

Summary
El ARN celular total proporciona una plantilla pobres para el estudio de los cambios a corto plazo en la síntesis de ARN y la descomposición, así como la cinética de procesamiento del ARN. A continuación, describimos marcaje metabólico de ARN recién transcrito con 4-tiouridina seguido por biotinilación específico de tiol y la purificación de ARN recién transcrito que permiten superar estas limitaciones.
Abstract
El desarrollo de microarrays de todo el transcriptoma y de secuenciación de nueva generación ha revolucionado nuestra comprensión de la complejidad de la expresión génica celular. Junto con una mejor comprensión de los mecanismos moleculares implicados, las mediciones precisas de la cinética subyacentes se han convertido en cada vez más importante. Aquí, estas metodologías poderosas se enfrentan a grandes limitaciones debido a las propiedades intrínsecas de las muestras de plantilla que estudian, es decir, el ARN celular total. En muchos casos, los cambios en el ARN celular total se producen ya sea demasiado lento o demasiado rápido para representar los eventos moleculares subyacentes y su cinética con suficiente resolución. Además, la contribución de las alteraciones en la síntesis de ARN, el procesamiento, y la decadencia no se diferencian fácilmente.
Recientemente hemos desarrollado alta resolución-perfiles de expresión génica para superar estas limitaciones. Nuestro enfoque se basa en el marcaje metabólico de los recién transcrito de ARN con 4-thiouricenar (por lo tanto también se conoce como 4SU-etiquetado) seguido por rigurosa purificación de ARN recién transcrito utilizando biotinilación específico de tiol y perlas magnéticas recubiertas con estreptavidina. Es aplicable a una amplia gama de organismos, incluyendo vertebrados, Drosophila, y levadura. Hemos aplicado con éxito 4SU-etiquetado para estudiar la cinética en tiempo real de las actividades de factor de transcripción, proporcionar mediciones precisas de ARN vidas medias, y obtener nuevos conocimientos sobre la cinética de procesamiento del ARN. Por último, el modelado computacional se puede emplear para generar un análisis integrado, integral de los mecanismos moleculares subyacentes.
Introduction
Perfiles de expresión génica es una herramienta clave usada para estudiar los procesos celulares y la red asociada a la interacción compleja. Los estudios sobre la abundancia de ARNm han sido por lo general el método de elección para obtener conocimientos básicos sobre los mecanismos moleculares subyacentes. El desarrollo de microarrays de todo el transcriptoma 1 y, más recientemente, de secuenciación de nueva generación de ARN (ARN-ss) 2-4 alimentó este enfoque. Aunque estas tecnologías han revolucionado nuestra comprensión de la complejidad de la expresión génica celular, se enfrentan a limitaciones importantes debido a las propiedades intrínsecas de la muestra de la plantilla, es decir, ARN celular total. Primero, los cambios a corto plazo en los niveles de ARN total no se ajustan a los cambios en las tasas de transcripción, pero son inherentemente dependiente en la vida media de los respectivos transcripciones de ARN. Mientras que cinco veces la inducción de una transcripción de corta duración, por ejemplo de codificación para un factor de transcripción, será fácilmente detectable en ARN totaldentro de una hora, la misma inducción de una transcripción de larga duración, por ejemplo, que codifica para una enzima metabólica, permanecerá prácticamente invisible. Además, incluso una completa desconexión (> 1.000 veces baja regulación) de la tasa de transcripción de un gen normal, con una vida media durante cinco horas ARN simplemente se llevará cinco horas para sus niveles de ARN total disminuirá en sólo dos veces . Por lo tanto, el análisis de ARN total favorece la detección de un máximo de regulación de las transcripciones de corta vida, muchos de los cuales codifican para factores de transcripción y genes con funciones de regulación 5. Además, la verdadera cascada de cinética de la regulación se oscurece y eventos de señalización primaria no puede ser diferenciada de la secundaria. Ambos, a su vez, puede dar lugar a un sesgo sustancial en posteriores análisis de la bioinformática. En segundo lugar, las alteraciones en los niveles de ARN total no se pueden atribuir a los cambios en la síntesis de ARN o la caries. Las mediciones de esta última requieren enfoques invasivos de células, por ejemplo, el bloqueo transcriptisobre el uso de actinomicina D 6, y el seguimiento prolongado de curso RNA decadencia en el tiempo. Con un ARNm media de la semivida en células de mamífero de 5 - 10 h 5,7, los niveles de mRNA de la mayoría de los genes sólo se han disminuido en menos de dos veces después de varias horas de detención transcripcional. Estas más bien pequeñas diferencias dan como resultado mediciones imprecisas groseramente de mRNA vida media para la mayoría de los genes celulares debido a la naturaleza exponencial de las ecuaciones matemáticas subyacentes. Por último, mientras que el ARN-ss de ARN celular total reveló que aproximadamente la mitad de nuestros genes están sujetos a eventos alternativos de corte y empalme 8, la cinética subyacentes, así como los mecanismos dinámicos rectores de tejido y el contexto específico de regulación de procesamiento del ARN siguen siendo poco conocidos. Además, la contribución de procesamiento del ARN para la expresión diferencial de genes, en particular para los ARN no codificantes, queda por determinar. En conjunto, estas limitaciones constituyen importantes obstáculos paramodelización cinética bioinformático de los mecanismos moleculares subyacentes.
Recientemente hemos desarrollado un enfoque, perfiles de expresión génica denominada de alta resolución, para superar estos problemas 5,7,9. Se basa en el marcaje metabólico de ARN recién transcrito utilizando 4-tiouridina (4SU-etiquetado), un derivado de uridina que ocurre naturalmente, y proporciona acceso directo a las transcripciones recién transcrito con una interferencia mínima en el crecimiento celular y la expresión génica (véase la Figura 1) 5, 10-12. La exposición de células eucariotas a los resultados de 4SU en su rápida absorción, la fosforilación de 4SU-trifosfato, y la incorporación en el ARN recién transcrito. Después del aislamiento del ARN celular total, la fracción de ARN 4SU-marcado es biotinilado tiol-específicamente la generación de un enlace disulfuro entre la biotina y el ARN recién transcrito. "Total RNA celular" puede entonces ser cuantitativamente separado en la etiqueta ('recién transcrita') y sin marcar ("pre-existing ') ARN con alta pureza usando perlas magnéticas recubiertas con estreptavidina. Por último, ARN marcado se recupera de las perlas por simple adición de un agente reductor (por ejemplo, ditiotreitol) escindir el enlace disulfuro y la liberación de los ARN recién transcrito a partir de las perlas.
Recientemente transcrito de ARN representa la actividad transcripcional de cada gen durante el marco de tiempo de la exposición 4SU. 4SU-etiquetado en el plazo de minutos proporciona así una imagen instantánea de la expresión de genes eucariotas y una plantilla ideal para los análisis bioinformáticas aguas abajo (por ejemplo, el análisis de promotor). En los casos en que se puede suponer condiciones de estado estable, las relaciones entre el recién transcrito / total, recién transcritas / sin etiqueta y sin etiqueta / ARN total proporcionan acceso no invasivo para RNA precisos vida media 7,13. Además, es importante tener en cuenta que los transcritos de ARN recientemente purificadas después de tan poco como 5 minutos de 4SU-etiquetado (5 min 4SU-ARN) es menor de 15 y 60 min 4SU-ARN.Al llevar a cabo tanto de ultra-corta y progresivamente más largo 4SU-etiquetado en un solo entorno experimental combinado con ARN-ss, la cinética de procesamiento del ARN se revelan a una resolución de 9 nucleótidos. Por último, los análisis de tiempo-luego de ARN recién transcrito y totales combinados con modelos computacionales permiten un análisis integral de la síntesis de ARN y la decadencia 14.
En conclusión, este enfoque permite el análisis directo de la dinámica de la síntesis de ARN, el procesamiento, y la degradación en las células eucariotas. Es aplicable en todos los principales organismos modelo incluidos los mamíferos, insectos (Drosophila), anfibios (Xenopus), y la levadura 5,15,16. Es directamente compatible con el análisis de microarrays 5,17, ARN-ss 9,13,14, y es aplicable en vivo 12,15. A continuación, detallamos la metodología para etiquetar, aislar y purificar el ARN recién transcrito en células de mamífero cultivadas. Además, potenciómetroSe discuten los problemas y dificultades al.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. Marcaje metabólico con 4-tiouridina
Haga un plan detallado de la configuración experimental / calendario, por ejemplo, cuándo agregar la 4SU de cultivo celular y cuándo cosechar las muestras. Plan de al menos 5 minutos entre cada condición. Sólo el tratamiento de células de un estado a la vez. Maneje max. 3 - 5 platos en un momento dado. Manipular las células tan rápidamente como sea posible para reducir al mínimo los cambios en la temperatura y los niveles de CO 2. Evite la exposición de las células a la luz brillante después de añadir 4SU ya que esto puede dar lugar a la reticulación de RNA 4SU marcados a las proteínas celulares.
Inicio de etiquetado
- Descongelar 4-tiouridina (4SU) justo antes de uso y la pipeta cantidad requerida de 4SU para cada condición en un tubo Falcon estéril.
- Tomar la cantidad requerida de medio de cultivo celular (5 ml por placa de 10 cm) de los platos y añadir a tubo Falcon 4SU que contiene y mezclar bien. Retire y deseche el medio restante de los platos. <li> Aplicar 4SU medio que contiene de nuevo a los platos.
Fin de etiquetado
- Retire el medio de cultivo celular de las células. Añadir 5 ml de Trizol a cada placa. Para los experimentos de complejos incluyendo múltiples puntos de tiempo o condiciones, este paso se lleva a cabo mejor por dos personas, una eliminación del medio, el otro la adición de Trizol y cosechar el lisado.
- Incubar durante 5 min a temperatura ambiente para la lisis celular completa.
- Utilice una pipeta de 10 ml para enjuagar la placa cuidadosamente con el Trizol añadido. Esto ayuda a la lisis celular completa y recuperación de la muestra. Maneje con cuidado ya Trizol es extremadamente peligroso al entrar en contacto con la piel o los ojos! Tener un antídoto para las quemaduras de fenol en la mano (por ejemplo, polietilenglicol 300 o 400 en alcoholes metilados industriales (70:30)). Transferir las muestras a tubos de polipropileno. Tenga en cuenta que los tubos Falcon estándar no resisten estas elevadas fuerzas g). Las muestras pueden ser almacenadas a -20 ° C durante al menos un mes hasta el ARN total is preparado.
2. Preparación de ARN usando Modificado Protocolo de Trizol
- Añadir 1 ml de cloroformo (0,2 ml por ml de Trizol) y agitar vigorosamente durante 15 segundos. Incubar a temperatura ambiente durante 2 - 3 min.
- Centrifugar a 13.000 × g durante 15 min a 4 ° C.
- Transferir fase superior acuosa (que contiene el ARN) a un nuevo tubo de 15 ml de polipropileno.
- Añadir ½ del volumen de reacción de precipitación de amortiguación tanto ARN e isopropanol (por ejemplo, a 3 ml de sobrenadante añadir 1,5 ml de tampón de precipitación ARN y 1,5 ml de isopropanol).
- Mezclar bien. Incubar a temperatura ambiente durante 10 min.
- Centrifugar a 13.000 × g durante 10 min a 4 ° C. Desechar el sobrenadante.
- Centrifugar brevemente (5000 × g durante 30 segundos) y retirar el isopropanol residual con 200 l pipeta.
- Añadir un volumen igual de etanol al 75% y agitar tubo hasta que se desenganche el pellet. Evite romper en muchos pedazos pequeños como esto puede hacer que la eliminación de secuelasl etanol difícil.
- Centrifugar a 13.000 × g durante 10 min a 4 ° C. Desechar el sobrenadante.
- Centrifugar brevemente ARN y eliminar el etanol restante con una pipeta de 200. Repita el paso y retirar el etanol restante con una pipeta de 20. Después de estos dos pasos, se debe realizar no más secado del pellet.
- Añadir 100 l de H 2 O por 100 g de ARN rendimiento esperado y mezclar bien pipeteando arriba y abajo 5 - 6 veces para ayudar a disolver el ARN.
- Disolver y desnaturalizar el ARN mediante calentamiento a 65 ° C durante 10 minutos (agitador) y colocar inmediatamente en hielo.
- Medir la concentración de ARN a 260 nm utilizando un espectrofotómetro NanoDrop, siguiendo las instrucciones del fabricante. Este ARN se puede almacenar a -80 ° C durante al menos un mes.
3. Biotinilación tiol específica de recién transcritos de ARN
- Comience con 60-80 g de ARN celular total.
- Constituir el etiquetado de reacción. Pipetear en el siguienteorden (por mu g de ARN):
- 1 l de tampón 10x Biotinilación
- 7 l de ARN (que contienen 1 RNA mu g diluidos en nucleasa libre de H 2 O)
- 2 l de biotina-HPDP (1 mg / ml de DMF)
Siempre agregue la biotina HPDP pasado y mezclar inmediatamente con la pipeta. En caso de que la biotina precipita, contenido de DMF se puede aumentar a una concentración final de 40%.
- Incubar a temperatura ambiente durante 1,5 horas con rotación.
- Añadir un volumen igual de cloroformo. Mezcle vigorosamente. Incubar durante 2 - 3 minutos hasta que las fases comienzan a separarse y burbujas comienzan a desaparecer.
- Se centrifuga a 20000 × g durante 5 min a 4 ° C. Transferir con cuidado la fase acuosa superior a un nuevo tubo.
- Repetir los pasos 3.4 y 3.5 una vez. Es posible que desee llevar a cabo este paso en 2 ml de fase Lock Gel tubos pesados para reducir la pérdida de RNA.
- ARN precipitación: añadir 1/10 del volumen de NaCl 5 M y un volumen igual deisopropanol a la fase acuosa.
- Se centrifuga a 20000 × g durante 20 min a 4 ° C. Desechar el sobrenadante.
- Añadir un volumen igual de 75% de etanol, se centrifuga a 20.000 xg durante 10 min a 4 ° C, desechar el sobrenadante.
- Girar brevemente y eliminar el etanol residual con 200 l pipeta.
- Girar brevemente y eliminar el etanol residual con 20 l pipeta.
- No permita que el ARN se sequen. Vuelva a suspender en 50 a 100 l H 2 O (~ 1 l por 1 entrada g ARN). Mezclar bien con la pipeta hacia arriba y abajo 5-6 veces.
- Comprobar la calidad del ARN por análisis electroforético de excluir la degradación del ARN.
4. Dot Blot análisis de 4SU incorporación (Opcional)
Incorporación 4SU puede ser fácilmente determinada por análisis de transferencia de punto de ARN con biotina. Este es un paso opcional que permite la resolución de problemas y la estimación de las tasas de incorporación 4SU relativas a un control oligo ADN biotinilado. Para este ensayo se recommend usando yodoacetil-biotina en lugar de biotina-HPDP para la biotinilación de ARN 4SU marcados en el paso 3.2. Esto resulta en una biotinilación irreversible de 4SU-ARN. Por lo tanto, los métodos basados en columnas (por ejemplo RNeasy) se pueden utilizar para la recuperación de cantidades mucho más pequeñas de ARN biotinilado (por ejemplo, 5 g). Mientras que el uso de ARN biotinilado biotina-HPDP también es adecuado para este ensayo, la señal resultante es más débil y la relación señal-ruido menos favorable (Figura 3).
- Siga el protocolo para 4SU el etiquetado y el aislamiento de ARN celular total como se describe en las secciones 1 y 2.
- Biotinilar 4SU marcado RNA como se describe en la sección 3 reemplazando biotina HPDP con yodoacetilo-biotina y realizar dos extracciones cloroformo para eliminar completamente excesivos residuos yodoacetilo-biotina.
- Recuperar ARN con biotina por isopropanol / precipitación con etanol como se describe o utilizando un enfoque basado en columnas (por ejemplo RNeasy) en el caso de pequeñas cantidades de ARN (<10 g) Se utilizan.
- Incubar la membrana Zeta en agua libre de nucleasa con agitación durante 10 min.
- Tome la membrana fuera del agua libre de nucleasa y eliminar el exceso de líquidos mediante la colocación de la membrana en entre dos toallas de papel limpias y presión firmemente. El secado al aire de la membrana durante 5 minutos se traducirá en puntos más agradables.
- Para cada muestra, preparar 20 l de 200 ng de ARN / l blot utilizando tampón de unión fría del punto de hielo (NaOH 10 mM, EDTA 1 mM). Aplicar 5 l de esta dilución (es decir, 1 g de ARN), así como tres diluciones de 10 veces posteriores (es decir, 100, 10, y 1 ng de ARN, respectivamente) a la membrana Zeta por pipeteado. Pipeteado a través de un rack vacío de puntas de pipeta puede ser empleada para proporcionar una separación distribuido uniformemente. Alternativamente, utilizar un aparato de transferencia de puntos de acuerdo con las instrucciones del fabricante.
- Aplicar 5 l de la biotina marcada con oligo de ADN a concentraciones que van desde 20 ng / l a 20 pg / l (es decir, 100 a 0,1 ng de oligo) como un cont positivorol a la membrana mediante pipeteo. Utilice una muestra biotinilado, 4SU-ingenuo como control negativo.
- Aire secar la membrana durante 5 min.
- Se incuba la membrana durante 30 min en 40 ml de tampón de bloqueo con balanceo.
- Incubar la membrana con 10 ml de 1:1000 de estreptavidina-peroxidasa de rábano picante durante 15 minutos (5 ml de PBS + 5 ml de SDS al 20% + 10 l de estreptavidina peroxidasa de rábano picante)
- Lavar la membrana dos veces en 40 ml de PBS + 10% de SDS (20 ml de PBS + 20 ml de SDS al 20%) durante 5 min.
- Lavar la membrana dos veces en 40 ml de PBS + 1% de SDS (38 ml de PBS + 2 ml de SDS al 20%) durante 5 min.
- Lavar la membrana dos veces en 40 ml de PBS + 0,1% de SDS (40 ml de PBS + 200 l 20% de SDS) durante 5 min.
- Retire el exceso de líquido mediante la colocación de la membrana entre dos toallas de papel limpias y presionando firmemente.
- Visualizar HRP unida a la membrana utilizando ECL según las instrucciones del fabricante.
- Colocar la membrana en papel de aluminio / bolsa de plástico, eliminar las burbujas de aire y se incuba durante 2 minutos en la oscuridad.
- Exponga membranapelícula durante 1 - 5 min.
5. La separación de RNA con y sin etiqueta usando perlas magnéticas recubiertas de estreptavidina
- Calor tampón de lavado (3 ml por muestra) a 65 ° C en un baño de agua.
- Prepare fresco ditiotreitol 100 mM (DTT) en nucleasa libre de H 2 O. Hacerlo por decantación 15 - 30 mg de polvo de TDT en un tubo Falcon de 50 ml limpio colocado en la escala ultra-fino. Pese y agregue la cantidad necesaria de nucleasa libre de H 2 O.
- Calor biotinilado muestras de ARN a 65 º C durante 10 min para desnaturalizar e inmediatamente se coloca sobre hielo.
- Lugar μMacs columnas en el soporte magnético. Se recomienda no procesar más de 12 muestras a la vez (6-8 muestras son óptimas).
- Columnas Pre-equilibre Miltenyi con 1 ml de tampón de lavado temperatura ambiente. Esto tomará alrededor de 15 min.
- Mientras tanto, añadir 100 l de perlas de estreptavidina a 50 - 100 l de ARN con biotina. Incubar a temperatura ambiente durante 15 minutos con rotación. < li> Si cualquiera de las columnas no se ha iniciado el drenaje por ahora esto puede facilitarse por presionando suavemente en la parte superior de la columna con un dedo enguantado. Una vez que el flujo ha comenzado las columnas drenan fácilmente.
- Aplicar los ARN / los granos para las columnas. Deseche el flujo continuo a menos que quiera recuperar la fracción de ARN sin etiqueta (ver sección 7).
- Lavar tres veces con 0,9 ml de 65 ° C de tampón de lavado (1 ml puntas de pipeta se encogen al pipetear tampones a 65 ° C).
- Lavar tres veces con 0,9 ml de tampón de lavado temperatura ambiente.
- Pipetear 700 l RLT Buffer (Kit de Limpieza MinElute RNeasy, Qiagen) en nuevos tubos de 2 ml y se colocan debajo de las columnas.
- Se eluye el ARN recién transcrito en el búfer RLT mediante la adición de 100 l de DTT 100 mM a las columnas.
- Lleve a cabo una ronda de elución segundos 3 minutos más tarde en el mismo tubo mediante la adición de otros 100 l de DTT 100 mM.
6. La recuperación de recién ARN transcrito
ontenido "> Continuar con la limpieza MinElute RNeasy (Qiagen) de protocolo siguiendo las instrucciones del fabricante. Elute en 25 l de H 2 O. Medir concentraciones de ARN sin nucleasa usando un espectrofotómetro Nanodrop. Para evitar la necesidad de descongelar y volver a congelar RNA antes de presentar a un ensayo de alto rendimiento, se recomienda la preparación de ADNc inmediatamente después de la recién transcrito de ARN se purifica. Utilizar 2,5 l de los ARN recién transcrito en 20 l de la mezcla de síntesis de ADNc para la síntesis de ADNc, siguiendo las instrucciones del fabricante. Realizar controles de QRT-PCR utilizando 1 : 10 diluciones de la mezcla de ADNc de la tienda de ARN a -80 ° C..7. Recuperación de sin rotular, consolidar RNA (Opcional)
En caso de que el ARN no unido necesita ser recuperado; recolectar y combinar el flujo a través (después de añadir la solución de perlas de estreptavidina-ARN a las columnas) y del primer lavado para su posterior precipitación. Por lo general, es suficiente para precipitar sólo el 50% del ARN no unido como this contendrá> 80% del material de partida.
- Añadir un volumen igual de isopropanol (no necesita ninguna sal que se añade como el tampón de lavado ya contiene 1 M de NaCl).
- Se centrifuga a 20000 × g durante 20 min a 4 ° C. Desechar el sobrenadante.
- Añadir un volumen igual de 75% de etanol, se centrifuga a 20.000 xg durante 10 min a 4 ° C, desechar el sobrenadante.
- Girar brevemente y eliminar el etanol residual con 200 l pipeta.
- Girar brevemente y eliminar el etanol residual con 20 l pipeta.
- No permita que el ARN se sequen. Resuspender en 100 l H 2 O. Mezclar bien con la pipeta hacia arriba y abajo 5-6 veces. Incubar a 65 ° C durante 10 min con agitación y transferir directamente a hielo.
- Comprobar la calidad del ARN por análisis electroforético de excluir la degradación del ARN.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
1. Material de Partida y rendimientos esperados
Después de 1 hora (h) de 4SU-exposición recién transcrito de ARN representa cerca de 1 - 4% del total de ARN celular. Este será menor en crecimiento detenido células ya que ya no sintetizan ARN para tener en cuenta para el crecimiento celular / replicación. Al etiquetar durante 1 hora, se recomienda iniciar el ensayo con 60 a 80 g de ARN total. A partir de menos de 30 g del total de ARN resultados en pequeños gránulos de ARN que son difíciles de ver después de la etapa de biotinilación y por lo tanto puede ser fácilmente perdido. Los niveles de ARN de entrada se pueden aumentar hasta un máximo de 150 g para las duraciones muy cortas de etiquetado (por ejemplo, 5 - 10 min). Cuando la duración de ARN etiquetado se acorta de 1 hr a 5 min, la contribución de las secuencias intrónicas de corta duración en incrementos de ARN recién transcrito a partir de 60 ~% a 80 ~% 9. Como intrones son sustancialmente más largo en comparación con las secuencias de codificación así como 5'-y 3'-UTRs, la cantidad de recién transcritoARN, que puede purificarse siguiendo a corto o ultracorto 4SU-tagging, no cae linealmente. Como tal, hemos obtenido> 0,5% del total de ARN tras 5 min de 4SU-etiquetado en humanos no adherentes líneas de células B 9. Debe, sin embargo, tener en cuenta que una mayor concentración de las duraciones 4SU y un poco más largo de etiquetado puede ser necesaria para alcanzar velocidades de incorporación 4SU similares en células adherentes. Mientras que incluso una baja tasa 4SU-incorporación permitirá la captura eficiente y la purificación de las transcripciones grandes, ricos en uridina, transcripción muy corto con bajo contenido de uridina (por ejemplo, miRNAs) son propensos a escapar de purificación incluso cuando se utilizan concentraciones 4SU altas (> 1 mM). En fibroblastos murinos NIH-3T3, 1 hr de 200 mM exposición 4SU etiquetados ARN recién transcrito con alrededor de un 4SU residuos por 50 - 100 nucleótidos (nt) 5. Esto debería permitir la recuperación altamente eficiente de las transcripciones> 500 - 1000 nt de longitud. En consecuencia, sólo se observó un menor tamaño de transcripciónsesgo al etiquetar durante 1 hora utilizando 200 mu M 4SU en ambos fibroblastos murinos y células B humanos 7. Mientras que 1 hora de 200 4SU mu M no resultó en alteraciones significativas en los niveles de transcripciones celulares en fibroblastos murinos, la exposición prolongada de las células a ≥ 200 mM 4SU da lugar a un déficit de crecimiento medible dentro de 24 horas (datos no publicados). Por lo tanto, tanto la duración de etiquetado y la 4SU-concentración empleada debe ser minimizado para evitar los efectos tóxicos o ectópicos. Una manera fácil de determinar la 4SU-concentración mínima requerida para la recuperación eficiente de los recién transcrito de ARN es para purificar ARN recién transcrito siguientes 4SU-etiquetado con concentraciones crecientes de 4SU (por ejemplo, 50 a 1600 mM). Como se muestra en las Figuras 2A y 2B, la recuperación de ARN recién transcrito marcados durante 1 hora en fibroblastos humanos primarios aumentaron sustancialmente de 50 a 200 mM 4SU pero luego comenzaron a la meseta.
2. PuntoSeque Cuantificación de Incorporación 4SU (opcional)
En algunos casos puede ser de interés para medir la cantidad de incorporación 4SU en ARN total. Esto se hace mejor por análisis de transferencia de puntos en los ARN con biotina utilizando un conjugado de estreptavidina. Debido a su naturaleza química yodoacetil-biotina es más reactivo a grupos tiol que biotina-HPDP que resulta en la biotinilación de prácticamente todos los residuos 4SU en recién transcrito de ARN. Es importante tener en cuenta que, como biotina-HDPD, yodoacetilo-biotina no es soluble en agua y por lo tanto se elimina de manera eficiente por extracción con cloroformo tal como se realizó por la biotina-HPDP. Por lo tanto, idénticas condiciones de reacción y las concentraciones se pueden emplear como cuando se utiliza biotina-HPDP. Sin embargo, yodoacetilo-biotina no es reversible. Es por lo tanto no puede ser utilizado para la purificación de ARN recién transcrito en los enfoques basados en columna. Mientras que el uso de yodoacetil-biotina permite cuantificar 4SU-incorporación, las mediciones basadas en biotina-HPDP consideran tanto4SU-incorporación y la eficiencia de biotinilación. El empleo de los dos reactivos de biotinilación a la misma muestra permite la medición de la eficacia de biotinilación de ARN-incorporado 4SU. Eficiencia biotinilación de biotina-HPDP para 4SU-ARN marcado parece ser aproximadamente tres veces menor que la de yodoacetil-biotina lo que indica que sólo aproximadamente uno de cada tres 4SU residuos en los recién transcrito de ARN es en realidad biotinilados por biotina-HPDP (Figura 3). Mediante la comparación de las intensidades de señal de muestra con el control oligo de ADN biotinilado, las densidades de biotinilación se pueden medir. Para la mayoría de las líneas celulares de mamífero una señal positiva todavía debe ser detectable en 10 ng de ARN biotinilado después de 1 hora de 200 mM etiquetado 4SU. Una señal de fondo débil es generalmente detectable para la concentración más alta (1 g) de ARN no marcados.
3. La purificación de ARN transcrito Recientemente
La recuperación de recién transcrito de ARN es altamente Quancuantitativa. Si ha iniciado con la misma concentración de ARN que puede esperar obtener la misma cantidad de ARN recién transcrito para todas las muestras. Al igual que muchos ensayos basados en columnas, colección de ARN recién transcrito utilizando el kit RNeasy MinElute puede resultar en absorción adicional en 230 a 260 nm (presencia de detergentes derivados de los tampones de lavado) que pueden interferir con las mediciones de DO 260. Esto se ve en menor medida cuando se utiliza un tubo de recogida de 2 ml fresca para cada etapa de la centrifugación. Sin embargo, cualquier medición de DO indebidamente altos (> 2 veces mayor que otras muestras) se deben considerar con cuidado, sobre todo si OD 260/280 proporciones son <1,7. Para los análisis de aguas abajo es por lo tanto a menudo mejor utilizar la misma cantidad de volumen de plantilla de ARN para todas las muestras. En los casos en que los rendimientos de ARN marcado son más bajos que se espera de verificación para detectar signos de degradación del ARN por análisis electroforético. ARN recién transcrito contiene cantidades significativamente mayores de grandes transcripciones unspliced, con las bandas de rRNA típicos siendo mucho menos prominente (Figura 4).
4. La cuantificación de ARN transcrito Recientemente
Finalmente, las tasas de incorporación de 4SU en el ARN recién transcrito se pueden cuantificar directamente mediante análisis espectrofotométrico basado en el máximo de absorción de 4SU a 330 nm y la OD 330/260 ratio de 5,18. Esto requiere> 3 g de ARN marcados se concentró en un pequeño volumen (10 - 20 l) de isopropanol / precipitación con etanol. Para evitar la pérdida de la pequeña sedimento de RNA de co-precipitación con 30 g de glucógeno libre de nucleasa (Fermentas, # R0551) se debe realizar. Un pico adicional es visible a 330 nm que reflejan la tasa de incorporación de 4SU en el ARN recién transcrito (Figura 5).
/ Files/ftp_upload/50195/50195fig1highres.jpg "/>
Figura 1. . Principio de marcaje metabólico con 4-tiouridina (4SU) 4SU se añade a las células para la deseada (5-120 min) tiempo seguido de la preparación de ARN celular total. Tras biotinilización tiol específica, el ARN celular total se divide en 4SU marcados, ARN recién transcrito y sin etiqueta, ARN pre-existentes usando perlas magnéticas recubiertas con estreptavidina. Recién transcrito de ARN se recupera de las perlas usando un agente reductor que rompe los enlaces disulfuro que unen al ARN recién transcrito a las perlas. Haz clic aquí para ver más grande la figura .
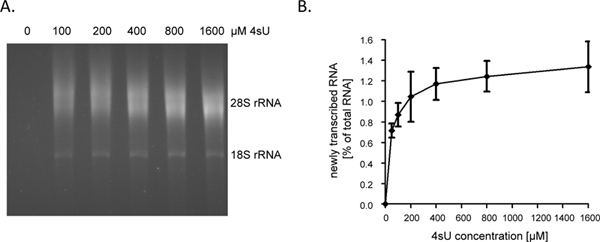
Figura 2. La recuperación de recién transcrito de ARN siguiendo concentraciones crecientes de 4SU. (A) fibroblastos primarios de prepucio humano (HFF) se incubaron con 100, 200, 400, 800 o 1.600 M de 4SU. ARN recién transcrito se purificó a partir de 50 g de ARN celular total y se sometieron a análisis electroforético. Como era de esperar, se observó un aumento dependiente de la concentración en recuperados ARN recién transcrito que comenzó a estabilizarse en concentraciones más altas. (B) Las cantidades de ARN purificado recién transcrito se cuantificaron utilizando el software ImageJ 1.45s. Los datos combinados de cuatro experimentos independientes en las cantidades de ARN recién transcrito se recuperaron después de diferentes concentraciones de 4SU etiquetado desde cualquiera de 50-800 mM 4SU (n = 2) o 100 -. 1,600 mM 4SU (n = 2) se muestran clic aquí para ver más grande la figura .
upload/50195/50195fig3.jpg "alt =" Figura 3 "fo: content-width =" 4.5in "fo: src =" / files/ftp_upload/50195/50195fig3highres.jpg "/>
Figura 3. Estimación de la incorporación en el ARN total de 4SU 4SU-etiquetados utilizando análisis de transferencia de puntos. El ARN total se aisló a partir de fibroblastos murinos NIH-3T3 o fibroblastos de prepucio humano (HFF) incubadas con 200 mM 4SU durante una hora. No hay 4SU se añadió a un plato como control negativo. Por tanto HFF contacto inhibe (n = células no crecen) y las células en crecimiento (y) fueron incluidos. Se aisló el ARN usando reactivo Trizol y, posteriormente, conjugado con biotina-HPDP o yodoacetilo-biotina. La concentración de cada muestra se ajustó a 200 ng / l y 5 l de esta dilución (es decir, 1 g de ARN), así como tres diluciones de 10 veces posteriores (es decir, 100, 10, y 1 ng de ARN, respectivamente), fueron todos visto en un pedazo de membrana Zeta. 5 l de diluciones marcado con biotina oligo de ADN se colocaron sobre la membrana como controles positivos en conceniones que van desde 20 ng / l a 20 pg / l (es decir, 100 a 0,1 ng, respectivamente). Densidad de biotina se sondeó utilizando un conjugado de peroxidasa de rábano-estreptavidina.
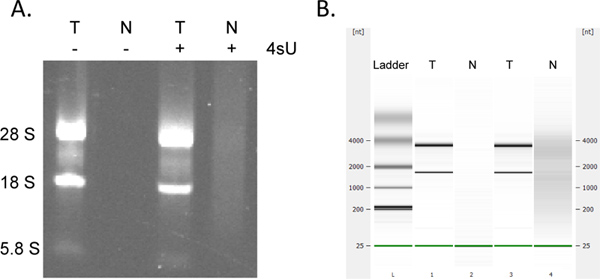
La Figura 4. Análisis electroforético de ARN recién transcrito y total. ARN total (T) y recién transcrito de ARN (N) preparada a partir de fibroblastos NIH-3T3 murinas cultivadas tanto en presencia y ausencia de 500 mM 4SU durante 1 h se analizó por electroforesis en gel de agarosa (A) y (en el mismo orden) usando el Bioanalyser Agilent (B). No se ARN se recuperó sin tratamiento 4SU de las células. Purificada ARN recién transcrito contiene una mayor cantidad de ARNm de alto peso molecular y rRNAs significativamente menos maduros que el total deRNA tan notable entre el 28S, 18S y 5.8S rRNA bandas. Haz clic aquí para ver más grande la figura .
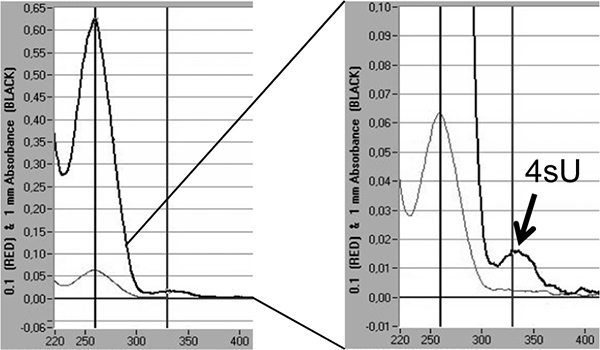
Figura 5. La cuantificación de la incorporación en el ARN 4SU recién transcrito mediante análisis espectrofotométrico. ARN recién transcrito purificadas a partir de 2 x 100 mg de ARN total después de 1 h de 200 mM 4SU en fibroblastos NIH-3T3 murinos. ARN recién transcrito se precipitó con isopropanol / etanol después de añadir 30 g de glucógeno libre de nucleasa. Se muestra el análisis espectrofotométrico de ARN recién transcrito obtenidos por un espectrofotómetro Nanodrop 1000. Las líneas de color gris claro representan las mediciones a 0,1 mm, mientras que las líneas gruesas de color gris oscuro representan valores en la columna de fluido 1 mm. A la derecha, un aumento del pico de extinción que representa tincorporó 4SU-residuos se muestran. Sobre la base del coeficiente de extinción de 18 4SU las velocidades de incorporación de 4SU se pueden estimar.
| Duración del etiquetado [min] | Concentración 4SU Recomendado [mM] |
| 120 | 100-200 |
| 60 | 200 - 500 |
| 15 - 30 | 500 - 1000 |
| <10 | 500 - 2000 |
Tabla 1. Concentraciones 4SU recomendado.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Metabólicas etiquetado de recién transcrito RNA aumenta sustancialmente el poder de las tecnologías de alto rendimiento como los microarrays y RNA-Seq, proporcionando plantillas más adecuadas para hacer frente a la cuestión biológica de interés. El presente protocolo se sometió a una amplia optimización. Se permite el enriquecimiento> 1.000 veces de ARN recién transcrito y proporciona resultados altamente reproducibles.
El diseño experimental de un experimento de 4SU-etiquetado es de importancia crucial, ya ARN recién transcrito se representar la actividad transcripcional en tiempo real sólo durante el tiempo de exposición de las células a 4SU. Si los cambios reales en las tasas de transcripción después de un estímulo ya han disminuido, estos se perdieron en el análisis de ARN recién transcrito a pesar de los cambios en los niveles de ARN total pueden todavía ser detectable. Por lo tanto, una buena comprensión de la biología subyacente es importante para definir la configuración experimental, así como la óptima o períodosf tiempo de exposición 4SU. A continuación, ofrecemos recomendaciones y las formas para evitar los errores comunes de los pasos más cruciales.
Preparación de las soluciones madre y artículos de plástico
Todas las soluciones madre deben prepararse con agua libre de nucleasa. El uso de la casa agua purificada desionizada puede dar lugar a problemas si el agua contiene agentes reductores. En un caso, esto resultó en la pérdida completa de todos los ARN marcado. Por lo tanto, le recomendamos comprar pre-hechos nucleasa libre de NaCl, Tris-Cl, EDTA, citrato de sodio y agua. Garantizar condiciones de nucleasa libre en todo momento. Dimetilformamida (DMF) se disuelve algunos materiales plásticos. Hemos encontrado que el uso de 25 ml de cultivo de células pipetas de plástico para transferir DMF de su botella de vidrio stock a tubos Falcon de 50 ml para preparar la solución madre de biotina-HPDP era suficiente para reducir sustancialmente los rendimientos de ARN recién transcrito a partir de todo el ensayo. Curiosamente, esto no afectó negativamente a la biotinileficiencia ación (como se probó por dot blot), pero resultó en un 75 a> 90% de pérdida de ARN recién transcrito que podrían ser recuperados de las perlas. La pérdida fue más pronunciado cuando la duración de etiquetado se redujo de 60 a 30 min o menos. Lo más probable, una sustancia eluida de las pipetas de plástico por el DMF destruyó parcialmente el recubrimiento de las perlas de estreptavidina. Por lo tanto, el uso de materiales plásticos no se sabe que son compatibles con DMF se debe evitar por todos los medios. Por las mismas razones, raspadores de células no deben ser utilizados para mejorar la recuperación de muestras de Trizol de placas de cultivo celular. Es interesante observar que las sustancias putativo eluidas de los plásticos por el DMF o Trizol fueron aparentemente ni eliminan por extracción con cloroformo ni isopropanol / precipitación con etanol.
Cultivo de células
La densidad celular en las placas es de importancia crucial. En un experimento en el que las células parecían ser un poco demasiado confluentes (90 -100%), se trataron fibroblastos murinos NIH-3T3 durante 30 minutos con 100 U / ml de interferón (IFN) α o γ. En menos células confluentes incluso 15 min de tratamiento con IFN ya resultó en un 5 - a 8 veces la inducción de genes como IRF1 o SOCS3 5. Con las células siendo ligeramente el análisis de microarrays demasiado confluente no mostró ninguna inducción de genes inducibles por IFN, incluso para los más rápidamente inducible genes como IRF1 o SOCS3. Por lo tanto, la densidad celular es un factor crucial para los experimentos 4SU-etiquetado y todas las placas de cultivo celular debe ser examinado cuidadosamente antes de comenzar el etiquetado.
4SU es un ribonucleósido fotoactivable y 4SU que contiene RNA se entrecruza de manera eficiente a las proteínas después de la exposición a la fuente de luz 365 nm. 4SU células tratadas deben ser cultivadas en la oscuridad y la exposición a la luz brillante se debe evitar. Después de la eliminación de las proteínas celulares mediante el aislamiento de ARN Trizol se reduce sustancialmente este riesgo.
<p class = "jove_content"> 4SU no se incorpora en el ADN celular. Debe, sin embargo, tener en cuenta que el total de ARN se todavía contienen pequeñas cantidades de ADN celular. Al utilizar 4SU-tagging y análisis Q-RT-PCR para estudiar la expresión génica viral en la infección por citomegalovirus nos vimos en la necesidad de incluir un paso DNaseI resumen en el protocolo para eliminar los genomas virales concatemeric 19. Esto probablemente no es necesario cuando se utilizan protocolos de aguas abajo que no son sensibles a la presencia de ADN.Velocidades de incorporación 4SU y la concentración óptima 4SU
4SU es fácilmente absorbido por las células con niveles intra y extracelular probablemente equilibrado en menos de un minuto 9,16. Captación e incorporación tasas de 4SU son dependiente de la concentración. Por lo tanto, la concentración 4SU puede ser convenientemente ajustado de acuerdo a la duración empleada de etiquetado. Tabla 1 proporciona asesoramiento sobre las concentraciones 4SU en relción a la duración de etiquetado basado en nuestra mejor experiencia personal. Para 1 h de etiquetado 4SU en células de mamíferos, 200 4SU mu M serán suficientes para la mayoría de aplicaciones que resultan en cerca de un residuo 4SU por cada 50 a 100 nucleótidos en el ARN recién transcrito en fibroblastos.
En el último par de años, hemos aplicado 4SU-etiquetado para una amplia gama de tipos de células de origen humano y murino, incluyendo fibroblastos, células endoteliales, células epiteliales, células del estroma de médula ósea, macrófagos y células T. Además, se utilizaron con éxito células de Drosophila y Xenopus. En todos estos experimentos, se encontró que la incorporación 4SU a ser altamente eficiente que requiere un mínimo de ajustes en la concentración de 4SU para los diferentes tipos de células. Al establecer el método para nuevos tipos de células, recomendamos a las células de etiquetas con el aumento de las concentraciones de 4SU (por ejemplo, que van desde 50 hasta 1600 m) y analizar la relación entre purificada recién transcrita RNA a los 4SU-concentraciones aplicadas (véase la figura 2A / B). El 4SU-concentración a la que la cantidad de transcrito de ARN recién purificada entra en una meseta debe ser elegido.
En los casos en que se utilizan, las células inhibidas contacto altamente confluentes, recomendamos usar concentraciones 4SU ligeramente más altos (por ejemplo, 500 en lugar de 200 M) para garantizar la eficacia incorporación 4SU. Además, en los casos en que la captura de muy corto transcripciones recién transcritas (<200 nt) es de particular interés, también puede ser necesario aumentar la concentración 4SU. Esto no se debe combinar con tiempos prolongados de etiquetado (por ejemplo,> 1 hora) con el fin de evitar los efectos ectópicos o toxicidad. Finalmente, hemos encontrado que el uso de demasiado pequeño un volumen de medio de cultivo celular puede reducir la eficacia de incorporación 4SU. Por lo tanto, recomendamos el uso de 5 ml o 10 ml de medio por 10 cm o 15 cm de plato, respectivamente.
Preparación de ARN celular total
Para el éxito de este protocolo es crucial para obtener ARN celular total limpios, libres de RNasa. Uso de 5 ml de Trizol por 15 cm de plato produce ARN limpios libres de nucleasas. Le recomendamos que utilice el protocolo Trizol modificada por Chomczynski et al. 20. En primer lugar, es más adecuado para aislar grandes cantidades de ARN (> 100 g) como la fuerza centrífugas resultados mejorados en gránulos más firmes que son más fáciles de manejar durante las etapas de lavado. Sin embargo, esto requiere el uso de tubos y adaptadores especiales de polipropileno como las regulares 15 ml tubos Falcon de laboratorio no sobreviven más de 6000 x g. En segundo lugar, mejora la eliminación de ADN y glicoproteínas. Esto se hace particularmente evidente cuando la preparación de ARN a partir de órganos o tejidos. En tercer lugar, no se limita la máxima cantidad de ARN total que se pueden aislar. Aunque también encontramos métodos de aislamiento de ARN basados en columnas RNeasy (por ejemplo) para proporcionar ARN de calidad adecuada, columnas estándar are sólo es capaz de capturar hasta 100 g de ARN total, limitando así la cantidad de material de partida. Por último, mediante la eliminación del etanol restante dos veces con una pipeta, el secado de la ARN para eliminar el etanol residual ya no es necesario. Esto elimina el riesgo de un exceso de secado del ARN, que puede ser difícil de disolver de nuevo después. En principio, 4SU-etiquetado es aplicable in vivo, por ejemplo, mediante inyección iv de los ratones. Sin embargo, hemos observado que la pureza del ARN representa un problema importante que requiere la purificación de las transcripciones polyA antes de la purificación de los recién transcrito de ARN (datos no publicados).
Biotinilación y la eliminación de la biotina no unida
Biotina-HPDP es 100% específico de tiol y forma un enlace disulfuro entre el residuo de biotina y tiol-etiquetados moléculas de ARN recién transcrito. La biotinilación de la eficiencia 4SU-ARN marcado es de aproximadamente 30% según se determinó por análisis de transferencia de puntos 5. Como biotina-HPDP no es soluble en aguase puede eliminar de manera eficiente por extracción con cloroformo. Mientras que una sola etapa de extracción de cloroformo es suficiente para eliminar la inmensa mayoría de biotina no unida repetimos regularmente este paso para asegurar la eliminación completa. Para reducir la pérdida de ARN durante la etapa de extracción con cloroformo 2 ml Lock Gel tubos fase pesada (Eppendorf) puede ser utilizado siguiendo las instrucciones del fabricante. Por lo general, se utilizan los tubos de bloqueo de fase sólo para la segunda etapa de extracción con cloroformo como los volúmenes de la plantilla de la primera etapa son a menudo demasiado alto para ser directamente compatible con estos tubos. Después de la eliminación del no unido biotina-HPDP, el ARN se recupera mediante isopropanol / precipitación con etanol. Es importante tener en cuenta que los kits basados en columnas comerciales para recuperar el ARN biotinilado (por ejemplo, de QIAGEN RNeasy) no debe ser usado ya que contienen agentes reductores en los tampones proporcionados, que escinden el enlace disulfuro y eliminar la biotina a partir de la recién transcrito de ARN .
Purificación de nuevoLY ARN transcrito
No agregue más de 100 l de ARN con biotina a 100 perlas de estreptavidina l. Adición de menos volumen se prefiere. Sin embargo, se debe agregar la misma cantidad de RNA para todas las muestras. Ajuste el volumen de entrada de ARN (entre muestras) que se agrega a las perlas de estreptavidina simplemente añadiendo el volumen requerido de 1x TE a las perlas. Una manera fácil de hacer mM DTT libre de nucleasa 100 dulce es decantar una cantidad suficiente de polvo de la TDT en un tubo Falcon colocado en una escala ultra-sensible y luego añadir la cantidad necesaria de nucleasa libre de H 2 O para generar DTT 100 mM (64,8 l de agua por DTT 1 mg).
Durante el desarrollo de 4SU-etiquetado probamos streptavidina bolas de distintos proveedores. Varios de ellos genera una gran cantidad de antecedentes. Por lo tanto, se recomienda utilizar las cuentas de estreptavidina Miltenyi ya que, hasta ahora, nunca hemos tenido ningún problema con el traspaso de ARN sin etiqueta de tejido cLas muestras de ARN ulture-derivados. De esta manera, tan poco como 150 ng de ARN marcado se puede purificar específicamente de 150 g de ARN biotinilado (en 100 l de agua) usando 100 l de perlas de estreptavidina. El equilibrado de las perlas con el tampón de equilibrado suministrado con las perlas se puede realizar y puede mejorar ligeramente las tasas de captura 13.
Los controles de calidad
Se recomienda la realización de controles de q-RT-PCR sobre ARN recién transcrito antes de someterlo a análisis de alto rendimiento. Esto puede incluir la cuantificación de varios genes de referencia se sabe son regulados diferencialmente en el entorno experimental dado. En los casos en los que se emplea 4SU-etiquetado para estudiar las tasas de descomposición de ARN, lo recomendamos para cuantificar la transcripción de corta duración (por ejemplo myc, fos) y una larga duración de un (por ejemplo GAPDH), tanto en el total y recientemente ARN transcrito. La proporción de ARN recién transcrito / total debe ser sustancialmente mayor (~ 5 - a 10 veces)de las transcripciones de corta duración. En base a la vida media de un gen de referencia ARN, ARN vidas medias se pueden determinar. Si se analizan las tres fracciones de ARN (ARN total, ARN recién transcrito y sin etiqueta de ARN pre-existente) durante cuatro o más genes, la normalización de los diferentes subgrupos de ARN se puede realizar mediante el análisis de regresión lineal y las puntuaciones de control de calidad se puede determinar como se describe 7 , 21.
Para el análisis de q-RT-PCR, se recomienda utilizar 2,5 l de ARN marcado en 20 l de la mezcla de síntesis de ADNc. Para la comparación óptima de los resultados Q-RT-PCR congelar el ADNc en alícuotas de 5 l antes del primer uso. Descongelar los tubos justo antes de su uso, añadir 45 l de H 2 O y con sujeción 5 l de las diluciones a análisis q-RT-PCR. Esto mejora significativamente a la comparabilidad entre las diferentes carreras de PCR.
Las muestras de ARN recién transcrito se debe comprobar si hay signos de degradación del ARN utilizando el Bioanalyser Agilent antes de someterlos aanálisis de alto rendimiento (microarrays o RNA-seq). Debe, sin embargo, tener en cuenta que las bandas adicionales a veces se observan por el Bioanalyser Agilent. El significado biológico de esta sigue siendo poco clara. Como recién transcrito RNA contiene RNA ribosomal mucho menos, estas muestras de vez en cuando fallan los controles de calidad Bioanalyser Agilent. Si esto no es debido a la degradación del ARN muestras visibles de calidad aceptable son generalmente muy bien ser sometido a análisis de alto rendimiento.
Compatibilidad de los recién transcrito de ARN con análisis de aguas abajo
ARN recién transcrito ARNm contiene sustancialmente más que el total de ARN. Esto se debe principalmente a las cantidades más grandes de secuencias intrónicas en el ARN recién transcrito que aumentan cuando se acorta la duración de 4SU-etiquetado. Por lo tanto, no nos comprometemos con regularidad el agotamiento de ARNr a partir de muestras de ARN recién transcrito, ya que requiere grandes cantidades de material de partida, al tiempo que proporcionación y no poca ganancia (~ doble) en la no-rRNA lee. Por último, queda por señalar que el mayor porcentaje de unspliced, transcripciones de alto peso molecular presentes en el ARN recién transcrito puede requerir fragmentación adicional en la preparación de bibliotecas de ADNc para la secuenciación de próxima generación. Los resultados de la etapa de fragmentación tamaño deben, por tanto, la calidad cuidadosamente controlados.
La normalización de datos de ARN mediciones medio-vivos
El enfoque estándar para normalizar los datos experimentales para las mediciones de la vida media de ARN es para normalizar todos los datos de la vida media de un gen de mantenimiento de la casa bien caracterizado ARN o la vida media en un determinado tipo de células determinado en experimentos anteriores mediana de ARN. En células de mamíferos, esta última se encuentra en el rango de 5 a 10 h 6,7. Si bien este enfoque también trabaja muy bien para mediciones 4SU basados, se requieren otros medios para la normalización si la mediana de la semivida de ARN no se conoce o si may aún verse afectada por alteraciones en el sistema celular en estudio, por ejemplo, por el knock-out de una vía RNA decadencia. 4SU-etiquetado ofrece una forma única de la estimación de la mediana de la semivida de ARN basado en el análisis de las tres fracciones de ARN, es decir, el ARN celular total, recién transcrito de ARN, y no marcados ARN pre-existentes. Como el ARN celular total se separa en las dos últimas fracciones de ARN de un modelo de regresión lineal simple se puede emplear para normalizar las tres fracciones de ARN entre sí y determinar la vida media de 7,16 mediana de ARN. Un paquete de software está disponible en línea para llevar a cabo estos análisis 22.
Captura ineficiente de las transcripciones con bajo contenido de uridina puede afectar las mediciones de la semivida de ARN que resulta en tasas artificialmente bajas ARN recién transcrito / total y ARN vida media prolongada. La magnitud de este problema se puede evaluar mediante el trazado de ARN vidas medias o registro (recién transcrito / proporciones de ARN total) en contra de la uridicontenidos ne de todas las transcripciones 7,15. Esto también proporciona un buen control de calidad para evaluar las diferencias en las tasas de 4SU-incorporación entre diferentes muestras o condiciones. En los casos en que se observa una correlación sustancial al contenido uridina puede ser corregida por bioinformático medios 15. Sin embargo, cabe señalar que la contribución de los transcritos maduros en recién transcrito de ARN no puede diferenciarse fácilmente de la mucho más grande y por lo tanto mucho más precursores de uridina-ricos. Ser que se sepa la cinética de transformación de una transcripción dada (la que por lo general no lo son), simplemente corrigiendo bajo contenido de uridina (captura ineficiente) puede distorsionar groseramente ARN vida media. Por lo tanto, encontramos recientemente procesamiento de la mayoría de snoRNAs humanos para ser altamente ineficientes 9. Si hubiéramos corregido las proporciones de ARN recién transcrito / total por el bajo contenido de uridina de la más pequeña (70 a 300 nt) snoRNAs, esto habría dado lugar a muy corto snoRNA medio-lives (<5 min) con numerosas relaciones de ARN recién transcrito / total de más de 100%. Por lo tanto, por lo general, no se recomienda la corrección de bajo contenido de uridina en la medición de ARN vida media.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Los autores declaran que no tienen intereses financieros en competencia.
Acknowledgments
Nos gustaría dar las gracias a Amie Regan de una cuidadosa lectura del manuscrito. Este trabajo fue apoyado por NGFN Plus subvención # 01GS0801, MRC comunión subvención y subvención G1002523 NHSBT WP11-05 al LD y DFG subvención FR2938/1-1 de CCF
Materials
| Name | Company | Catalog Number | Comments |
| 4-thiouridine | Carbosynth | T4509 | Prepare 50 mM stock in sterile H2O, store at -20 °C in aliquots of 50-500 μl, discard unused reagent, do not refreeze. |
| Trizol | Invitrogen | 15596026 (100 ml), 15596018 (200 ml) | WARNING - CORROSIVE and HAZARDOUS TO HEALTH! Ensure immediate access to Phenol antidote (PEG-Methanol); Store at 4 °C. |
| Chloroform | Sigma | 372978 | WARNING - HAZARDOUS TO HEALTH |
| Isopropanol | Sigma | 650447 | |
| Sodium citrate, nuclease-free | Sigma | C8532 | Prepare 1.6 M stock solution using nuclease-free water. |
| 5M nuclease-free NaCl | Sigma | 71386 | Stock solution |
| Nuclease-free H2O | Sigma | W4502 | Make 1 ml aliquots in nuclease-free tubes. |
| RNA precipitation buffer | 1.2 M NaCl, 0.8 M sodium citrate in nuclease free water. Prepare in advance under strictly nuclease-free conditions. Store at room temperature in 50 ml falcon tubes. | ||
| Ethanol | Sigma | 459844 | Use with nuclease-free water to prepare 80% ethanol, store at -20 °C. |
| 1 M nuclease-free Tris Cl, pH 7.5 | Lonza | 51237 | Stock solution |
| 500 mM nuclease-free EDTA, pH 8.0 | Invitrogen | 15575-020 | Stock solution |
| 10x Biotinylation Buffer (BB) | 100 mM Tris pH 7.4, 10 mM EDTA in nuclease-free water, make aliquots of 1 ml. | ||
| Dimethylformamide (DMF) | Sigma | D4551 | |
| EZ-Link biotin-HPDP | Pierce | 21341 | Prepare 1 mg/ml stock solution by dissolving 50 mg biotin-HPDP in 50 ml DMF. Gentle warming enhances solubilisation. Store at 4 °C in aliquots of 1 ml. |
| Phase Lock Gel Heavy tubes 2.0 ml | Eppendorf | 0032 005.152 | Optional for the chloroform extraction step. |
| Zeta membrane | BIORAD | 162-0153 | |
| 10x Dot blot binding buffer | 100 mM NaOH, 10 mM EDTA | ||
| Biotin-oligo | 5'-biotin, 25 nucleotides, any sequence | ||
| Sodium dodecyl sulphate | Fisher | BPE9738 | For 100 ml 20% stock solution, add 20 g SDS to 80 ml PBS pH 7-8 and adjust volume to 100 ml. Keep all high-percentage SDS solutions above 20 °C. Warm the solutions slightly should SDS precipitate. |
| EZ-Link Iodoacetyl-LC-Biotin | Pierce | 21333 | Prepare 1 mg/ml stock solution by dissolving 50 mg iodoacetyl-biotin in 50 ml DMF. Gentle warming enhances solubilisation. Store at 4 °C in aliquots of 1 ml. Generates irreversible, thiol-specific biotinylation. |
| Phosphate buffer saline | Gibco | 10010-015 | |
| Dot blot blocking buffer | Mix 20 ml 20% SDS with 20 ml 1 x PBS pH 7-8 and add EDTA to the final concentration of 1 mM. | ||
| Streptavidin-horseradish peroxidase | Vector Laboratories | SA5004 | Store at -20 °C. Mix 10 ml 20% SDS with 10 ml 1 x PBS. Add 20 μl Streptavidin-HRP before use. |
| ECL reagent | GE Healthcare | RNP2109 | Use following the manufacturer's instructions. |
| Super RX, X-RA Film, 18x24 cm | Fujifilm | 47410 19236 | |
| μMacs Streptavidin Kit | Miltenyi | 130-074-101 | Store the beads at 4 °C. |
| Tween 20 | Sigma | P1379 | |
| Washing buffer | 100 mM Tris pH 7.4, 10 mM EDTA, 1 M NaCl, 0.1% Tween 20 in nuclease-free H2O. | ||
| Dithiothreitol (DTT) | Sigma | 43817 | Prepare as 100 mM DTT in nuclease-free H2O, always prepare fresh before use. |
| RNeasy MinElute Kit | Qiagen | 74204 | Store columns at 4 °C, remaining components of the kit at room temperature. |
| 1.5 ml screw-top polypropylene tubes | Sarstedt | 72.692.005 | Compatible with Dimethylformamide |
| 2.0 ml screw-top polypropylene tubes | Sarstedt | 72.694.005 | Compatible with Dimethylformamide |
| 15 ml tubes | BD Falcon | 352096 | Compatible with Dimethylformamide |
| 50 ml tubes | BD Falcon | 352070 | Compatible with Dimethylformamide |
| All solutions/reagents should be stored at room temperature unless otherwise specified. | |||
| Equipment | |||
| UV/VIS spectrophotometer | Thermo Scientific | NanoDrop 1000 | Or equivalent. Use low volume (1-2 μl) for measurements of low RNA concentrations to avoid excessive sample loss. |
| Polypropylene 15 ml centrifuge tubes | VWR International | 525-0153 | In contrast to standard 15 ml tubes, these tolerate up to 15,000 × g |
| High-speed centrifuge | Beckman Coulter | Avanti J-25 | Or equivalent equipment capable of reaching 13,000×g |
| High-speed rotor | Beckman Coulter | JLA-16250 | Or equivalent equipment capable of reaching 13,000×g |
| Adaptors for 15 ml tubes | Laborgeräte Beranek | 356964 | Or equivalent equipment capable of reaching 13,000×g |
| Refrigerated table-top centrifuge | Eppendorf | 5430 R | Or equivalent. |
| Thermomixer | Eppendorf | Thermomixer compact | Or equivalent. |
| Magnetic stand | Miltenyi Biotec | 130-042-109 | One stand holds 8 μMacs columns. |
| Waterbath | Grant | SUB Aqua 5 | Or equivalent. |
| Ultra-fine scale | A&D | GR-202 | Or equivalent. |
| E-Gel iBase Power System | Invitrogen | G6400UK | For RNA gels; or equivalent. |
| E-Gel EX 1% agarose precast gels | Invitrogen | G4020-01 | For RNA gels; or equivalent. |
References
- Brown, P. O., Botstein, D. Exploring the new world of the genome with DNA microarrays. Nature Genetics. 21, 33-37 (1999).
- Mortazavi, A., Williams, B. A., Mccue, K., Schaeffer, L., Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nature Methods. 5, 621-628 (2008).
- Nagalakshmi, U., Wang, Z., et al. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science. 320, 1344-1349 (2008).
- Wilhelm, B. T., Marguerat, S., et al. Dynamic repertoire of a eukaryotic transcriptome surveyed at single-nucleotide resolution. Nature. 453, 1239-U1239 (2008).
- Dölken, L., Ruzsics, Z., et al. High-resolution gene expression profiling for simultaneous kinetic parameter analysis of RNA synthesis and decay. RNA. 14, 1959-1972 (2008).
- Yang, E., van Nimwegen, E., et al. Decay rates of human mRNAs: correlation with functional characteristics and sequence attributes. Genome Res. 13, 1863-1872 (2003).
- Friedel, C. C., Dölken, L., Ruzsics, Z., Koszinowski, U. H., Zimmer, R. Conserved principles of mammalian transcriptional regulation revealed by RNA half-life. Nucleic Acids Res. 37, e115 (2009).
- Nilsen, T. W., Graveley, B. R. Expansion of the eukaryotic proteome by alternative splicing. Nature. 463, 457-463 (2010).
- Windhager, L., Bonfert, T., et al. Ultra short and progressive 4sU-tagging reveals key characteristics of RNA processing at nucleotide resolution. Genome Res. , (2012).
- Melvin, W. T., Milne, H. B., Slater, A. A., Allen, H. J., Keir, H. M. Incorporation of 6-thioguanosine and 4-thiouridine into RNA. Application to isolation of newly synthesised RNA by affinity chromatography. Eur. J Biochem. 92, 373-379 (1978).
- Cleary, M. D., Meiering, C. D., Jan, E., Guymon, R., Boothroyd, J. C. Biosynthetic labeling of RNA with uracil phosphoribosyltransferase allows cell-specific microarray analysis of mRNA synthesis and decay. Nature Biotechnology. 23, 232-237 (2005).
- Kenzelmann, M., Maertens, S., et al. Microarray analysis of newly synthesized RNA in cells and animals. Proc. Natl. Acad. Sci. U.S.A. 104, 6164-6169 (2007).
- Schwanhäusser, B., Busse, D., et al. Global quantification of mammalian gene expression control. Nature. 473, 337-342 (2011).
- Rabani, M., Levin, J. Z., et al. Metabolic labeling of RNA uncovers principles of RNA production and degradation dynamics in mammalian cells. Nat. Biotechnol. , (2011).
- Miller, M. R., Robinson, K. J., Cleary, M. D., Doe, C. Q. TU-tagging: cell type-specific RNA isolation from intact complex tissues. Nat. Methods. 6, 439-441 (2009).
- Miller, C., Schwalb, B., et al. Dynamic transcriptome analysis measures rates of mRNA synthesis and decay in yeast. Mol. Syst. Biol. 7, 458 (2011).
- Weintz, G., Olsen, J. V., et al. The phosphoproteome of toll-like receptor-activated macrophages. Mol. Syst. Biol. 6, 371 (2010).
- Lipsett, M. N. The isolation of 4-thiouridylic acid from the soluble ribonucleic acid of Escherichia coli. Journal of Biological Chemistry. 240, 3975-3978 (1965).
- Marcinowski, L., Liedschreiber, M., et al. Real-time Transcriptional Profiling of Cellular and Viral Gene Expression during Lytic Cytomegalovirus Infection. PLoS Pathog. 8, e1002908 (2012).
- Chomczynski, P., Mackey, K. Short technical reports. Modification of the TRI reagent procedure for isolation of RNA from polysaccharide- and proteoglycan-rich sources. Biotechniques. 19, 942-945 (1995).
- Friedel, C. C., Dölken, L. Metabolic tagging and purification of nascent RNA: implications for transcriptomics. Mol. Biosyst. 5, 1271-1278 (2009).
- Friedel, C. C., Kaufmann, S., Dölken, L., Zimmer, R. HALO - A Java framework for precise transcript half-life determination. Bioinformatics. , (2010).
