appena trascritto RNA rappresenta circa il 1-4% del totale RNA cellulare. Questa sarà la più bassa in cellule di crescita-arrestati in quanto sintetizzano più RNA per conto per la crescita cellulare / replica. Quando l'etichettatura per 1 ora, si consiglia di iniziare il test con 60 - 80 mg di RNA totale. Partendo con meno di 30 microgrammi di RNA totale risultati in piccoli pellet di RNA che sono difficili da vedere dopo il passo biotinylation e quindi può essere facilmente perso. Livelli di RNA di ingresso può essere aumentata a un massimo di 150 mg per brevi durate di etichettatura (ad esempio 5 - 10 min). Quando la durata del RNA etichettatura è ridotto da 1 ora a 5 min il contributo di breve durata sequenze introniche in aumento di RNA appena trascritto dal ~ 60% ~ 80% 9. Come introni sono sostanzialmente più lunga rispetto alle sequenze codificanti e 5'-e 3'-UTR, la quantità di trascritto di recenteRNA, che può essere purificato a seguito di breve o anche ultra-breve 4SU-tagging, non scende linearmente. Come tale, abbiamo ottenuto> 0,5% di RNA totale dopo 5 min di 4SU-tagging in non-aderenti linee di cellule B umane 9. Va, tuttavia, osservato che maggiore concentrazione di durate 4SU e leggermente più lungo di etichettatura può essere richiesto per ottenere simili tassi di incorporazione 4SU in cellule aderenti. Mentre anche un tasso 4SU-incorporazione basso permetterà di acquisizione efficienti e purificazione di grandi dimensioni, uridina ricchi di trascrizioni, molto breve trascrizione a basso contenuto di uridina (es. miRNA) sono suscettibili di sfuggire purificazione anche utilizzando alte concentrazioni 4SU (> 1 mm). In NIH-3T3 fibroblasti murini, 1 ora di 200 micron esposizione 4SU etichettato RNA appena trascritto con circa un 4SU residuo per 50 - 100 nucleotidi (nt) 5. Questo dovrebbe consentire il recupero ad alta efficienza di trascrizioni> 500 - 1000 nt di lunghezza. Di conseguenza, abbiamo osservato solo una dimensione di trascrizione minorepregiudizio per l'etichettatura per 1 ora con 200 micron 4SU in entrambi i fibroblasti murini e B-cellule umane 7. Mentre 1 ora di 200 4SU mM non ha comportato alterazioni significative dei livelli trascrizioni cellulari in fibroblasti murini, l'esposizione prolungata di cellule a ≥ 200 micron 4SU fa seguito a un deficit di crescita misurabile entro 24 ore (dati non pubblicati). Pertanto, sia la durata di etichettatura e la 4SU-concentrazione di lavoratori dovrebbero essere ridotti al minimo per evitare effetti ectopiche o tossici. Un modo semplice per determinare il minimo 4SU-concentrazione richiesta per il recupero efficiente di nuovi RNA trascritto è quello di purificare l'RNA appena trascritto seguenti 4SU-etichettatura con concentrazioni crescenti di 4SU (es. 50-1600 microM). Come mostrato nelle Figure 2A e 2B, il recupero di RNA trascritto recentemente etichettati per 1 ora in fibroblasti umani primari notevolmente aumentate da 50 a 200 pM 4SU ma poi iniziato a plateau.
2. DotAsciugare Quantificazione di Incorporazione 4SU (opzionale)
In alcuni casi può essere di interesse per misurare la quantità di incorporazione 4SU in RNA totale. Questo è fatto meglio con l'analisi dot blot sugli RNA biotinilate utilizzando un coniugato streptavidina. Per la sua natura chimica iodoacetyl-biotina è più reattivo a tiolo-gruppi di biotina-HPDP conseguente biotinilazione di quasi tutti i residui 4SU in appena trascritto RNA. È importante notare che, come biotina-HDPD, iodoacetyl-biotina non è solubile in acqua e viene così efficacemente rimosso mediante estrazione con cloroformio interpretato per biotina-HPDP. Pertanto, condizioni di reazione identiche e concentrazioni possono essere impiegati come quando si usa biotina-HPDP. Tuttavia, iodoacetyl-biotina non è reversibile. Non può pertanto essere utilizzato per la purificazione di RNA trascritto in recente approcci basati colonna. Mentre l'uso di iodoacetyl-biotina consente di quantificare 4SU-incorporazione, le misurazioni basate biotina HPDP considerano entrambi4SU-incorporazione e l'efficienza biotinilazione. Impiegando i due reagenti biotinylation allo stesso campione consente la misura dell'efficienza biotinilazione di RNA-costituita 4SU. Efficienza biotinilazione di biotina-HPDP per 4SU RNA marcate sembra essere di circa tre volte inferiore a quella di iodoacetyl-biotina indicando che solo uno su tre 4SU residui nei nuovi RNA trascritto è effettivamente biotinilato da biotina-HPDP (Figura 3). Confrontando le intensità di segnale di esempio con il controllo biotinilato DNA oligo, densità biotinilazione possono essere misurati. Per la maggior parte delle linee cellulari di mammiferi un segnale positivo dovrebbe essere ancora rilevabile in 10 ng di RNA biotinilate dopo 1 ora di 200 micron etichettatura 4SU. Un segnale di fondo debole è solitamente rilevabile per la più alta concentrazione (1 pg) di RNA non marcati.
3. Purificazione di recente RNA trascritto
Recupero di appena trascritto RNA è altamente Quantitativo. Se hai iniziato con la stessa concentrazione di RNA si può aspettare di ottenere le stesse quantità di RNA appena trascritto per tutti i campioni. Come molti saggi in colonne, raccolta di RNA appena trascritto utilizzando il kit RNeasy MinElute può comportare ulteriori assorbimento a 230-260 nm (presenza di detergenti derivati dai tamponi di lavaggio) che possono interferire con OD 260 misurazioni. Questo è visto in misura minore quando si utilizza un nuovo tubo di raccolta 2 ml per ogni fase di centrifugazione. Tuttavia, eventuali misure irragionevolmente alti OD (> 2 volte superiori rispetto agli altri campioni) devono essere considerati con cautela, soprattutto se OD 260/280 rapporti sono <1.7. Per le analisi a valle è quindi spesso preferibile utilizzare la stessa quantità di volume RNA modello per tutti i campioni. Nei casi in cui i rendimenti di RNA etichettati inferiori assegno previsto per i segni di degradazione dell'RNA mediante analisi electrophoretical. RNA appena trascritto contiene quantità significativamente più grandi di grandi dimensioni, trascritti unspliced con le bande tipiche rRNA essere molto meno prominente (Figura 4).
4. Quantificazione di recente RNA trascritto
Infine, i tassi di incorporazione di 4SU a RNA appena trascritto possono essere quantificati direttamente da analisi spettrofotometrica basata sul massimo assorbimento di 4SU a 330 nm e la OD 330/260 rapporto di 5,18. Ciò richiede> 3 mg di RNA etichettati concentrate in un piccolo volume (10 - 20 l) da isopropanolo / precipitazione in etanolo. Per evitare di perdere il piccolo pellet di RNA di co-precipitazione con 30 mg di glicogeno nucleasi (Fermentas, # R0551) deve essere eseguita. Un ulteriore picco è visibile a 330 nm riflettono il tasso di incorporazione 4SU in appena trascritto RNA (Figura 5).
/ Files/ftp_upload/50195/50195fig1highres.jpg "/>
Figura 1. . Principio di marcatura metabolica con 4 thiouridine (4SU) 4SU viene aggiunta alle cellule per la (5-120 min) richiesto tempo seguita da preparazione di RNA cellulare totale. Seguendo biotinilazione tiolo-specifico, RNA cellulare totale è suddiviso in 4SU etichettati-RNA, appena trascritte e senza etichetta, RNA pre-esistenti utilizzando biglie magnetiche rivestite di streptavidina. Recentemente trascritto RNA viene recuperato dalle perline usando un agente riducente che scinde i legami disolfuro che legano l'RNA appena trascritto ai talloni. Clicca qui per ingrandire la figura .
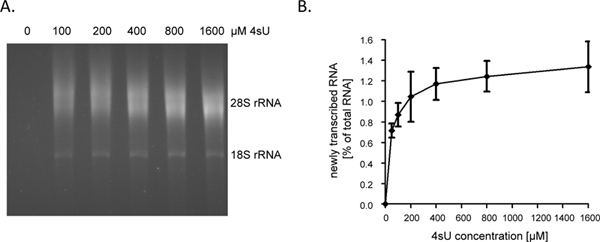
Figura 2. Recupero di nuovi RNA trascritto in seguito concentrazioni crescenti di 4SU. (A) fibroblasti prepuzio umano primarie (HFF) sono state incubate con 100, 200, 400, 800 o 1.600 mM di 4SU. RNA trascritto recentemente è stato purificato da 50 pg RNA cellulare totale e sottoposti ad analisi electrophoretical. Come previsto, una concentrazione dipendente della recuperati RNA appena trascritto è stato osservato che ha iniziato a stabilizzarsi a concentrazioni più elevate. (B) Importi di RNA purificato appena trascritto sono stati quantificati utilizzando il software ImageJ 1.45s. Dati combinati di quattro esperimenti indipendenti sulle quantità di RNA appena trascritto recuperati in seguito a diverse concentrazioni di 4SU-etichettatura che vanno da uno 50-800 micron 4SU (n = 2) o 100 -. 1.600 mM 4SU (n = 2) sono mostrati Clicca qui per ingrandire la figura .
upload/50195/50195fig3.jpg "alt =" Figura 3 "fo: contenuti-width =" 4.5in "fo: src =" / files/ftp_upload/50195/50195fig3highres.jpg "/>
Figura 3. Stima di incorporazione 4SU in RNA totale 4SU-marcati usando l'analisi dot blot. RNA totale è stato isolato da fibroblasti murini NIH-3T3 o fibroblasti prepuzio umano (HFF) incubati con 200 pM 4SU per un'ora. Nessun 4SU stato inserito un piatto come controllo negativo. Per HFF sia contatto inibito (n = cellule non-crescita) e le cellule che crescono (y) sono stati inclusi. L'RNA è stato isolato usando il reagente Trizol e successivamente coniugato con biotina-HPDP o iodoacetyl-biotina. Concentrazione di ogni campione è stato regolato a 200 ng / ml e 5 ml di questa diluizione (cioè 1 mg di RNA), nonché tre successive diluizioni di 10 volte (cioè 100, 10, e 1 ng di RNA, rispettivamente), erano tutti avvistato su un pezzo di membrana Zeta. 5 diluizioni microlitri di DNA marcato con biotina oligo sono stati collocati sulla membrana come controlli positivi a concenioni comprese tra 20 ng / ml fino a 20 pg / ml (cioè 100-,1 ng, rispettivamente). Densità di biotina è stato sondato con un coniugato streptavidina-perossidasi di rafano.
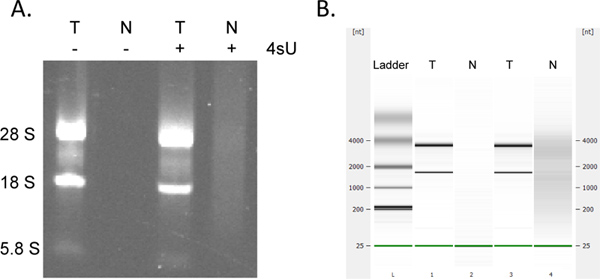
Figura 4. Electrophoretical analisi di RNA trascritto recente e totale. RNA totale (T) e recentemente trascritto RNA (N) preparata da fibroblasti murini NIH-3T3 coltivate sia in presenza che in assenza di 500 pM 4SU per 1 ora è stato analizzato mediante elettroforesi su gel di agarosio (A) e (nello stesso ordine) utilizzando il Bioanalyser Agilent (B). No RNA è stato recuperato senza trattamento 4SU delle cellule. Purificato nuova trascritto RNA contiene una maggiore quantità di alta mRNA peso molecolare e rRNA significativamente meno maturo rispetto totaleRNA come notevole tra il 28S, 18S e 5.8S rRNA bande. Clicca qui per ingrandire la figura .
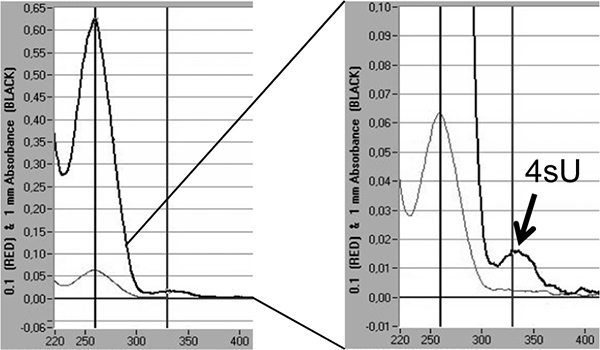
Figura 5. Quantificazione di incorporazione 4SU in RNA appena trascritto da analisi spettrofotometrica. RNA recentemente trascritto purificati da 2 x 100 mcg RNA totale dopo 1 ora di 200 micron 4SU in fibroblasti murini NIH-3T3. RNA appena trascritto è stato precipitato con isopropanolo / etanolo dopo l'aggiunta di 30 mg di glicogeno nucleasi. Viene mostrata l'analisi spettrofotometrica di RNA appena trascritto ottenuti da un NanoDrop 1000 spettrofotometro. Le linee grigio chiaro rappresentano misurazioni a 0.1 mm mentre le linee più spesse, di colore grigio scuro rappresentano misurazioni a 1 mm di colonna fluida. Sulla destra, un ingrandimento del picco di estinzione che rappresenta tha incorporato 4SU-residui è mostrato. Basato sul estinzione coefficiente di 4SU 18 i tassi di incorporazione di 4SU possono essere stimati.
| Durata di etichettatura [min] | Concentrazione 4SU consigliata [micron] |
| 120 | 100 - 200 |
| 60 | 200 - 500 |
| 15-30 | 500 - 1000 |
| <10 | 500 - 2000 |
Tabella 1. Consigliato concentrazioni 4SU.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Marcatura metabolica di nuova trascritto RNA aumenta sensibilmente la potenza delle tecnologie high-throughput, come microarrays e RNA-Seq fornendo modelli più adeguati per affrontare la questione biologica di interesse. Il presente protocollo ha subito ottimizzazione vasta. Permette> 1.000 volte arricchimento di RNA appena trascritto e fornisce risultati altamente riproducibili.
Il disegno sperimentale di un esperimento 4SU-tagging è di importanza cruciale come RNA trascritto appena si raffigurano l'attività trascrizionale tempo reale soltanto durante il tempo di esposizione delle cellule a 4SU. Se i movimenti dei tassi di trascrizione a seguito di stimolo hanno già placata, questi saranno perse quando si analizza l'RNA appena trascritto anche se i cambiamenti nei livelli totali di RNA possono essere ancora rilevabile. Pertanto, una buona comprensione della biologia di base è importante definire il setup sperimentale, nonché il periodo ottimale otempo f per l'esposizione 4SU. Di seguito, forniamo consigli e modi per evitare gli errori più comuni per i passi più importanti.
Preparazione delle soluzioni madre e articoli in plastica
Tutte le soluzioni madre devono essere preparati utilizzando acqua priva di nucleasi. Utilizzando in-house acqua purificata deionizzata può causare problemi se l'acqua contiene agenti riducenti. In un caso, ciò ha determinato la perdita completa di tutti gli RNA etichettati. Pertanto, si consiglia vivamente l'acquisto di pre-fatto priva di nucleasi NaCl, Tris-Cl, EDTA, citrato di sodio e acqua. Garantire condizioni di nucleasi in ogni momento. Dimetilformammide (DMF) scioglie alcuni materiali plastici. Abbiamo trovato che utilizzando 25 ml di coltura cellulare pipette in plastica per trasferire DMF dal suo magazzino bottiglia di vetro da 50 ml provette Falcon per preparare la soluzione madre biotina-HPDP sono sufficienti a ridurre notevolmente le rese di RNA appena trascritto da tutta dosaggio. È interessante notare che questo non ha influenzato negativamente la Biotinylefficienza zione (come testato da dot blot), ma portato a un 75 per> la perdita del 90% di RNA appena trascritto che potrebbero essere recuperati dalle perline. La perdita era più pronunciato quando la durata di etichettatura è stato ridotto da 60 a 30 minuti o meno. Molto probabilmente, una sostanza eluita dalla pipetta in plastica dal DMF parzialmente distrutto il rivestimento delle perline streptavidina. Pertanto, l'uso di materiali plastici non noti per essere compatibile con DMF dovrebbe essere evitato con ogni mezzo. Per le stesse ragioni, raschietti cellulari non dovrebbero essere utilizzati per migliorare il recupero di campioni Trizol dalle piastre di coltura cellulare. È interessante notare che le sostanze putativi eluito dalla plastica entro il DMF o Trizol erano apparentemente né rimosse mediante estrazione cloroformio né isopropanolo / precipitazione con etanolo.
Coltura cellulare
Densità delle cellule sulle piastre è di cruciale importanza. In un esperimento in cui le cellule sembravano essere un po 'troppo confluenti (90 -100%), abbiamo trattato NIH-3T3 fibroblasti murini per 30 minuti con 100 U / ml di interferone (IFN) α o γ. Nelle cellule confluenti meno anche 15 minuti di trattamento con IFN già determinato un 5 - a 8 volte induzione di geni come IRF1 o SOCS3 5. Con cellule essendo leggermente microarray troppo confluenti non ha mostrato alcuna induzione di geni IFN-inducibili per persino inducibile più rapidamente geni come IRF1 o SOCS3. Pertanto, la densità delle cellule è un fattore cruciale per esperimenti 4SU-etichettatura e tutte le piastre di coltura cellulare dovrebbe essere esaminato con attenzione prima di iniziare l'etichettatura.
4SU è un ribonucleoside fotoattivabile e 4SU contenente RNA è efficacemente reticolato a proteine dopo esposizione a 365 nm sorgente luminosa. Cellule 4SU trattati dovrebbero essere coltivati al buio e l'esposizione alla luce dovrebbe essere evitato. Dopo la rimozione delle proteine cellulari da Trizol isolamento dell'RNA questo rischio è sostanzialmente ridotta.
<p class = "jove_content"> 4SU non viene incorporato nel DNA cellulare. Va tuttavia osservato che l'RNA totale conterranno ancora piccole quantità di DNA cellulare. Quando si utilizza 4SU-tagging e analisi q-RT-PCR per lo studio dell'espressione genica virale nell'infezione da citomegalovirus abbiamo trovato necessario includere un DNaseI digest passo nel protocollo per rimuovere i genomi virali concatemeric 19. Questo probabilmente non è necessario quando si usano protocolli a valle che non sono sensibili alla presenza di DNA.Tassi di incorporazione 4SU e ottimale concentrazione 4SU
4SU è prontamente ripreso da cellule con livelli intra-ed extra-cellulare molto probabilmente equilibrante in meno di un minuto 9,16. Assorbimento e incorporazione tassi di 4SU sono concentrazione-dipendente. Pertanto, la concentrazione 4SU può essere regolata facilmente in base alla durata impiegato di etichettatura. Tabella 1 fornisce un parere sulle concentrazioni 4SU in relzione per la durata di etichettatura in base alla nostra migliore esperienza personale. Per 1 ora di etichettatura 4SU in cellule di mammiferi, 200 4SU microM saranno sufficienti per la maggior parte delle applicazioni con conseguente circa un residuo 4SU per 50 a 100 nucleotidi di RNA appena trascritto nei fibroblasti.
Negli ultimi due anni, abbiamo applicato 4SU-tagging per una vasta gamma di tipi di cellule di origine umana e murina inclusi i fibroblasti, cellule endoteliali, cellule epiteliali, cellule dello stroma del midollo osseo, macrofagi e cellule T. Inoltre, sono stati utilizzati con successo cellule dal Drosophila e Xenopus. In tutti questi esperimenti, incorporazione 4SU è risultato essere molto efficace richiede aggiustamenti minimi in concentrazione 4SU per i diversi tipi cellulari. Quando si imposta il metodo per i nuovi tipi di cellule, si consiglia di etichettare le cellule con l'aumentare 4SU-concentrazioni (ad esempio, che vanno 50-1600 microM) e analizzare la relazione di purificata appena trascritto RNA-4SU alle concentrazioni applicati (vedere la Figura 2A / B). Il 4SU-concentrazione alla quale la quantità di RNA purificato appena trascritto entra un plateau dovrebbe essere scelto.
Nei casi in cui vengono utilizzati, a contatto cellule inibito fortemente confluenti, consigliamo di usare un po 'più alte concentrazioni 4SU (ad esempio 500 invece di 200 micron) per garantire efficiente incorporazione 4SU. Inoltre, nei casi in cui l'acquisizione di brevi trascrizioni appena trascritte (<200 nt) è di particolare interesse, la concentrazione 4SU può anche avere bisogno di essere aumentata. Questo non dovrebbe essere combinato con tempi prolungati di etichettatura (ad esempio> 1 ora), al fine di evitare effetti ectopici o tossicità. Infine, abbiamo trovato che usando troppo piccolo un volume di mezzo di coltura cellulare può ridurre 4SU efficienza di incorporazione. Si consiglia pertanto di utilizzare 5 ml o 10 ml di terreno per 10 cm o 15 centimetri piatto, rispettivamente.
Preparazione di RNA cellulare totale
Per il successo di questo protocollo è fondamentale avere puliti RNA cellulare totale, RNase-free. Utilizzando 5 ml Trizol per 15 centimetri piatto produce RNA puliti gratuitamente nucleasi. Si consiglia di utilizzare il protocollo Trizol modificato da Chomczynski et al. 20. In primo luogo, è più adatto per isolare grandi quantità di RNA (> 100 mg) per il miglioramento della centrifuga vigenti determina pellet più solide, che sono più facili da gestire durante le fasi di lavaggio. Tuttavia, questo richiede l'uso di speciali tubi in polipropilene e adattatori come i normali 15 ml tubi Falcon da laboratorio non sopravvivono più di 6.000 × g. In secondo luogo, migliora la rimozione di DNA e glicoproteine. Questo diventa particolarmente evidente quando si prepara l'RNA da organi o tessuti. In terzo luogo, esso non limita la massima quantità di RNA totale, che possono essere isolati. Anche se abbiamo anche trovato metodi di isolamento di RNA colonna-based (ad esempio RNeasy) per fornire RNA di qualità adeguata, colonne standard di are solo in grado di acquisire fino a 100 mg di RNA totale limitando così la quantità di materiale di partenza. Infine, rimuovendo l'etanolo residuo due volte con una pipetta, essiccamento del RNA per eliminare l'etanolo residuo non è più richiesta. Questo elimina il rischio di sovra-essiccazione del RNA, che può essere difficile da sciogliere nuovamente in seguito. In linea di principio, 4SU-tagging è applicabile in vivo, ad esempio mediante iniezione endovenosa di topi. Tuttavia, abbiamo notato che la purezza RNA rappresenta un importante problema che richiede la purificazione di poliA trascritti prima purificazione appena trascritto RNA (dati non pubblicati).
Biotinilazione e la rimozione di non legato biotina
Biotin-HPDP è 100% tiolo-specifici e forma un legame disolfuro tra il residuo biotina e tiolo-RNA marcate nuove molecole trascritti. Efficienza biotinilazione di 4SU RNA marcate è circa il 30%, come determinato da analisi dot blot 5. Come biotina-HPDP non è solubile in acquapuò essere rimosso in modo efficiente da estrazione con cloroformio. Mentre un singolo passaggio di estrazione cloroformio è sufficiente a rimuovere la maggior parte dei non legato biotina ripetiamo regolarmente questo passaggio per assicurare la rimozione completa. Per ridurre le perdite di RNA durante la fase di estrazione cloroformio 2 ml Blocco Gel tubi pesanti fase (Eppendorf) può essere utilizzato seguendo le istruzioni del produttore. Di solito usiamo i tubi phase lock solo per la seconda fase di estrazione cloroformio come i volumi dei modelli della prima fase sono spesso troppo alti per essere direttamente compatibile con questi tubi. Dopo la rimozione di non legato biotina-HPDP, RNA viene recuperato da isopropanolo / precipitazione in etanolo. E 'importante notare che i kit commerciali colonna a base di recuperare l'RNA biotinilato (es. RNeasy da QIAGEN) non devono essere utilizzate in quanto contengono agenti riducenti nei buffer forniti, che unirà il legame disolfuro e rimuovere la biotina dalla nuova RNA trascritto .
Purificazione di nuovoly trascritto RNA
Non aggiungere più di 100 microlitri di RNA biotinilate a 100 l perline streptavidina. Aggiunta minore volume è preferito. Tuttavia, lo stesso volume di RNA deve essere aggiunta per tutti i campioni. Regolare il volume di ingresso dell'RNA (tra i campioni), che si aggiungono ai talloni streptavidina semplicemente aggiungendo il volume richiesto di 1x TE ai talloni. Un modo semplice per fare fresco nucleasi 100 mM DTT è a decantare una quantità sufficiente di polvere DTT in un tubo falcon posto su scala ultra-sensibile e quindi aggiungere la quantità richiesta di nucleasi H 2 O per generare 100 mM DTT (64,8 microlitri di acqua per 1 mg DTT).
Durante lo sviluppo di 4SU-tagging abbiamo testato perline streptavidina da vari fornitori. Un certo numero di loro ha generato una grande quantità di sfondo. Pertanto, si consiglia vivamente di utilizzare i MILTENYI streptavidina perle come, finora, non abbiamo mai avuto problemi con il riporto di RNA senza etichetta di tessuto ccampioni di RNA ultura-derivati. In questo modo, il meno 150 ng di RNA etichettati può essere specificamente purificato da 150 mg RNA biotinilato (in 100 ml di acqua) utilizzando 100 pl di perline streptavidina. Equilibrazione delle perline con il tampone di equilibratura in dotazione con le perline può essere effettuata e può leggermente aumentare tassi di cattura 13.
I controlli di qualità
Si consiglia di eseguire controlli q-RT-PCR su RNA appena trascritto prima di sottoporlo alle analisi high-throughput. Ciò può comprendere quantificazione di diversi geni di riferimento noti per essere differenzialmente regolati nel contesto dato sperimentale. Nei casi in cui 4SU-tagging è occupato di studiare tassi di decadimento di RNA, si consiglia di quantificare una trascrizione di breve durata (es. myc, fos) e una più lunga durata (ad esempio GAPDH) in RNA sia totale e recentemente trascritta. Il rapporto di RNA trascritto nuova / totale dovrebbe essere sostanzialmente più elevati (~ 5 - a 10 volte)per le trascrizioni di breve durata. Sulla base del RNA emivita di un gene di riferimento, RNA emivita può essere determinato. Se tutte e tre le frazioni di RNA (RNA, RNA appena trascritto e non marcato RNA pre-esistente) a livello di quattro o più geni, la normalizzazione dei diversi sottoinsiemi di RNA può essere eseguita mediante analisi di regressione lineare e punteggi di controllo di qualità può essere determinato come descritto 7 , 21.
Per l'analisi q-RT-PCR, si consiglia di utilizzare 2,5 ml di RNA etichettati in 20 microlitri mix sintesi del DNA. Per confronto ottimale dei risultati q-RT-PCR congelare il cDNA in aliquote di 5 ml prima del primo utilizzo. Tubi Scongelare prima dell'uso, aggiungere 45 ml di H 2 O e soggetto 5 ml delle diluizioni alle analisi q-RT-PCR. Questo migliora significativamente la comparabilità tra diverse esecuzioni di PCR.
Campioni di RNA appena trascritto devono essere controllati per i segni di degradazione dell'RNA utilizzando il Bioanalyser Agilent prima di sottoporli aanalisi high-throughput (microarrays o RNA-Seq). Si deve, tuttavia, rilevare che le bande addizionali a volte sono osservate dalla Bioanalyser Agilent. Il significato biologico di questo rimane poco chiaro. Come appena trascritto RNA contiene RNA ribosomiale significativamente meno, questi campioni di tanto in tanto riescono i controlli di qualità Bioanalyser Agilent. Se questo non è dovuto al visibile RNA campioni di degradazione di qualità accettabile sono solitamente multa da sottoporre ad analisi ad alta produttività.
Compatibilità dei nuovi RNA trascritto con analisi a valle
RNA appena trascritto contiene sostanzialmente più mRNA di RNA totale. Ciò è dovuto principalmente alle grandi quantità di sequenze introniche in RNA trascritto di recente che aumentano quando la durata di 4SU-tagging viene abbreviato. Pertanto, noi non assumiamo regolarmente l'esaurimento di rRNA di campioni di RNA appena trascritto come questo richiede grandi quantità di materiale di partenza al tempo stesso l'ing piuttosto piccolo guadagno (~ duplice) in non-rRNA legge. Resta infine da notare che la maggiore percentuale di unspliced, elevati trascrizioni peso molecolare presenti nel RNA trascritto recente può richiedere ulteriore frammentazione nel preparare librerie di cDNA per il sequenziamento di prossima generazione. Risultati della frammentazione passo dimensioni devono quindi essere di qualità controllati attentamente.
Normalizzazione dei dati per le misure a metà dal vivo di RNA
L'approccio standard per normalizzare i dati sperimentali per RNA misurazioni emivita è di normalizzare tutti i dati per l'RNA emivita di un gene house-keeping ben caratterizzati o la mediana RNA emivita in un dato tipo di cellula con esperimenti precedenti. In cellule di mammifero, quest'ultimo si trova nella gamma da 5 a 10 ore 6,7. Anche se questo approccio funziona anche abbastanza bene per misure 4SU-based, sono necessari altri mezzi per la normalizzazione se la mediana RNA emivita non è noto o se mAy anche essere influenzata da alterazioni del sistema cellulare in studio, ad esempio mediante il knock-out di un decadimento pathway di RNA. 4SU-tagging offre un modo unico di stimare la mediana RNA emivita basata sull'analisi di tutte tre frazioni RNA, cioè RNA cellulare totale, appena trascritto RNA, RNA e senza etichetta preesistenti. Come RNA cellulare totale viene separato in queste ultime due frazioni RNA un semplice modello di regressione lineare può essere impiegato per normalizzare le tre frazioni di RNA tra loro e determinare la mediana RNA emivita 7,16. Un pacchetto software è disponibile online per eseguire queste analisi 22.
Cattura inefficiente delle trascrizioni a basso contenuto di uridina può influenzare RNA misurazioni emivita conseguente artificialmente rapporti di RNA appena trascritto / totale bassi e prolungati RNA emivita. La portata di questo problema può essere valutata tracciando RNA dimezzamento o di log (appena trascritto / rapporti di RNA totale) contro il uridicontenuto ne di tutte le trascrizioni 7,15. Questo fornisce anche un buon controllo di qualità per valutare le differenze nei tassi di 4SU-incorporazione tra campioni o condizioni diverse. Nei casi in cui si osserva una correlazione sostanziale al contenuto uridina questo può essere corretto dalla bioinformatica significa 15. Tuttavia, va notato che il contributo di trascritti maturi in appena trascritto RNA non facilmente distinguibile dal molto più grande e quindi molto più ricchi di precursori di uridina. A meno che la cinetica di trasformazione di un dato trascrizione sono noti (che di solito non sono) semplicemente correggendo basso contenuto di uridina (cattura inefficiente) possono gravemente alterare RNA emivita. Come tale, abbiamo recentemente scoperto l'elaborazione della maggior parte snoRNAs umani per essere altamente inefficiente 9. Se avessimo corretto i rapporti di RNA appena trascritto / totale per il basso contenuto di uridina della piuttosto piccola (70-300 nt) snoRNAs, questo avrebbe comportato estremamente breve snoRNA mezza lives (<5 min) con numerosi rapporti di RNA appena trascritto / totale superiore al 100%. Pertanto, in genere non è consigliabile la correzione per il basso contenuto di uridina quando si misura RNA emivita.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Gli autori dichiarano di non avere interessi finanziari in competizione.
Acknowledgments
Vorremmo ringraziare Amie Regan per un'attenta lettura del manoscritto. Questo lavoro è stato sostenuto da NGFN Inoltre concessione # 01GS0801, MRC comunione concessione G1002523 e NHSBT concessione WP11-05 al LD e DFG concessione FR2938/1-1 alla CCF
Materials
| Name | Company | Catalog Number | Comments |
| 4-thiouridine | Carbosynth | T4509 | Prepare 50 mM stock in sterile H2O, store at -20 °C in aliquots of 50-500 μl, discard unused reagent, do not refreeze. |
| Trizol | Invitrogen | 15596026 (100 ml), 15596018 (200 ml) | WARNING - CORROSIVE and HAZARDOUS TO HEALTH! Ensure immediate access to Phenol antidote (PEG-Methanol); Store at 4 °C. |
| Chloroform | Sigma | 372978 | WARNING - HAZARDOUS TO HEALTH |
| Isopropanol | Sigma | 650447 | |
| Sodium citrate, nuclease-free | Sigma | C8532 | Prepare 1.6 M stock solution using nuclease-free water. |
| 5M nuclease-free NaCl | Sigma | 71386 | Stock solution |
| Nuclease-free H2O | Sigma | W4502 | Make 1 ml aliquots in nuclease-free tubes. |
| RNA precipitation buffer | 1.2 M NaCl, 0.8 M sodium citrate in nuclease free water. Prepare in advance under strictly nuclease-free conditions. Store at room temperature in 50 ml falcon tubes. | ||
| Ethanol | Sigma | 459844 | Use with nuclease-free water to prepare 80% ethanol, store at -20 °C. |
| 1 M nuclease-free Tris Cl, pH 7.5 | Lonza | 51237 | Stock solution |
| 500 mM nuclease-free EDTA, pH 8.0 | Invitrogen | 15575-020 | Stock solution |
| 10x Biotinylation Buffer (BB) | 100 mM Tris pH 7.4, 10 mM EDTA in nuclease-free water, make aliquots of 1 ml. | ||
| Dimethylformamide (DMF) | Sigma | D4551 | |
| EZ-Link biotin-HPDP | Pierce | 21341 | Prepare 1 mg/ml stock solution by dissolving 50 mg biotin-HPDP in 50 ml DMF. Gentle warming enhances solubilisation. Store at 4 °C in aliquots of 1 ml. |
| Phase Lock Gel Heavy tubes 2.0 ml | Eppendorf | 0032 005.152 | Optional for the chloroform extraction step. |
| Zeta membrane | BIORAD | 162-0153 | |
| 10x Dot blot binding buffer | 100 mM NaOH, 10 mM EDTA | ||
| Biotin-oligo | 5'-biotin, 25 nucleotides, any sequence | ||
| Sodium dodecyl sulphate | Fisher | BPE9738 | For 100 ml 20% stock solution, add 20 g SDS to 80 ml PBS pH 7-8 and adjust volume to 100 ml. Keep all high-percentage SDS solutions above 20 °C. Warm the solutions slightly should SDS precipitate. |
| EZ-Link Iodoacetyl-LC-Biotin | Pierce | 21333 | Prepare 1 mg/ml stock solution by dissolving 50 mg iodoacetyl-biotin in 50 ml DMF. Gentle warming enhances solubilisation. Store at 4 °C in aliquots of 1 ml. Generates irreversible, thiol-specific biotinylation. |
| Phosphate buffer saline | Gibco | 10010-015 | |
| Dot blot blocking buffer | Mix 20 ml 20% SDS with 20 ml 1 x PBS pH 7-8 and add EDTA to the final concentration of 1 mM. | ||
| Streptavidin-horseradish peroxidase | Vector Laboratories | SA5004 | Store at -20 °C. Mix 10 ml 20% SDS with 10 ml 1 x PBS. Add 20 μl Streptavidin-HRP before use. |
| ECL reagent | GE Healthcare | RNP2109 | Use following the manufacturer's instructions. |
| Super RX, X-RA Film, 18x24 cm | Fujifilm | 47410 19236 | |
| μMacs Streptavidin Kit | Miltenyi | 130-074-101 | Store the beads at 4 °C. |
| Tween 20 | Sigma | P1379 | |
| Washing buffer | 100 mM Tris pH 7.4, 10 mM EDTA, 1 M NaCl, 0.1% Tween 20 in nuclease-free H2O. | ||
| Dithiothreitol (DTT) | Sigma | 43817 | Prepare as 100 mM DTT in nuclease-free H2O, always prepare fresh before use. |
| RNeasy MinElute Kit | Qiagen | 74204 | Store columns at 4 °C, remaining components of the kit at room temperature. |
| 1.5 ml screw-top polypropylene tubes | Sarstedt | 72.692.005 | Compatible with Dimethylformamide |
| 2.0 ml screw-top polypropylene tubes | Sarstedt | 72.694.005 | Compatible with Dimethylformamide |
| 15 ml tubes | BD Falcon | 352096 | Compatible with Dimethylformamide |
| 50 ml tubes | BD Falcon | 352070 | Compatible with Dimethylformamide |
| All solutions/reagents should be stored at room temperature unless otherwise specified. | |||
| Equipment | |||
| UV/VIS spectrophotometer | Thermo Scientific | NanoDrop 1000 | Or equivalent. Use low volume (1-2 μl) for measurements of low RNA concentrations to avoid excessive sample loss. |
| Polypropylene 15 ml centrifuge tubes | VWR International | 525-0153 | In contrast to standard 15 ml tubes, these tolerate up to 15,000 × g |
| High-speed centrifuge | Beckman Coulter | Avanti J-25 | Or equivalent equipment capable of reaching 13,000×g |
| High-speed rotor | Beckman Coulter | JLA-16250 | Or equivalent equipment capable of reaching 13,000×g |
| Adaptors for 15 ml tubes | Laborgeräte Beranek | 356964 | Or equivalent equipment capable of reaching 13,000×g |
| Refrigerated table-top centrifuge | Eppendorf | 5430 R | Or equivalent. |
| Thermomixer | Eppendorf | Thermomixer compact | Or equivalent. |
| Magnetic stand | Miltenyi Biotec | 130-042-109 | One stand holds 8 μMacs columns. |
| Waterbath | Grant | SUB Aqua 5 | Or equivalent. |
| Ultra-fine scale | A&D | GR-202 | Or equivalent. |
| E-Gel iBase Power System | Invitrogen | G6400UK | For RNA gels; or equivalent. |
| E-Gel EX 1% agarose precast gels | Invitrogen | G4020-01 | For RNA gels; or equivalent. |
References
- Brown, P. O., Botstein, D. Exploring the new world of the genome with DNA microarrays. Nature Genetics. 21, 33-37 (1999).
- Mortazavi, A., Williams, B. A., Mccue, K., Schaeffer, L., Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nature Methods. 5, 621-628 (2008).
- Nagalakshmi, U., Wang, Z., et al. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science. 320, 1344-1349 (2008).
- Wilhelm, B. T., Marguerat, S., et al. Dynamic repertoire of a eukaryotic transcriptome surveyed at single-nucleotide resolution. Nature. 453, 1239-U1239 (2008).
- Dölken, L., Ruzsics, Z., et al. High-resolution gene expression profiling for simultaneous kinetic parameter analysis of RNA synthesis and decay. RNA. 14, 1959-1972 (2008).
- Yang, E., van Nimwegen, E., et al. Decay rates of human mRNAs: correlation with functional characteristics and sequence attributes. Genome Res. 13, 1863-1872 (2003).
- Friedel, C. C., Dölken, L., Ruzsics, Z., Koszinowski, U. H., Zimmer, R. Conserved principles of mammalian transcriptional regulation revealed by RNA half-life. Nucleic Acids Res. 37, e115 (2009).
- Nilsen, T. W., Graveley, B. R. Expansion of the eukaryotic proteome by alternative splicing. Nature. 463, 457-463 (2010).
- Windhager, L., Bonfert, T., et al. Ultra short and progressive 4sU-tagging reveals key characteristics of RNA processing at nucleotide resolution. Genome Res. , (2012).
- Melvin, W. T., Milne, H. B., Slater, A. A., Allen, H. J., Keir, H. M. Incorporation of 6-thioguanosine and 4-thiouridine into RNA. Application to isolation of newly synthesised RNA by affinity chromatography. Eur. J Biochem. 92, 373-379 (1978).
- Cleary, M. D., Meiering, C. D., Jan, E., Guymon, R., Boothroyd, J. C. Biosynthetic labeling of RNA with uracil phosphoribosyltransferase allows cell-specific microarray analysis of mRNA synthesis and decay. Nature Biotechnology. 23, 232-237 (2005).
- Kenzelmann, M., Maertens, S., et al. Microarray analysis of newly synthesized RNA in cells and animals. Proc. Natl. Acad. Sci. U.S.A. 104, 6164-6169 (2007).
- Schwanhäusser, B., Busse, D., et al. Global quantification of mammalian gene expression control. Nature. 473, 337-342 (2011).
- Rabani, M., Levin, J. Z., et al. Metabolic labeling of RNA uncovers principles of RNA production and degradation dynamics in mammalian cells. Nat. Biotechnol. , (2011).
- Miller, M. R., Robinson, K. J., Cleary, M. D., Doe, C. Q. TU-tagging: cell type-specific RNA isolation from intact complex tissues. Nat. Methods. 6, 439-441 (2009).
- Miller, C., Schwalb, B., et al. Dynamic transcriptome analysis measures rates of mRNA synthesis and decay in yeast. Mol. Syst. Biol. 7, 458 (2011).
- Weintz, G., Olsen, J. V., et al. The phosphoproteome of toll-like receptor-activated macrophages. Mol. Syst. Biol. 6, 371 (2010).
- Lipsett, M. N. The isolation of 4-thiouridylic acid from the soluble ribonucleic acid of Escherichia coli. Journal of Biological Chemistry. 240, 3975-3978 (1965).
- Marcinowski, L., Liedschreiber, M., et al. Real-time Transcriptional Profiling of Cellular and Viral Gene Expression during Lytic Cytomegalovirus Infection. PLoS Pathog. 8, e1002908 (2012).
- Chomczynski, P., Mackey, K. Short technical reports. Modification of the TRI reagent procedure for isolation of RNA from polysaccharide- and proteoglycan-rich sources. Biotechniques. 19, 942-945 (1995).
- Friedel, C. C., Dölken, L. Metabolic tagging and purification of nascent RNA: implications for transcriptomics. Mol. Biosyst. 5, 1271-1278 (2009).
- Friedel, C. C., Kaufmann, S., Dölken, L., Zimmer, R. HALO - A Java framework for precise transcript half-life determination. Bioinformatics. , (2010).
