

Summary
Totalt cellulärt RNA ger en dålig mall för att studera kortsiktiga förändringar i RNA-syntes och förfall samt kinetiken för RNA-bearbetning. Här beskriver vi metabolisk märkning av nyligen transkriberat RNA med 4-tiouridin följt av tiol-specifik biotinylering och rening av nyligen transkriberat RNA gör det möjligt att övervinna dessa begränsningar.
Abstract
Utvecklingen av hela transkriptom mikromatriser och nästa generations sekvensering har revolutionerat vår förståelse av komplexiteten av cellens genuttryck. Tillsammans med en bättre förståelse av de inblandade molekylära mekanismer, har exakta mätningar av de underliggande kinetik blivit allt viktigare. Här, dessa kraftfulla metoder inför stora begränsningar på grund av inneboende egenskaper mall prover de ska studera, dvs totala cellulära RNA. I många fall förändringar i totalt cellulärt RNA ske antingen för långsamt eller för snabbt för att representera de underliggande molekylära händelser och deras kinetik med tillräcklig upplösning. Dessutom är bidraget av förändringar i RNA-syntes, bearbetning, och förfall ej lätt differentierade.
Vi utvecklade nyligen högupplösta profilering av genuttryck för att övervinna dessa begränsningar. Vår strategi bygger på metabolisk märkning av nyligen transkriberade RNA med 4-thiouridine (således även kallad 4SU-taggning) följt av rigorös rening av nyligen transkriberade RNA med användning av tiol-specifik biotinylering och streptavidin-belagda magnetiska pärlor. Det är tillämpligt på ett brett spektrum av organismer, inklusive ryggradsdjur, Drosophila och jäst. Vi tillämpade framgångsrikt 4SU-taggning för att studera i realtid kinetik aktiviteter transkriptionsfaktor, ge exakta mätningar av RNA halveringstider, och få nya insikter i kinetik RNA bearbetning. Slutligen kan datormodellering användas för att generera ett integrerat, heltäckande analys av de bakomliggande molekylära mekanismerna.
Introduction
Gene expression profiling är ett viktigt verktyg som används för att studera cellulära processer och tillhörande komplexa samspelet nätverk. Studier av mRNA överflöd har normalt varit en lämplig metod för att få grundläggande insikter i de bakomliggande molekylära mekanismerna. Utvecklingen av hela transkriptom mikromatriser 1 och, mer nyligen, nästa generations sekvensering av RNA (RNA-seq) 2-4 underblåst detta tillvägagångssätt. Även om dessa tekniker har revolutionerat vår förståelse av komplexiteten av cellens genuttryck, möter de stora begränsningar på grund av inneboende egenskaper deras mall prov, dvs totalt cellulärt RNA. Först kortsiktiga förändringar i den totala RNA-nivåer motsvarar inte förändringar i transkription priser, men är i sig beroende av RNA halveringstiden av respektive transkript. Medan en femfaldig induktion av en kortlivad transkriptet, t.ex. kodning för en transkriptionsfaktor, kommer att vara lätt detekterbara i totalt RNAinom en timme, samma induktion av en långlivad transkriptet, t.ex. kodning för ett metaboliskt enzym, kommer att förbli praktiskt taget osynlig. Dessutom, till och med en fullständig avstängning (> 1000-faldig ned-reglering) i transkriptionen satsen för en genomsnittlig genen med ett RNA-halveringstid på fem timmar kommer helt enkelt ta fem timmar för alla de RNA-nivåer för att minska med endast tvåfaldig . Därför gynnar analys av total RNA påvisande av uppreglering av kortlivade transkript, varav många kodar för transkriptionsfaktorer och gener med reglerande funktioner 5. Dessutom är den sanna kinetiska kaskad reglering fördunklas och primära signalering händelser kan inte skiljas från sekundärt. Både i sin tur kan resultera i betydande bias i nedströms bioinformatik analyser. Det andra, kan förändringar i totala RNA-nivåer inte kan hänföras till förändringar i RNA-syntes eller nedbrytning. Mätningar av de sistnämnda kräver cellen invasiva metoder, t.ex. blockerar transcriptipå att använda actinomycin D 6, och utökad övervakning av pågående RNA förfall över tid. Med en genomsnittlig mRNA halveringstid i däggdjursceller av 5 - 10 HR 5,7, kommer mRNA-nivåer av de flesta gener har bara minskat med mindre än dubbelt efter flera timmar av transkriptionell gripandet. Dessa ganska små skillnader leda till grovt oprecisa mätningar av mRNA-halveringstider för de flesta cellulära gener på grund av den exponentiella karaktären hos de underliggande matematiska ekvationer. Slutligen, medan RNA-seq av totalt cellulärt RNA visade att ungefär hälften av våra gener är föremål för alternativa splitsar händelser 8, underliggande kinetik samt dynamiska mekanismer som styr vävnads-och kontext-specifik reglering av RNA bearbetning Oklara. Dessutom bidrag RNA bearbetning till differentiell genexpression, i synnerhet för icke-kodande RNA, återstår att fastställa. Sammantaget dessa begränsningar utgör stora hinder förbioinformatic kinetisk modellering av de underliggande molekylära mekanismer.
Vi har nyligen utvecklat en metod, benämnd högupplöst profilering av genuttryck för att övervinna dessa problem 5,7,9. Den är baserad på metabolisk märkning av nyligen transkriberade RNA med 4-tiouridin (4SU-taggning), ett naturligt förekommande uridine derivat, och ger direkt tillgång till nyligen transkriberade utskrifter med minimal inblandning i celltillväxt och genuttryck (se figur 1) 5, 10-12. Exponering av eukaryota celler till 4SU resulterar i dess snabba upptag, fosforylering till 4SU-trifosfat, och inkorporering i nyligen transkriberade RNA. Efter isolering av totalt cellulärt RNA, är det 4SU-märkt RNA-fraktion tiol-specifikt biotinyleras generera en disulfidbindning mellan biotin och de nyligen transkriberade RNA. "Totalt cellulärt RNA" kan sedan kvantitativt separeras i märkt ("nyligen transkriberade) och omärkt (" pre-existing ') RNA med hög renhet med streptavidinbelagda magnetiska pärlor. Slutligen är märkt RNA återvinns från kulorna genom att helt enkelt lägga ett reduktionsmedel (t.ex. ditiotreitol) klyva disulfidbindningen och släppa de nyligen transkriberade RNA från kulorna.
Nyligen transkriberas RNA skildrar transkriptionsaktiviteten av varje gen under tidsramen för 4SU exponering. 4SU-taggning i tidsskalan minuter ger således en ögonblicksbild av eukaryot genexpression och en perfekt mall för nedströms bioinformatiska analyser (t.ex. promotor analys). I de fall där steady-state kan förutsättas, förhållandena nyligen transkriberade / totalt, nyligen transkriberas / omärkt och omärkta / totala RNA tillhandahålla icke-invasiv tillgång till exakta RNA halveringstider 7,13. Dessutom är det viktigt att notera att nyproducerade transkriberat RNA renade efter så lite som 5 min av 4SU-taggning (5 min 4SU-RNA) är yngre än 15 och 60 min 4SU-RNA.När du utför både ultrakorta och progressivt längre 4SU-taggning i en enda experimentell inställning kombinerad med RNA-seq är kinetiken av RNA bearbetning avslöjade vid nukleotid upplösning 9. Slutligen, tidsförlopp analyser av nyligen transkriberade och total RNA kombination med datormodellering möjliggöra en integrerad analys av RNA-syntes och förfall 14.
Sammanfattningsvis tillåter detta tillvägagångssätt för direkt analys av dynamiken i RNA-syntes, bearbetning, och nedbrytning i eukaryota celler. Det gäller i alla större modell organismer, inklusive däggdjur, insekter (Drosophila), amfibier (Xenopus), och jäst 5,15,16. Det är direkt kompatibel med microarray analys 5,17, RNA-seq 9,13,14, och är tillämplig i vivo 12,15. Här, vi detalj metoden att märka, isolera och rena nyligen transkriberade RNA i odlade däggdjursceller. Dessutom potentioal problem och fallgropar diskuteras.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
Ett. Metabolisk märkning med 4-tiouridin
Gör en detaljerad plan för experimentuppställning / schema, t ex när man ska lägga till 4SU till cellodling och då för att skörda proverna. Planera för minst 5 minuter mellan varje skick. Endast behandla celler av ett villkor i taget. Handtag max. 3 - 5 rätter vid en given tidpunkt. Handtag celler så snabbt som möjligt för att minimera förändringar i temperatur och CO 2 nivåer. Undvik att utsätta cellerna för starkt ljus efter 4SU tillsätts som detta kan resultera i tvärbindning av 4SU-märkta RNA till cellulära proteiner.
Börja om märkning
- Thaw 4-tiouridin (4SU) strax före användning och pipett önskad mängd 4SU för varje tillstånd i en steril Falcon rör.
- Ta önskad mängd cellodlingsmedium (5 ml per 10 cm skål) från disken och lägg till 4SU-innehållande Falcon röret och blanda väl. Avlägsna och kassera den kvarvarande medium från disken. <li> Applicera 4SU-medium tillbaka till disken.
Slut på märkning
- Avlägsna cellodlingsmedium från celler. Tillsätt 5 ml Trizol till varje platta. För komplexa experiment inklusive flera tidpunkter eller villkor, är detta steg görs bäst av två personer, en avlägsnande av mediet, den andra lägger Trizol och skörda lysatet.
- Inkubera under 5 min vid rumstemperatur för fullständig cellys.
- Använd en 10 ml pipett för att skölja plåten försiktigt med den extra Trizol. Detta underlättar fullständig cellysering och provutbyte. Hanteras varsamt som Trizol är extremt farligt när att komma i kontakt med hud eller ögon! Ha motgift för fenol brännskador på handen (t.ex. polyetylenglykol 300 eller 400 i industriell denaturerad sprit (70:30)). Överför proverna till polypropenrör. Observera att vanliga Falcon rör inte motstå dessa höga g-krafter). Prover kan förvaras vid -20 ° C under minst en månad tills totalt RNA is förberedd.
2. RNA Förberedelse Använda Ändrad Trizol Protocol
- Tillsätt 1 ml kloroform (0,2 ml per ml Trizol) och skaka kraftigt i 15 sekunder. Inkubera vid rumstemperatur under 2-3 min.
- Centrifugera vid 13.000 x g under 15 min vid 4 ° C.
- Överför vattenhaltiga övre fasen (innehållande RNA) till en ny 15 ml polypropylenrör.
- Lägg ½ reaktionen volym både RNA nederbörd buffert och isopropanol (t.ex. till 3 ml flytande tillsätt 1,5 ml RNA nederbörd buffert och 1,5 ml isopropanol).
- Blanda väl. Inkubera vid rumstemperatur under 10 min.
- Centrifugera vid 13.000 x g under 10 min vid 4 ° C. Kassera supernatanten.
- Spinn ner kort (5,000 × g under 30 sek) och avlägsna kvarvarande isopropanol med 200 l pipett.
- Tillsätt en motsvarande volym av 75% etanol och skaka röret tills pelleten lossnar. Undvik att bryta den i många små bitar som detta kan göra avlägsnandet av residual etanol svårt.
- Centrifugera vid 13.000 x g under 10 min vid 4 ° C. Kassera supernatanten.
- Spinn ner RNA kort och ta bort kvarvarande etanol med en 200 l pipett. Upprepa steg och ta bort kvarvarande etanol med en 20 l pipett. Efter dessa två steg, bör inga ytterligare torkning av pellets ske.
- Lägg 100 | il H2O per 100 | ig förväntade RNA avkastning och blanda väl genom att pipettera upp och ned 5-6 gånger för att hjälpa till vid upplösning av RNA.
- Lös upp och denaturera RNA genom upphettning till 65 ° C under 10 min (skakare) och omedelbart placera på is.
- Mät RNA koncentration vid 260 nm med användning av en NanoDrop spektrofotometer, enligt tillverkarens instruktioner. Detta RNA kan förvaras vid -80 ° C under minst en månad.
Tre. Tiol-specifik Biotinylering av Newly transkriberat RNA
- Börja med 60-80 mikrogram av det totala cellulära RNA.
- Utgör märkningsreaktionen. Pipettera i följandeordning (per mikrogram RNA):
- 1 l 10x Biotinylering Buffer
- 7 il RNA (innehållande 1 ^ g RNA utspädda i nukleasfritt H2O)
- 2 | il biotin-HPDP (1 mg / ml DMF)
Tillsätt alltid biotin-HPDP sista och blanda genast genom att pipettera. Om biotin fällningar kan DMF halten ökas till en slutlig koncentration av 40%.
- Inkubera vid rumstemperatur under 1,5 timmar med rotation.
- Lägg en lika stor volym kloroform. Blanda kraftigt. Inkubera i 2 - 3 minuter tills faserna börjar att separera och bubblor börjar försvinna.
- Centrifugera vid 20.000 x g under 5 min vid 4 ° C. Försiktigt överföra den övre vattenfasen till ett nytt rör.
- Upprepa steg 3.4 och 3.5 en gång. Du kanske vill utföra detta steg i 2 Lock ml Fas Gel Tunga rör för att minska förlusten av RNA.
- RNA nederbörd: add 1/10 av volymen av 5 M NaCl och en lika stor volymisopropanol till vattenfasen.
- Centrifugera vid 20.000 x g under 20 min vid 4 ° C. Kassera supernatanten.
- Tillsätt en motsvarande volym av 75% etanol, centrifugera vid 20.000 x g under 10 min vid 4 ° C, kassera supernatanten.
- Snurra kort och ta bort rester av etanol med 200 l pipett.
- Snurra kort och ta bort rester av etanol med 20 l pipett.
- Låt inte RNA torka. Återsuspendera det i 50 till 100 l H2O (~ 1 l per 1 mikrogram ingång RNA). Blanda väl genom att pipettera upp och ned 5-6 gånger.
- Kontrollera RNA kvalitet genom electrophoretical analys för att utesluta RNA nedbrytning.
4. Dot blot-analys av 4SU-inkorporering (tillval)
4SU inkorporering kan lätt bestämmas genom dot blot-analys av biotinylerade RNA. Detta är ett valfritt steg som gör felsökning och uppskattning av 4SU inblandningshalter förhållande till ett biotinylerat DNA oligo kontroll. För denna analys vi recommend använda jodacetyl-biotin i stället för biotin-HPDP för biotinylering av 4SU-märkt RNA i steg 3.2. Detta resulterar i en irreversibel biotinylering av 4SU-RNA. Därför (t.ex. RNeasy) kolonn-baserade metoder kan användas för återvinning av mycket mindre mängder biotinylerad RNA (t.ex. 5 | ig). Medan RNA biotinylerades med användning av biotin-HPDP är också lämplig för denna analys, är den resulterande signalen svagare och signal-brus-förhållande är mindre gynnsam (Figur 3).
- Följ protokollet för 4SU-märkning och isolering av totalt cellulärt RNA som beskrivs i avsnitt 1 och 2.
- Biotinylera 4SU-märkt RNA som beskrivs i avsnitt 3 ersätter biotin-HPDP med jodacetyl-biotin och utför två kloroformextraktioner att helt ta bort överdrivna jodacetyl-biotinrester.
- Återvinna biotinylerade RNA från isopropanol / etanol fällning som beskrivits eller använda en kolonn synsätt (t.ex. RNeasy) om små mängder RNA (<10 pg) Används.
- Inkubera Zeta membranet i nukleasfritt vatten med gungande under 10 min.
- Ta membranet ur nukleasfritt vatten och ta bort överflödig vätska genom att placera membranet i mellan två rena pappershanddukar och trycka stadigt. Luft-torkning av membranet under 5 minuter kommer att resultera i trevligare prickar.
- För varje prov, förbereda 20 pl av 200 ng / | il RNA med användning av iskall dot blöt bindningsbuffert (10 mM NaOH, 1 mM EDTA). Applicera 5 | il av denna spädning (det vill säga 1 mikrogram av RNA) såväl som tre efterföljande 10-faldiga utspädningar (dvs. 100, 10, och 1 ng RNA, respektive) till Zeta membranet genom pipettering. Pipettering genom ett tomt rack av pipettspetsar kan användas för att åstadkomma jämnt fördelade mellanrum. Alternativt kan man använda en dot blot-apparat enligt tillverkarens instruktioner.
- Applicera 5 pl av biotin-märkt DNA-oligo i koncentrationer från 20 ng / ul till 20 pg / il (dvs. från 100 till 0,1 ng oligo) som en positiv fortsrol till membranet genom pipettering. Använd en biotinylerad, 4SU-naiva prov som negativ kontroll.
- Lufttorka membranet under 5 min.
- Inkubera membranet under 30 min i 40 ml blockerande buffert med gungning.
- Inkubera membranet med 10 ml av 1:1000 streptavidin-pepparrotsperoxidas under 15 min (5 ml PBS + 5 ml 20% SDS + 10 ^ il streptavidin-pepparrotsperoxidas)
- Tvätta membranet två gånger i 40 ml PBS + 10% SDS (20 ml PBS + 20 ml 20% SDS) under 5 min.
- Tvätta membranet två gånger i 40 ml PBS + 1% SDS (38 ml PBS + 2 ml 20% SDS) under 5 min.
- Tvätta membranet två gånger i 40 ml PBS + 0,1% SDS (40 ml PBS + 200 | il 20% SDS) under 5 min.
- Ta bort överdriven vätska genom att placera membranet mellan två rena pappershanddukar och trycka på dem ordentligt.
- Visualisera membranbundet HRP användning av ECL enligt tillverkarens instruktioner.
- Placera membranet i plastfolie / påse, ta bort luftbubblor och inkubera i 2 minuter i mörker.
- Exponera membran tillfilm i 1 - 5 min.
Fem. Separation av märkta och omärkta RNA användning av streptavidin-belagda magnetiska pärlor
- Heat tvättbuffert (3 ml per prov) till 65 ° C i ett vattenbad.
- Bered en färsk 100 mM ditiotreitol (DTT) i nukleasfritt H2O Gör det genom dekantering 15-30 mg DTT pulver i en ren 50 ml Falcon-rör som placerats på det ultrafina skala. Väg och tillsätt erforderlig mängd nukleasfritt H2O
- Heat biotinylerad RNA-prover till 65 ° C under 10 min för att denaturera och omedelbart placera på is.
- Placera μMacs kolumner i den magnetiska monter. Vi rekommenderar att inte behandla mer än 12 prover åt gången (6 - 8 prover är optimal).
- Pre-jämvikt Miltenyi kolonnerna med 1 ml rumstemperatur tvättbuffert. Detta kommer att ta ca 15 min.
- Under tiden, tillsätt 100 pl av streptavidinpärloma till 50-100 | il av biotinylerade RNA. Inkubera vid rumstemperatur under 15 min med rotation. < li> Om någon av kolumnerna inte har initierat dränering nu detta kan underlättas genom att försiktigt trycka på toppen av kolonnen med en handske finger. När flödet har startat kolumnerna rinna lätt.
- Applicera RNA / pärlor till kolumnerna. Kassera genomströmning om du vill återställa den omärkta RNA fraktionen (se avsnitt 7).
- Tvätta tre gånger med 0,9 ml av 65 ° C tvätt-buffert (1 ml pipettspetsar krympa vid pipettering buffertar vid 65 ° C).
- Tvätta tre gånger med 0,9 ml rumstemperatur tvättbuffert.
- Pipettera 700 l buffert RLT (RNeasy MinElute Cleanup Kit, Qiagen) i nya 2 ml rör och placera dem under kolumnerna.
- Eluera de nyligen transkriberade RNA till RLT buffert genom att tillsätta 100 ul av 100 mM DTT till kolumnerna.
- Utför en andra eluering omgång 3 minuter senare i samma rör genom att lägga till ytterligare 100 ^ 100 mM DTT.
6. Återvinning av Newly transkriberat RNA
ontent "> Fortsätt med RNeasy MinElute Cleanup (Qiagen) protokollet enligt tillverkarens anvisningar. Elute i 25 l nukleasfritt H2O Mät RNA koncentrationer med en Nanodrop spektrofotometer. För att undvika behovet av att tina och återfrysa RNA innan du skickar den till en hög genomströmning analys vi rekommenderar förbereder cDNA omedelbart efter den nyligen transkriberade RNA renas. Använd 2,5 pl av de nyligen transkriberade RNA i 20 | il cDNA-syntes mix för cDNA-syntes genom att följa tillverkarens instruktioner. Utför QRT-PCR-kontroller med hjälp av en : 10 utspädningar av cDNA-mix Store RNA vid -80 ° C..7. Återvinning av omärkt Obundet RNA (tillval)
I fall den obundna RNA måste återvinnas, samla och kombinera genomströmning (efter tillsats av RNA-streptavidinpärloma lösning kolumnerna) och den första tvätten för efterföljande utfällning. Vanligtvis är det tillräckligt för att fälla ut endast 50% av det obundna RNA såsom This kommer att innehålla> 80% av utgångsmaterialet.
- Tillsätt en motsvarande volym isopropanol (inget salt behöver tillsättas som tvättbufferten redan innehåller 1 M NaCl).
- Centrifugera vid 20.000 x g under 20 min vid 4 ° C. Kassera supernatanten.
- Tillsätt en motsvarande volym av 75% etanol, centrifugera vid 20.000 x g under 10 min vid 4 ° C, kassera supernatanten.
- Snurra kort och ta bort rester av etanol med 200 l pipett.
- Snurra kort och ta bort rester av etanol med 20 l pipett.
- Låt inte RNA torka. Resuspendera den i 100 | il H2O Blanda väl genom att pipettera upp och ned 5-6 gånger. Inkubera vid 65 ° C under 10 min med skakning och överföra direkt till is.
- Kontrollera RNA kvalitet genom electrophoretical analys för att utesluta RNA nedbrytning.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Ett. Utgångsmaterial och förväntad avkastning
Efter 1 timme (h) i 4SU-exponering nyligen transkriberade RNA utgör cirka 1 - 4% av totalt cellulärt RNA. Detta kommer att vara lägre i tillväxtföretag arresterade-celler eftersom de inte längre syntetisera RNA redogöra för celltillväxt / replikation. När märkning för 1 timme, rekommenderar vi börjar analysen med 60-80 mikrogram av det totala RNA. Från och med mindre än 30 mikrogram av det totala RNA resulterar i små RNA pellets som är svåra att se efter biotinylerings steget och därmed lätt kan förloras. Input RNA-nivåer kan ökas till så mycket som 150 mikrogram för mycket korta löptider för etikettering (t.ex. 5 - 10 min). När varaktigheten av RNA märkning förkortas från 1 timme till 5 min bidraget från kortlivade intronsekvenser i nyligen transkriberade RNA ökar från ~ 60% till ~ 80% 9. Eftersom introner betydligt längre jämfört med kodande sekvenser samt 5'-och 3'-UTRs, mängden transkriberas nyligenRNA, som kan renas efter kort eller ens ultrakort 4SU-taggning, inte sjunker linjärt. Som sådan fick vi> 0,5% av det totala RNA efter 5 min av 4SU-taggning i icke vidhäftande humana B-cellinjer 9. Det bör dock noteras att högre koncentration av 4SU och något längre löptider för märkning kan krävas för att uppnå liknande 4SU inblandningshalter i vidhäftande celler. Medan även en låg 4SU-upptagningsförmågan möjliggör effektiv insamling och rening av stora, uridinproducerande rika transkript, mycket kort utskrift med låg uridin innehåll (t.ex. miRNA) är sannolikt att undkomma rening även när du använder höga 4SU koncentrationer (> 1 mm). I NIH-3T3 murina fibroblaster, märkta 1 hr av 200 iM 4SU exponering nyligen transkriberade RNA med omkring en 4SU rest per 50-100 nukleotider (nt) 5. Detta bör möjliggöra mycket effektiv återvinning av avskrifter> 500 - 1000 nt i längd. Därför observerade vi endast en mindre transkript storlekpartiskhet när märkningen i 1 timme med 200 ìm 4SU i både murina fibroblaster och humana B-celler 7. Medan 1 hr av 200 iM 4SU inte medföra några väsentliga förändringar i cellulära avskrifter nivåer i murina fibroblaster, långvarig exponering av celler till ≥ 200 iM 4SU leder till en mätbar tillväxt underskott inom 24 tim (opublicerade data). Därför bör både varaktigheten av märkning och den anställde 4SU-koncentrationen minimeras för att undvika ektopisk eller toxiska effekter. Ett enkelt sätt att bestämma den minimala 4SU-koncentration som krävs för effektiv återvinning av nyligen transkriberade RNA är att rena nyligen transkriberade RNA efter 4SU-märkning med ökande koncentrationer av 4SU (t.ex. 50-1600 iM). Som visas i figurerna 2A och 2B, återvinning av nyligen transkriberade RNA märkta för 1 timme i primära humana fibroblaster ökade kraftigt från 50 till 200 iM 4SU men sedan började platå.
2. PunktBlot Kvantifiering av 4SU Incorporation (tillval)
I vissa fall kan det vara av intresse att mäta mängden 4SU inkorporering i totalt RNA. Detta görs bäst genom dot blot analys på de biotinylerade RNA med en streptavidinkonjugat. På grund av sin kemiska natur jodacetyl-biotin är mer reaktiv till tiol-grupper än biotin-HPDP resulterar i biotinyleringen av praktiskt taget alla 4SU rester i nyligen transkriberade RNA. Det är viktigt att notera att, såsom biotin-HDPD är jodacetyl-biotin inte vattenlösliga och därmed effektivt bort av kloroform extraktion som utförs för biotin-HPDP. Därför kan samma reaktionsbetingelser och koncentrationer användas som vid användning av biotin-HPDP. Dock är jodacetyl-biotin inte reversibel. Det kan således inte användas för rening av nyligen transkriberade RNA i kolumn ansatser. Även om användningen av jodacetyl-biotin gör att kvantifiera 4SU-inkorporering, biotin-HPDP mätningar beakta både4SU-inkorporering och biotinylerings effektivitet. Under användning av två Biotinylering reagens till samma prov tillåter mätning av biotinylering effektiviteten av RNA-bildat 4SU. Biotinylering effektivitet biotin-HPDP för 4SU märkt RNA verkar vara ungefär tre gånger mindre än den för jodacetyl-biotin visar att endast omkring en i tre 4SU rester i nyligen transkriberade RNA faktiskt biotinylerades av biotin-HPDP (Figur 3). Genom att jämföra intensiteterna sampelsignal med biotinylerade kontroll DNA oligo, kan Biotinylering densiteter mätas. För de flesta däggdjurscellinjer en positiv signal bör fortfarande finnas kvar i 10 ng av biotinylerade RNA efter 1 timme av 200 iM 4SU märkning. En svag bakgrund signal är vanligtvis påvisas för den högsta koncentrationen (1 mikrogram) av omärkta RNA.
Tre. Rening av nyligen transkriberat RNA
Återvinning av nyligen transkriberade RNA är mycket kvantitativa. Om du började med samma RNA-koncentrationen du kan förvänta sig att få samma mängd nyligen transkriberade RNA för alla prover. Liksom många spalt-baserade analyser, kan insamlingen av nyligen transkriberade RNA använder RNeasy MinElute kit resultera i ytterligare absorption vid 230-260 nm (närvaro av rengöringsmedel som härrör från tvättbuffertar) som kan störa OD 260 mätningar. Detta ses i mindre utsträckning vid användning av ett färskt 2 ml uppsamlingsrör för varje centrifugeringssteg. Ändå bör alla orimligt höga OD mätningar (> 2-faldigt högre än andra prover) betraktas med försiktighet, särskilt om OD 260/280 nyckeltal är <1.7. För nedströms analyser är det därför ofta bäst att använda samma mängd mall RNA volymen för alla prover. I fall där utbytena av märkt RNA är lägre än väntat check för tecken på RNA-nedbrytning genom electrophoretical analys. Nyligen transkriberade RNA innehåller betydligt större mängder av stora, oskarvade transkript med de typiska rRNA-banden är mycket mindre framträdande (figur 4).
4. Kvantifiering av nyligen transkriberat RNA
Slutligen kan inblandningshalter av 4SU i nyligen transkriberade RNA direkt kvantifieras genom spektrofotometrisk analys baserad på absorption högst 4SU vid 330 nm och OD 330/260 förhållandet 5,18. Detta kräver> 3 ^ g av märkt RNA koncentrerade till en liten volym (10 - 20 | il) av isopropanol / etanolutfällning. För att undvika att förlora den lilla RNA pelleten samfällning med 30 pg nukleasfritt glykogen (Fermentas, # R0551) ska utföras. En ytterligare topp syns vid 330 nm speglar införlivandet andelen 4SU in nyligen transkriberas RNA (Figur 5).
/ Files/ftp_upload/50195/50195fig1highres.jpg "/>
Figur 1. . Principen om metabolisk märkning med 4-tiouridin (4SU) 4SU läggs till celler för erforderlig (5-120 min) tid följt av beredning av totalt cellulärt RNA. Efter tiol-specifik biotinylering, är totalt cellulärt RNA separeras i 4SU märkta, nyligen transkriberade RNA, och omärkta, pre-existerande RNA med streptavidinbelagda magnetiska pärlor. Nyligen transkriberas RNA utvinns från kulorna med ett reduktionsmedel som klyver disulfidbindningarna som förbinder de nyligen transkriberade RNA till pärlorna. Klicka här för att visa en större bild .
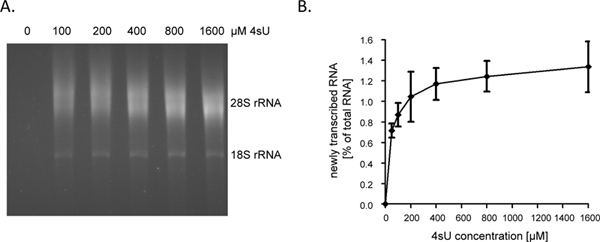
Figur 2. Återvinning av nyligen transkriberade RNA efter ökande koncentrationer av 4SU. (A) Primära humanförhudsfibroblaster (HFF) inkuberades med 100, 200, 400, 800 eller 1600 iM 4SU. Nyligen transkriberat RNA renades från 50 ^ g totalt cellulärt RNA och underkastades electrophoretical analys. Som förväntat, var en koncentrationsberoende ökning av återvunna nyligen transkriberade RNA som observerades som började plana ut vid högre koncentrationer. (B) mängder av renat nyligen transkriberat RNA kvantifierades med hjälp av ImageJ 1.45s programvara. Kombinerade data från fyra oberoende experiment på mängderna nyligen transkriberade RNA återkrävas efter olika koncentrationer av 4SU-märkning som sträcker sig från antingen 50-800 iM 4SU (n = 2) eller 100 -. 1.600 iM 4SU (n = 2) visas Klicka här att visa en större bild .
upload/50195/50195fig3.jpg "alt =" Bild 3 "fo: innehåll-width =" 4.5in "fo: src =" / files/ftp_upload/50195/50195fig3highres.jpg "/>
Figur 3. Uppskattning av 4SU inkorporering i 4SU-märkta totalt RNA med hjälp av dot blot-analys. Totalt RNA isolerades från NIH-3T3 murina fibroblaster eller humana fibroblaster förhudsfibroblaster (HFF) inkuberade med 200 pM 4SU under en timme. Inga 4SU sattes till en maträtt som negativ kontroll. För HFF både kontakt hämmade (n = icke-växande celler) och växande celler (y) ingick. RNA isolerades med användning av Trizol-reagens och därefter konjugerat till biotin-HPDP eller jodacetyl-biotin. Koncentration av varje prov justerades till 200 ng / ul och 5 | il av denna spädning (det vill säga 1 mikrogram av RNA), samt tre efterföljande 10-faldiga utspädningar (dvs. 100, 10, och 1 ng RNA, respektive), var alla fläckvis på en bit Zeta membran. 5 mikroliter utspädningar av biotin-märkt DNA oligo placerades på membranet som positiva kontroller vid koncentreratsjoner från 20 ng / l ned till 20 pg / l (dvs. 100 till 0,1 ng, respektive). Biotin täthet undersöktes med hjälp av ett streptavidin-pepparrotsperoxidaskonjugat.
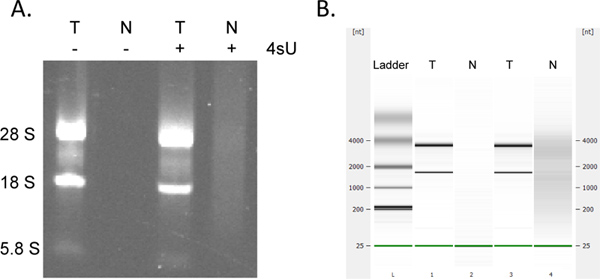
Figur 4. Electrophoretical analys av nyligen transkriberade och total-RNA. Total RNA (T) och nyligen transkriberade RNA (N) framställd från murina NIH-3T3-fibroblaster odlade både i närvaro och frånvaro av 500 | iM 4SU i 1 timme analyserades genom agarosgelelektrofores (A) och (i samma ordning) med Agilent Bioanalyser (B). Ingen RNA återvanns utan 4SU behandling av celler. Renat nyligen transkriberas RNA innehåller större mängder högmolekylära mRNA och betydligt mindre mogna rRNA än totaltRNA som anmärkningsvärt mellan 28S, 18S, och 5.8S rRNA-banden. Klicka här för att visa en större bild .
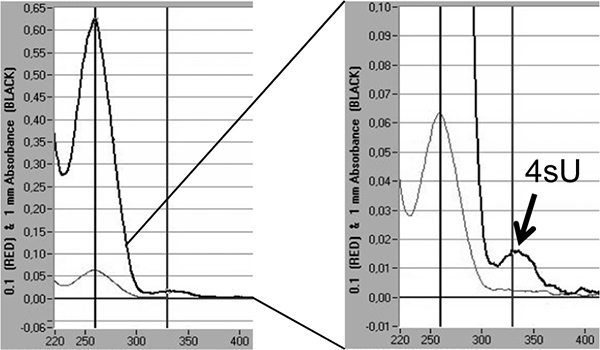
Figur 5. Kvantifiering av 4SU inkorporering i nyligen transkriberade RNA genom spektrofotometrisk analys. Nyligen transkriberade RNA renas från 2 x 100 mikrogram totalt RNA efter 1 timme av 200 iM 4SU i murina NIH-3T3-fibroblaster. Nyligen transkriberade RNA fälldes med isopropanol / etanol efter tillsats av 30 pg nukleasfritt glykogen. Spektrofotometrisk analys av nyligen transkriberade RNA som erhållits genom en Nanodrop 1000 spektrofotometer visas. De ljusgrå linjerna representerar mätningar vid 0,1 mm medan den tjockare, mörkgrå linjerna representerar mätningar vid 1 mm vätska kolumnen. Till höger en förstoring av toppen av utrotning representerar tHan införlivade 4SU-rester visas. Baserat på extinktionen koefficient av 4SU 18 Införlivandet klassar av 4SU kan uppskattas.
| Varaktighet för märkning [min] | Rekommenderad 4SU koncentration [iM] |
| 120 | 100-200 |
| 60 | 200-500 |
| 15-30 | 500 - 1000 |
| <10 | 500 - 2000 |
Tabell 1. Rekommenderad 4SU koncentrationer.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Metabolisk märkning av nyligen transkriberade RNA ökar väsentligt kraften av teknik med hög kapacitet som microarrays och RNA-Seq genom att ge mer lämpliga mallar för att hantera den biologiska frågan om ränta. Det nuvarande protokollet genomgick omfattande optimering. Den tillåter> 1000-faldig anrikning av nyligen transkriberade RNA och ger mycket reproducerbara resultat.
Den experimentella designen av en 4SU-taggning försöket är av avgörande betydelse som nyligen transkriberade RNA kommer skildra realtid transkriptionsaktiviteten endast under tiden för exponering av celler för 4SU. Om de faktiska förändringarna i transkription priser efter en stimulans har redan avtagit, kommer dessa att missa när man analyserar nyligen transkriberade RNA även om förändringar i den totala RNA-nivåer kan fortfarande finnas kvar. Därför är en god förståelse av den underliggande biologin viktigt att definiera experimentuppställning liksom den optimala perioder of tid för 4SU exponering. Nedan ger vi rekommendationer och sätt för att undvika vanliga fallgropar för de mest avgörande stegen.
Beredning av stamlösningar och plastartiklar
Alla stamlösningar måste beredas med nukleasfritt vatten. Använda in-house renat avjoniserat vatten kan leda till problem om vattnet innehåller reducerande ämnen. I ett fall resulterade detta i fullständig förlust av alla märkta RNA. Därför rekommenderar vi starkt att köpa pre-made nukleasfritt NaCl, Tris-Cl, EDTA, natriumcitrat och vatten. Säkerställ nukleasfritt villkor vid alla tidpunkter. Dimetylformamid (DMF) upplöser vissa plastmaterial. Vi fann att använda 25 ml cell pipetter kultur plast för att överföra DMF från sitt lager glasflaska till 50 ml Falcon rör för att förbereda biotin-HPDP stamlösning var tillräcklig för att avsevärt minska avkastningen av nyligen transkriberade RNA från hela analysen. Intressant, detta inte negativt påverka biotinylation effektivitet (som testats av dot blot) men resulterade i en 75 till> 90% förlust av nyligen transkriberade RNA som kunde återvinnas från kulorna. Förlusten var mest uttalad när varaktigheten av märkningen minskade från 60 till 30 minuter eller mindre. Troligtvis förstörde ett ämne elueras från plastpipetter av DMF delvis beläggning av streptavidinpärloma. Därför bör användningen av plastmaterial som inte är kända för att vara kompatibel med DMF undvikas med alla medel. Av samma skäl bör cellkoncentrationen skrapor inte användas för att öka återvinningen av Trizol prover från cellodlingsplattor. Det är intressant att notera att de förmodade ämnen eluerade från plasten av DMF eller Trizol uppenbarligen varken avlägsnades genom kloroformextraktion eller isopropanol / etanolutfällning.
Cellodling
Cell density på plattorna är av avgörande betydelse. I ett experiment där cellerna verkade vara något för sammanflytande (90 -100%), behandlades vi NIH-3T3 murina fibroblaster under 30 min med 100 U / ml av interferon (IFN) α eller γ. På mindre sammanflytande celler även 15 min av IFN-behandling resulterade redan i en 5 - till 8-faldig induktion av gener som irf1 eller SOCS3 5. Med celler är något alltför sammanflytande microarrayanalys inte visade någon induktion av IFN-inducerbara gener för även den mest snabbt inducerbara gener som irf1 eller SOCS3. Därför är celltäthet en avgörande faktor för 4SU-märkning experiment och alla cellplattorna kultur bör noggrant undersökas innan märkningen.
4SU är en fotoaktiverbar ribonukleosid och 4SU-innehållande RNA effektivt tvärbunden till proteiner efter exponering för 365 nm ljuskälla. 4SU-behandlade celler bör odlas i mörker och exponering för starkt ljus bör undvikas. Efter avlägsnande av cellulära proteiner genom Trizol RNA isolering denna risk minskas väsentligt.
<p class = "jove_content"> 4SU inte är införlivad i cellulärt DNA. Det bör dock noteras att de totala RNA fortfarande kommer att innehålla små mängder av cellulärt DNA. Vid användning 4SU-taggning och q-RT-PCR-analys för att studera viral genuttryck i cytomegalovirusinfektion vi funnit det nödvändigt att inkludera en DNasI digest steg i protokollet för att ta bort de concatemeric virala genom 19. Detta är förmodligen inte nödvändigt vid användning av efterföljande protokoll som inte är känsliga för förekomst av DNA.4SU inblandningshalter och optimal 4SU koncentration
4SU lätt tas upp av celler med intra-och extra-cellulära nivåer sannolikt jämvikt inom mindre än en minut 9,16. Upptag och inkorporering andelen 4SU är koncentrationsberoende. Därför kan 4SU koncentration kan enkelt justeras i enlighet med den använda varaktigheten av märkning. Tabell 1 ger råd om 4SU koncentrationer i relation till varaktigheten av märkning som grundar sig på vår bästa personliga erfarenhet. För 1 h av 4SU märkning i däggdjursceller, kommer 200 pM 4SU vara tillräckligt för de flesta användningar som resulterar i omkring en 4SU rest per 50 till 100 nukleotider i nyligen transkriberade RNA i fibroblaster.
Under de senaste åren har vi tillämpat 4SU-taggning till ett brett spektrum av celltyper av humant och murint ursprung inklusive fibroblaster, endotelceller, epitelceller, benmärgsceller stroma, makrofager och T-celler. Vidare har celler från Drosophila och Xenopus framgång använts. I alla dessa experiment 4SU inkorporering konstaterades vara effektiv kräver minimala justeringar i 4SU koncentration för de olika celltyper. När man sätter upp metoden för nya celltyper, rekommenderar vi att märka celler med ökande 4SU-koncentrationer (t.ex. intervallet 50-1600 iM) och analysera relationen mellan renade nyligen transkriberade RNA till tillämpade 4SU-koncentrationer (se fig 2A / B). Den 4SU-koncentration vid vilken mängd renat nyligen transkriberas RNA in en platå bör väljas.
I de fall där mycket sammanflytande, kontaktinhiberade celler används, rekommenderar vi att använda något högre 4SU koncentrationer (t.ex. 500 istället för 200 iM) för att säkerställa en effektiv 4SU inkorporering. Dessutom, i de fall där infångning av mycket korta nyligen transkriberade transkript (<200 nt) är av särskilt intresse, kan den 4SU koncentration måste också ökas. Detta bör inte kombineras med förlängda märkning gånger (t.ex.> 1 timme) för att undvika ektopisk effekter eller toxicitet. Slutligen fann vi att använda en för liten volym cellodlingsmedier kan minska 4SU inkorporering effektivitet. Vi rekommenderar därför att använda 5 ml eller 10 ml medium per 10 cm eller 15 cm skålen, respektive.
Framställning av totalt cellulärt RNA
För att lyckas med detta protokoll är det viktigt att få fram rena, RNase-fria totalt cellulärt RNA. Använda 5 ml Trizol per 15 cm skål producerar ren RNA gratis av nukleaser. Vi rekommenderar användning av den modifierade Trizol protokollet från Chomczynski et al. 20. Först, är det mer lämpligt att isolera stora mängder av RNA (> 100 ^ g) att det finns ökade centrifugalkraften resulterar i fastare pellets som är lättare att hantera under tvättningsstegen. Detta kräver dock att användningen av särskilda polypropylenrör och adaptrar som den vanliga 15 ml laboratorium Falcon rör inte överlever mer än 6,000 × g. Vidare förbättrar avlägsnandet av DNA och glykoproteiner. Detta blir särskilt tydligt när de förbereder RNA från organ eller vävnader. För det tredje begränsar dock inte den maximala mängden av totalt RNA som kan isoleras. Även om vi också funnit kolonn-baserade RNA Isolering metoder (t.ex. RNeasy) att ge RNA av lämplig kvalitet, standard kolumner are endast kunna fånga upp till 100 | ig av totalt RNA därigenom begränsa mängden av utgångsmaterial. Slutligen är genom att ta bort den återstående etanolen två gånger med en pipett, torkning av RNA för att avlägsna rester av etanol inte längre behövs. Detta eliminerar risken för över-torkning av RNA, vilket kan vara svårt att lösa upp igen efteråt. I princip är 4SU-tagging tillämplig in vivo, t.ex. genom intravenös injektion av möss. Däremot konstaterade vi att RNA renhet utgör ett stort problem som kräver rening av polyA transkript före rening av nyligen transkriberade RNA (opublicerade data).
Biotinylering och avlägsnande av obundet biotin
Biotin-HPDP är 100% tiol-specifika och bildar en disulfidbindning mellan biotin rester och tiol-märkt RNA nyligen transkriberade molekyler. Biotinylering effektivitet 4SU-märkt RNA är ca 30% bestämt genom dot blot-analys 5. Som biotin-HPDP inte är vattenlösligtdet effektivt kan avlägsnas genom kloroformextraktion. Medan en enda kloroformextraktion steg är tillräcklig för att avlägsna den stora majoriteten av obundet biotin vi upprepar regelbundet detta steg för att säkerställa fullständig borttagning. För att minska RNA förlust under kloroformextraktion steg 2 ml faslås Gel Tunga rör (Eppendorf) kan användas enligt tillverkarens instruktioner. Vanligtvis använder vi rören faslåsning endast för det andra kloroformextraktion steget som mall volymerna av det första steget är ofta för hög för att vara direkt kompatibla med dessa rör. Efter avlägsnande av obundet biotin-HPDP, RNA utvinns genom isopropanol / etanolutfällning. Det är viktigt att notera att kommersiella kolumn byggsatser att återvinna biotinylerade RNA (t.ex. RNeasy från QIAGEN) bör inte användas eftersom de innehåller reduktionsmedel i de tillhandahållna buffertar, vilket klyver disulfidbindningen och avlägsna biotin från den nyligen transkriberade RNA .
Rening av nyaly transkriberade RNA
Lägg inte till mer än 100 l biotinylerade RNA till 100 ^ streptavidinpärloma. Lägga mindre volym är att föredra. Dock bör samma volym av RNA läggas till för alla prover. Justera RNA input volym (mellan prover) som du lägger till streptavidinpärloma genom att bara lägga till erforderlig volym 1x TE till pärlorna. Ett enkelt sätt att göra färsk nukleasfritt 100 mM DTT är att dekantera en tillräcklig mängd av DTT pulvret i en falconrör placerad på en ultra-känslig skala och sedan lägga den erforderliga mängden nukleasfritt H 2 O för att generera 100 mM DTT (64,8 | il vatten per 1 mg DTT).
Under utvecklingen av 4SU-taggning vi testade streptavidinpärloma från olika leverantörer. Ett antal av dem genererade stora mängder bakgrunden. Därför rekommenderar vi starkt att använda Miltenyi streptavidinpärloma som, hittills, har vi aldrig upplevt några problem med överföring av omärkta RNA från vävnad culture-härledda RNA-prover. På detta sätt kan så lite som 150 ng av märkta RNA vara specifikt renas från 150 ^ g biotinylerad RNA (i 100 | il vatten) med användning av 100 pl av streptavidinpärloma. Ekvilibrering av pärlorna med jämviktsbuffert levereras med pärlorna kan utföras och kan något förhöja bindningsandel 13.
Kvalitetskontroller
Vi rekommenderar utför Q-RT-PCR-kontroller nyligen transkriberade RNA innan den utsätts för hög genomströmning analys. Detta kan innefatta kvantifiering av flera referens gener kända för att vara differentiellt regleras i viss experimentell inställning. I de fall 4SU-märkning används för att studera RNA avklingningshastigheter, skulle vi rekommendera att kvantifiera en kortlivad transkript (t.ex. myc, FOS) och ett långlivat en (t.ex. GAPDH) i både totalt och nyligen transkriberade RNA. Förhållandet av nyligen transkriberade / total-RNA bör vara väsentligen högre (~ 5 - till 10-faldig)för de kortlivade transkript. Baserat på RNA halveringstiden för en referens gen, kan RNA halveringstider bestämmas. Om alla tre RNA-fraktioner (totalt RNA, nyligen transkriberade RNA och omärkt befintlig RNA) analyseras för fyra eller fler gener, normalisering av olika RNA undergrupper kan göras med linjär regressionsanalys och kvalitetsresultat kontroll kan bestämmas såsom beskrivits 7 , 21.
För Q-RT-PCR-analys, rekommenderar vi att du använder 2,5 pl av märkta RNA i 20 ^ cDNA-syntes mix. För optimal jämförelse av Q-RT-PCR-resultat frysa cDNA i alikvoter om 5 | il före första användning. Tina rören strax före användning, tillsätt 45 pl H2O och föremål 5 mikroliter av utspädningar till Q-RT-PCR-analyser. Detta förbättrar avsevärt jämförbarheten mellan olika PCR-körningar.
Nyligen transkriberade RNA-prover bör kontrolleras för tecken på RNA nedbrytning med hjälp av Agilent Bioanalyser innan utsätter dem förhög genomströmning analys (mikromatriser eller RNA-Seq). Det bör dock noteras att ytterligare band ibland observeras av Agilent Bioanalyser. Den biologiska betydelsen av detta är fortfarande oklar. Som nyligen transkriberas RNA innehåller betydligt mindre ribosom RNA, dessa prover misslyckas ibland Agilent kontroller Bioanalyser kvalitet. Om detta inte beror på synliga RNA nedbrytning prover av acceptabel kvalitet är oftast bra att utsättas för hög genomströmning analys.
Kompatibilitet av nyligen transkriberade RNA med nedströms analyser
Nyligen transkriberade RNA innehåller betydligt mer mRNA än total RNA. Detta beror främst på de större mängder intronsekvenser i nyligen transkriberade RNA som ökar när varaktigheten av 4SU-taggning förkortas. Därför behöver vi inte regelbundet åta sig utarmning av rRNA från nyligen transkriberade RNA-prover som detta kräver större mängder av utgångsmaterial medan providIng ganska liten (~ tvåfaldigt) vinst i icke-rRNA läser. Slutligen återstår det att påpekas att den större andelen osplitsade, högmolekylära transkript närvarande i nyligen transkriberade RNA kan kräva ytterligare fragmentering när de förbereder cDNA-bibliotek för nästa generations sekvensering. Resultat av storlek fragmentering steget bör därför vara kvalitet kontrolleras noggrant.
Data normalisering för RNA halv-levande mätningar
Standarden metod att normalisera experimentella data för RNA halveringstid mätningar är att normalisera alla data till RNA halveringstiden i en väldefinierad house keeping gen eller medianvärdet RNA halveringstiden i en viss celltyp bestäms i tidigare experiment. I däggdjursceller, ligger den senare i området 5 till 10 h 6,7. Även om detta tillvägagångssätt även fungerar ganska bra för 4SU-baserade mätningar, är andra medel för normalisering krävs om median RNA halveringstiden inte är känd eller om det may även påverkas av förändringar i det cellulära systemet som studeras, t.ex. av knock-out av en RNA-sönderfall väg. 4SU-taggning erbjuder ett unikt sätt att skatta medianen RNA halveringstiden bygger på en analys av alla tre RNA-fraktionerna, dvs totalt cellulärt RNA, nyligen transkriberade RNA, och omärkta befintliga RNA. Som totalt cellulärt RNA separeras i de två sistnämnda RNA-fraktionerna en enkel linjär regressionsmodell kan användas för att normalisera de tre RNA-fraktionerna till varandra och bestämma medianen RNA halveringstiden 7,16. Ett programpaket är tillgängliga online för att utföra dessa analyser 22.
Ineffektiv fångst av transkript med låg uridin halt kan påverka RNA halveringstid mätningar resulterar i konstgjort låga nyligen transkriberade / total RNA förhållanden och förlängda RNA halveringstider. Omfattningen av detta problem kan bedömas genom att plotta RNA halveringstider eller log (nyligen transkriberade / totala RNA förhållanden) mot uridiNE halt av alla transkript 7,15. Detta ger också en god kvalitetskontroll för att bedöma skillnader i 4SU-inblandningshalter mellan olika prover eller villkor. I de fall där en betydande korrelation till uridin innehåll observerade detta kan rättas till genom bioinformatiska betyder 15. Emellertid bör det noteras att bidraget från mogna transkript i nyligen transkriberade RNA inte lätt kan skiljas från den mycket större och därmed mycket mer uridin-rika prekursorer. Om inte behandlingen kinetik en given transkript är kända (vilket de oftast inte) bara korrigera för låg uridin innehåll (ineffektiv capture) kan grovt förvränga RNA halveringstider. Som sådan, hittade vi nyligen bearbetning av de flesta mänskliga snoRNAs vara mycket ineffektiv 9. Om vi hade rättat de nyligen transkriberade / total RNA Nyckeltal för låga uridine innehållet i ganska små (70-300 nt) snoRNAs, skulle detta ha lett till extremt korta snoRNA halv-lives (<5 min) med många nya transkriberade / total RNA förhållanden överstiger 100%. Därför, vi generellt rekommenderar inte korrigera för låg uridin halt vid mätning RNA halveringstider.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Författarna förklarar att de inte har några konkurrerande ekonomiska intressen.
Acknowledgments
Vi vill tacka Amie Regan för noggrann läsning av manuskriptet. Detta arbete stöddes av NGFN Plus bevilja # 01GS0801, MRC gemenskap bidrag G1002523 och NHSBT bidrag WP11-05 till LD och DFG bidrag FR2938/1-1 till CCF
Materials
| Name | Company | Catalog Number | Comments |
| 4-thiouridine | Carbosynth | T4509 | Prepare 50 mM stock in sterile H2O, store at -20 °C in aliquots of 50-500 μl, discard unused reagent, do not refreeze. |
| Trizol | Invitrogen | 15596026 (100 ml), 15596018 (200 ml) | WARNING - CORROSIVE and HAZARDOUS TO HEALTH! Ensure immediate access to Phenol antidote (PEG-Methanol); Store at 4 °C. |
| Chloroform | Sigma | 372978 | WARNING - HAZARDOUS TO HEALTH |
| Isopropanol | Sigma | 650447 | |
| Sodium citrate, nuclease-free | Sigma | C8532 | Prepare 1.6 M stock solution using nuclease-free water. |
| 5M nuclease-free NaCl | Sigma | 71386 | Stock solution |
| Nuclease-free H2O | Sigma | W4502 | Make 1 ml aliquots in nuclease-free tubes. |
| RNA precipitation buffer | 1.2 M NaCl, 0.8 M sodium citrate in nuclease free water. Prepare in advance under strictly nuclease-free conditions. Store at room temperature in 50 ml falcon tubes. | ||
| Ethanol | Sigma | 459844 | Use with nuclease-free water to prepare 80% ethanol, store at -20 °C. |
| 1 M nuclease-free Tris Cl, pH 7.5 | Lonza | 51237 | Stock solution |
| 500 mM nuclease-free EDTA, pH 8.0 | Invitrogen | 15575-020 | Stock solution |
| 10x Biotinylation Buffer (BB) | 100 mM Tris pH 7.4, 10 mM EDTA in nuclease-free water, make aliquots of 1 ml. | ||
| Dimethylformamide (DMF) | Sigma | D4551 | |
| EZ-Link biotin-HPDP | Pierce | 21341 | Prepare 1 mg/ml stock solution by dissolving 50 mg biotin-HPDP in 50 ml DMF. Gentle warming enhances solubilisation. Store at 4 °C in aliquots of 1 ml. |
| Phase Lock Gel Heavy tubes 2.0 ml | Eppendorf | 0032 005.152 | Optional for the chloroform extraction step. |
| Zeta membrane | BIORAD | 162-0153 | |
| 10x Dot blot binding buffer | 100 mM NaOH, 10 mM EDTA | ||
| Biotin-oligo | 5'-biotin, 25 nucleotides, any sequence | ||
| Sodium dodecyl sulphate | Fisher | BPE9738 | For 100 ml 20% stock solution, add 20 g SDS to 80 ml PBS pH 7-8 and adjust volume to 100 ml. Keep all high-percentage SDS solutions above 20 °C. Warm the solutions slightly should SDS precipitate. |
| EZ-Link Iodoacetyl-LC-Biotin | Pierce | 21333 | Prepare 1 mg/ml stock solution by dissolving 50 mg iodoacetyl-biotin in 50 ml DMF. Gentle warming enhances solubilisation. Store at 4 °C in aliquots of 1 ml. Generates irreversible, thiol-specific biotinylation. |
| Phosphate buffer saline | Gibco | 10010-015 | |
| Dot blot blocking buffer | Mix 20 ml 20% SDS with 20 ml 1 x PBS pH 7-8 and add EDTA to the final concentration of 1 mM. | ||
| Streptavidin-horseradish peroxidase | Vector Laboratories | SA5004 | Store at -20 °C. Mix 10 ml 20% SDS with 10 ml 1 x PBS. Add 20 μl Streptavidin-HRP before use. |
| ECL reagent | GE Healthcare | RNP2109 | Use following the manufacturer's instructions. |
| Super RX, X-RA Film, 18x24 cm | Fujifilm | 47410 19236 | |
| μMacs Streptavidin Kit | Miltenyi | 130-074-101 | Store the beads at 4 °C. |
| Tween 20 | Sigma | P1379 | |
| Washing buffer | 100 mM Tris pH 7.4, 10 mM EDTA, 1 M NaCl, 0.1% Tween 20 in nuclease-free H2O. | ||
| Dithiothreitol (DTT) | Sigma | 43817 | Prepare as 100 mM DTT in nuclease-free H2O, always prepare fresh before use. |
| RNeasy MinElute Kit | Qiagen | 74204 | Store columns at 4 °C, remaining components of the kit at room temperature. |
| 1.5 ml screw-top polypropylene tubes | Sarstedt | 72.692.005 | Compatible with Dimethylformamide |
| 2.0 ml screw-top polypropylene tubes | Sarstedt | 72.694.005 | Compatible with Dimethylformamide |
| 15 ml tubes | BD Falcon | 352096 | Compatible with Dimethylformamide |
| 50 ml tubes | BD Falcon | 352070 | Compatible with Dimethylformamide |
| All solutions/reagents should be stored at room temperature unless otherwise specified. | |||
| Equipment | |||
| UV/VIS spectrophotometer | Thermo Scientific | NanoDrop 1000 | Or equivalent. Use low volume (1-2 μl) for measurements of low RNA concentrations to avoid excessive sample loss. |
| Polypropylene 15 ml centrifuge tubes | VWR International | 525-0153 | In contrast to standard 15 ml tubes, these tolerate up to 15,000 × g |
| High-speed centrifuge | Beckman Coulter | Avanti J-25 | Or equivalent equipment capable of reaching 13,000×g |
| High-speed rotor | Beckman Coulter | JLA-16250 | Or equivalent equipment capable of reaching 13,000×g |
| Adaptors for 15 ml tubes | Laborgeräte Beranek | 356964 | Or equivalent equipment capable of reaching 13,000×g |
| Refrigerated table-top centrifuge | Eppendorf | 5430 R | Or equivalent. |
| Thermomixer | Eppendorf | Thermomixer compact | Or equivalent. |
| Magnetic stand | Miltenyi Biotec | 130-042-109 | One stand holds 8 μMacs columns. |
| Waterbath | Grant | SUB Aqua 5 | Or equivalent. |
| Ultra-fine scale | A&D | GR-202 | Or equivalent. |
| E-Gel iBase Power System | Invitrogen | G6400UK | For RNA gels; or equivalent. |
| E-Gel EX 1% agarose precast gels | Invitrogen | G4020-01 | For RNA gels; or equivalent. |
References
- Brown, P. O., Botstein, D. Exploring the new world of the genome with DNA microarrays. Nature Genetics. 21, 33-37 (1999).
- Mortazavi, A., Williams, B. A., Mccue, K., Schaeffer, L., Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nature Methods. 5, 621-628 (2008).
- Nagalakshmi, U., Wang, Z., et al. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science. 320, 1344-1349 (2008).
- Wilhelm, B. T., Marguerat, S., et al. Dynamic repertoire of a eukaryotic transcriptome surveyed at single-nucleotide resolution. Nature. 453, 1239-U1239 (2008).
- Dölken, L., Ruzsics, Z., et al. High-resolution gene expression profiling for simultaneous kinetic parameter analysis of RNA synthesis and decay. RNA. 14, 1959-1972 (2008).
- Yang, E., van Nimwegen, E., et al. Decay rates of human mRNAs: correlation with functional characteristics and sequence attributes. Genome Res. 13, 1863-1872 (2003).
- Friedel, C. C., Dölken, L., Ruzsics, Z., Koszinowski, U. H., Zimmer, R. Conserved principles of mammalian transcriptional regulation revealed by RNA half-life. Nucleic Acids Res. 37, e115 (2009).
- Nilsen, T. W., Graveley, B. R. Expansion of the eukaryotic proteome by alternative splicing. Nature. 463, 457-463 (2010).
- Windhager, L., Bonfert, T., et al. Ultra short and progressive 4sU-tagging reveals key characteristics of RNA processing at nucleotide resolution. Genome Res. , (2012).
- Melvin, W. T., Milne, H. B., Slater, A. A., Allen, H. J., Keir, H. M. Incorporation of 6-thioguanosine and 4-thiouridine into RNA. Application to isolation of newly synthesised RNA by affinity chromatography. Eur. J Biochem. 92, 373-379 (1978).
- Cleary, M. D., Meiering, C. D., Jan, E., Guymon, R., Boothroyd, J. C. Biosynthetic labeling of RNA with uracil phosphoribosyltransferase allows cell-specific microarray analysis of mRNA synthesis and decay. Nature Biotechnology. 23, 232-237 (2005).
- Kenzelmann, M., Maertens, S., et al. Microarray analysis of newly synthesized RNA in cells and animals. Proc. Natl. Acad. Sci. U.S.A. 104, 6164-6169 (2007).
- Schwanhäusser, B., Busse, D., et al. Global quantification of mammalian gene expression control. Nature. 473, 337-342 (2011).
- Rabani, M., Levin, J. Z., et al. Metabolic labeling of RNA uncovers principles of RNA production and degradation dynamics in mammalian cells. Nat. Biotechnol. , (2011).
- Miller, M. R., Robinson, K. J., Cleary, M. D., Doe, C. Q. TU-tagging: cell type-specific RNA isolation from intact complex tissues. Nat. Methods. 6, 439-441 (2009).
- Miller, C., Schwalb, B., et al. Dynamic transcriptome analysis measures rates of mRNA synthesis and decay in yeast. Mol. Syst. Biol. 7, 458 (2011).
- Weintz, G., Olsen, J. V., et al. The phosphoproteome of toll-like receptor-activated macrophages. Mol. Syst. Biol. 6, 371 (2010).
- Lipsett, M. N. The isolation of 4-thiouridylic acid from the soluble ribonucleic acid of Escherichia coli. Journal of Biological Chemistry. 240, 3975-3978 (1965).
- Marcinowski, L., Liedschreiber, M., et al. Real-time Transcriptional Profiling of Cellular and Viral Gene Expression during Lytic Cytomegalovirus Infection. PLoS Pathog. 8, e1002908 (2012).
- Chomczynski, P., Mackey, K. Short technical reports. Modification of the TRI reagent procedure for isolation of RNA from polysaccharide- and proteoglycan-rich sources. Biotechniques. 19, 942-945 (1995).
- Friedel, C. C., Dölken, L. Metabolic tagging and purification of nascent RNA: implications for transcriptomics. Mol. Biosyst. 5, 1271-1278 (2009).
- Friedel, C. C., Kaufmann, S., Dölken, L., Zimmer, R. HALO - A Java framework for precise transcript half-life determination. Bioinformatics. , (2010).
