

Summary
L'ARN cellulaire total donne un mauvais modèle pour l'étude des changements à court terme dans la synthèse d'ARN et de la décomposition ainsi que les cinétiques de maturation de l'ARN. Ici, nous décrivons marquage métabolique des ARN transcrits récemment avec 4 thiouridine suivie par biotinylation et purification thiol-spécifique des ARN transcrits nouvellement permettant de surmonter ces limitations.
Abstract
Le développement de puces tout-transcriptome et le séquençage de prochaine génération a révolutionné notre compréhension de la complexité de l'expression des gènes cellulaires. Avec une meilleure compréhension des mécanismes moléculaires impliqués, des mesures précises de la cinétique sous-jacents sont devenus de plus en plus importante. Ici, ces méthodes puissantes font face à des limitations dues aux propriétés intrinsèques des échantillons de modèle à savoir l'étude des ARN cellulaires, ils totales. Dans de nombreux cas, des changements dans l'ARN cellulaire total surviennent trop lentement ou trop rapidement pour représenter les événements moléculaires sous-jacents et leur cinétique avec une résolution suffisante. En outre, la contribution des altérations de la synthèse de l'ARN, le traitement et la décadence sont pas facilement différencié.
Nous avons récemment développé à haute résolution de l'expression génique pour surmonter ces limitations. Notre approche est basée sur le marquage métabolique de l'ARN nouvellement transcrit avec 4 thiouridine (donc également dénommé 4SU-marquage) suivie d'une purification rigoureuse de l'ARN transcrit à l'aide de nouveaux biotinylation thiol-spécifique et des billes magnétiques revêtues de streptavidine. Il est applicable à un large éventail d'organismes, y compris les vertébrés, chez la drosophile et la levure. Nous avons appliqué avec succès 4SU-tagging pour étudier la cinétique en temps réel des activités de facteurs de transcription, fournir des mesures précises de l'ARN demi-vie, et obtenir de nouveaux aperçus sur les cinétiques de maturation de l'ARN. Enfin, la modélisation computationnelle peut être utilisée pour générer une analyse globale et intégrée des mécanismes moléculaires sous-jacents.
Introduction
Profil d'expression génique est un outil clé utilisé pour étudier les processus cellulaires et le réseau d'interaction complexe associé. Des études sur l'abondance de l'ARNm ont généralement été la méthode de choix pour obtenir un aperçu de base sur les mécanismes moléculaires sous-jacents. Le développement de puces tout-transcriptome 1 et, plus récemment, le séquençage de prochaine génération de l'ARN (RNA-Seq) 2-4 alimentée cette approche. Bien que ces technologies ont révolutionné notre compréhension de la complexité de l'expression du gène cellulaire, ils font face à des limitations dues aux propriétés intrinsèques de leur échantillon de modèle, à savoir l'ARN cellulaire total. Premiers changements à court terme des niveaux d'ARN totaux ne correspondent pas à l'évolution des taux de transcription, mais sont intrinsèquement dépendante de l'ARN demi-vie des transcriptions respectifs. Alors qu'une induction cinq fois d'une transcription, par exemple l'encodage de courte durée pour un facteur de transcription, sera facilement détectable dans l'ARN totalmoins d'une heure, le même induction de la transcription, par exemple l'encodage de longue durée pour une enzyme métabolique, restera pratiquement invisible. En outre, même un arrêt complet (> 1000 fois down-regulation) du taux de transcription d'un gène moyenne avec un ARN demi-vie de cinq heures seront simplement prendre cinq heures pour les niveaux d'ARN totaux diminuer de seulement double . Par conséquent, l'analyse de l'ARN totale favorise la détection d'une régulation de la transcription de courte durée, dont la plupart codent pour des facteurs de transcription et des gènes avec des fonctions de réglementation 5. En outre, la vraie cascade cinétique de la réglementation est obscurcie et événements de signalisation primaires ne peut être différenciée du secondaire. Les deux, à son tour, peut entraîner un biais important dans les analyses bioinformatiques aval. Deuxièmement, des modifications dans les niveaux d'ARN totaux ne peuvent pas être attribuées à des changements dans la synthèse d'ARN ou de pourriture. Les mesures de ce dernier nécessitent des approches invasives cellulaires, par exemple blocage transcriptisur l'utilisation de l'actinomycine D 6, et la surveillance prolongée de l'ARN décroissance continue au fil du temps. Avec un ARNm demi-vie moyenne dans les cellules de mammifères de 5 à 10 hr 5,7, les niveaux d'ARNm de la plupart des gènes seulement auront diminué de moins de double après plusieurs heures d'arrêt de la transcription. Ces plutôt petites différences se traduisent en mesures grossièrement imprécise des demi-vies d'ARNm pour la majorité des gènes cellulaires en raison de la nature exponentielle des équations mathématiques sous-jacents. Enfin, si l'ARN-seq de l'ARN cellulaire total a révélé que près de la moitié de nos gènes sont soumis à des événements d'épissage alternatifs 8, la cinétique sous-jacents ainsi que les mécanismes dynamiques directeurs tissus et spécifiques au contexte réglementaire de traitement de l'ARN restent mal compris. En outre, la contribution de l'ARN traitement à l'expression différentielle des gènes, en particulier pour les ARN non-codants, reste à déterminer. Au total, ces limitations représentent des obstacles majeurs pourmodélisation cinétique bioinformatique des mécanismes moléculaires sous-jacents.
Nous avons récemment développé une approche, appelée résolution gène profilage de haute expression, pour surmonter ces problèmes 5,7,9. Il est basé sur le marquage métabolique de l'ARN nouvellement transcrit en utilisant 4 thiouridine (4SU-tagging), un dérivé de l'uridine naturel, et offre un accès direct aux transcriptions nouvellement transcrites avec un minimum d'interférences dans la croissance cellulaire et l'expression des gènes (voir Figure 1) 5, 10-12. L'exposition des cellules eucaryotes aux résultats 4SU à son adoption rapide, la phosphorylation de 4SU-triphosphate, et incorporation dans l'ARN nouvellement transcrites. Après isolement de l'ARN cellulaire total, la fraction d'ARN 4SU-marqué est thiol-spécifique biotinylé à générer un pont disulfure entre la biotine et l'ARN nouvellement transcrites. 'ARN cellulaire total »peuvent ensuite être séparés quantitativement dans étiqueté (' nouvellement transcrit») et sans étiquette («pré-existinG ') d'ARN avec une grande pureté en utilisant des billes magnétiques revêtues de streptavidine. Enfin, l'ARN marqué est récupéré à partir des billes par simple addition d'un agent réducteur (par exemple le dithiothréitol) le clivage de la liaison disulfure et de libérer l'ARN nouvellement transcrites à partir des billes.
ARN nouvellement transcrit représente l'activité de transcription de chaque gène au cours de la période d'exposition 4SU. 4SU-tagging dans le calendrier de minutes fournit ainsi un instantané de l'expression des gènes eucaryotes et un modèle idéal pour les analyses bioinformatiques en aval (par exemple, l'analyse du promoteur). Dans les cas où l'état d'équilibre peut supposer, les ratios de nouveaux transcrits / total, nouvellement transcrites / non étiquetés et non étiquetés / ARN total fournissent un accès non-invasive à l'ARN précises des demi-vies 7,13. En outre, il est important de noter que les ARN transcrits nouvellement purifié après aussi peu que 5 min de 4SU-tagging (5 min 4SU-ARN) est plus jeune que 15 et 60 min 4SU-ARN.Lorsque vous effectuez à la fois ultra-court et progressivement plus 4SU-tagging dans un cadre expérimental unique combiné avec l'ARN-Seq, la cinétique de maturation de l'ARN sont révélés à la résolution de nucléotides 9. Enfin, les analyses temps-cours d'ARN nouvellement transcrites et total combiné avec la modélisation informatique permettent une analyse intégrative de la synthèse de l'ARN et de la décomposition 14.
En conclusion, cette approche permet l'analyse directe de la dynamique de la synthèse d'ARN, de la transformation et de la dégradation dans les cellules eucaryotes. Elle est applicable dans tous les grands organismes modèles y compris des mammifères, des insectes (drosophile), les amphibiens (Xenopus) et la levure 5,15,16. Il est directement compatible avec l'analyse des microréseaux 5,17, RNA-Seq 9,13,14, et est applicable in vivo 12,15. Ici, nous détaillons la méthodologie d'étiqueter, d'isoler et de purifier l'ARN nouvellement transcrites dans les cellules de mammifères en culture. En outre, potentiomètreproblèmes al et les pièges sont discutées.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. Marquage métabolique avec 4 thiouridine
Faire un plan détaillé du dispositif expérimental / calendrier, par exemple quand il faut ajouter le 4SU à la culture cellulaire et quand récolter des échantillons. Régime pendant au moins 5 minutes entre chaque état. Ne traiter que des cellules d'un état à la fois. Poignée max. 3-5 plats à un moment donné. Manipuler les cellules aussi rapidement que possible pour minimiser les changements dans 2 niveaux de température et CO. Évitez d'exposer les cellules à la lumière après 4SU est ajouté, car cela pourrait conduire à une réticulation de l'ARN 4SU marquées par des protéines cellulaires.
Début de l'étiquetage
- Dégel 4 thiouridine (4SU) juste avant l'utilisation et la pipette quantité requise de 4SU pour chaque état dans un tube Falcon stérile.
- Prenez le montant requis de milieu de culture cellulaire (5 ml par boîte de 10 cm) sur les assiettes et ajouter à tube Falcon 4SU contenant et bien mélanger. Retirez et jetez le milieu restant de la vaisselle. <li> Appliquer 4SU contenant dossier moyen pour les plats.
Fin de l'étiquetage
- Éliminer le milieu de culture cellulaire à partir de cellules. Ajouter 5 ml de Trizol à chaque plaque. Pour les expériences complexes incluant plusieurs points dans le temps ou des conditions, cette étape est mieux fait par deux personnes, une élimination du milieu, l'autre ajoutant Trizol et la récolte du lysat.
- Incuber pendant 5 min à la température ambiante pendant une lyse complète des cellules.
- Utiliser une pipette de 10 ml de rincer soigneusement la plaque avec le Trizol ajouté. Ceci facilite la lyse complète des cellules et la récupération de l'échantillon. Manipuler avec soin comme Trizol est extrêmement dangereux quand entrer en contact avec la peau ou les yeux! Avoir antidote pour les brûlures phénol à la main (par exemple le polyéthylène glycol 300 ou 400 dans l'alcool dénaturé industriel (70:30)). Transférer les échantillons dans des tubes en polypropylène. Veuillez noter que les tubes Falcon classiques ne résistent pas à ces grandes forces G). Les échantillons peuvent être conservés à -20 ° C pendant au moins un mois avant l'ARN total is préparé.
2. Préparation d'ARN utilisant protocole modifié Trizol
- Ajouter 1 ml de chloroforme (0,2 ml par ml Trizol) et agiter vigoureusement pendant 15 secondes. Incuber à température ambiante pendant 2 - 3 min.
- Centrifuger à 13000 g pendant 15 min à 4 ° C.
- Transférer la phase supérieure aqueuse (contenant de l'ARN) à un nouveau tube en polypropylène de 15 ml.
- Ajouter la moitié du volume de la réaction de précipitation à la fois un tampon d'ARN et de l'isopropanol (par exemple de 3 ml de surnageant ajouter 1,5 ml de tampon d'ARN précipitation et de 1,5 ml d'isopropanol).
- Mélangez bien. Incuber à température ambiante pendant 10 min.
- Centrifuger à 13000 g pendant 10 min à 4 ° C. Rejeter le surnageant.
- Centrifuger brièvement (5000 g pendant 30 sec) et retirer l'isopropanol résiduel avec pipette 200 pl.
- Ajouter un volume égal d'éthanol à 75% et agiter le tube jusqu'à ce que le culot se détache. Évitez le divisant en plusieurs petits morceaux comme cela peut rendre l'enlèvement de résidusl d'éthanol difficile.
- Centrifuger à 13000 g pendant 10 min à 4 ° C. Rejeter le surnageant.
- Isoler ARN brièvement et éliminer l'éthanol restant avec une pipette 200 pl. Répétez l'étape et éliminer l'éthanol restant avec une pipette 20 ul. Après ces deux étapes, aucune autre séchage du culot doivent être effectuées.
- Ajouter 100 ul de H 2 O par 100 ug de rendement en ARN attendu et bien mélanger par pipetage de haut en bas 5-6 fois pour aider à dissoudre l'ARN.
- Dissoudre et dénaturer l'ARN par chauffage à 65 ° C pendant 10 min (shaker) et placer immédiatement sur la glace.
- Mesurer l'ARN concentration à 260 nm en utilisant un spectrophotomètre NanoDrop, suivant les instructions du fabricant. Cet ARN peut être conservé à -80 ° C pendant au moins un mois.
3. Biotinylation thiol-spécifique de l'ARN récemment transcrits
- Commencez avec 60 à 80 ug d'ARN cellulaire total.
- Constituer étiquetage réaction. Pipette dans la suiteordre (ARN par ug d'):
- 1 pl de tampon de Biotinylation 10x
- 7 ARN ul (contenant 1 ug d'ARN dilués dans nucléase H 2 O)
- 2 pl biotine HPDP (1 mg / ml DMF)
Toujours ajouter la biotine HPDP dernier et mélanger immédiatement par pipetage. Dans le cas où la biotine précipite, contenu DMF peut être augmentée jusqu'à une concentration finale de 40%.
- Incuber à température ambiante pendant 1,5 heures avec rotation.
- Ajouter un volume égal de chloroforme. Mélanger vigoureusement. Incuber pendant 2-3 minutes jusqu'à ce que les phases commencent à se séparer et les bulles commencent à disparaître.
- Centrifuger à 20000 g pendant 5 min à 4 ° C. Soigneusement transférer la phase aqueuse supérieure dans un nouveau tube.
- Répétez les étapes 3,4 et 3,5 fois. Vous pouvez effectuer cette opération dans 2 ml à verrouillage de phase gel tubes lourds pour réduire la perte de l'ARN.
- ARN précipitations: ajouter 1/10 du volume de 5 M de NaCl et un volume égal d'l'isopropanol à la phase aqueuse.
- Centrifuger à 20000 g pendant 20 min à 4 ° C. Rejeter le surnageant.
- Ajouter un volume égal d'éthanol à 75%, centrifuger à 20000 g pendant 10 min à 4 ° C, éliminer le surnageant.
- Centrifuger brièvement et enlever des résidus d'éthanol avec 200 pipette ul.
- Centrifuger brièvement et enlever des résidus d'éthanol avec 20 pipette ul.
- Ne laissez pas l'ARN à sécher. Re-suspendre en 50 - 100 pi H 2 O (~ 1 pi par 1 ug d'ARN d'entrée). Bien mélanger par pipetage de haut en bas 5 - 6 fois.
- Vérifiez la qualité de l'ARN par analyse électrophorétique d'exclure dégradation de l'ARN.
4. Dot Blot Analyse des 4SU-incorporation (Facultatif)
Incorporation 4SU peut être facilement déterminée par analyse dot blot d'ARN biotinylé. Il s'agit d'une étape facultative qui permet le dépannage et l'estimation des taux d'incorporation 4SU rapport à un témoin oligo d'ADN biotinylé. Pour ce test nous avons recommend utilisant iodoacétyle-biotine au lieu de biotine HPDP pour biotinylation de l'ARN 4SU marqués à l'étape 3.2. Il en résulte une biotinylation irréversible de 4SU-ARN. Par conséquent, les méthodes basées sur colonne (par exemple RNeasy) peuvent être utilisés pour la récupération de plus petites quantités d'ARN biotinylé (par exemple 5 ug). Alors que l'ARN biotinylé en utilisant la biotine HPDP est également approprié pour cet essai, le signal résultant est plus faible et le rapport signal-bruit moins favorable (Figure 3).
- Suivre le protocole de 4SU-étiquetage et l'isolement de l'ARN cellulaire total, comme décrit dans les sections 1 et 2.
- Biotinyler 4SU-ARN marqué comme décrit dans la section 3, en remplaçant la biotine HPDP avec iodoacétyle-biotine et d'effectuer deux extractions chloroforme pour éliminer complètement les résidus iodoacétyle-biotine excessifs.
- Récupérer ARN biotinylé par isopropanol / éthanol précipitation comme décrit ou en utilisant une approche basée sur les colonnes (par exemple RNeasy) en cas de petites quantités d'ARN (<10 pg) Sont utilisés.
- Incuber la membrane Zeta dans l'eau sans nucléase à bascule pour 10 min.
- Prendre la membrane hors de l'eau sans nucléase et retirer les fluides excessifs en plaçant la membrane entre deux serviettes en papier propre et en appuyant fermement. Air-séchage de la membrane pendant 5 min se traduira par des points plus agréables.
- Pour chaque échantillon, préparer 20 pi d'ARN 200 ng / ul utilisant glacée tampon de liaison dot blot (NaOH 10 mM, EDTA 1 mM). Appliquer 5 ul de cette dilution (soit 1 ug d'ARN), ainsi que trois dilutions de 10 fois suivantes (soit 100, 10 et 1 ng d'ARN, respectivement) de la membrane Zeta par pipetage. Pipetage à travers un rack vide de pointes de pipette peut être utilisé pour fournir espacement uniformément répartie. Sinon, utilisez un appareil de dot blot selon les instructions du fabricant.
- Appliquer 5 pl de l'ADN oligo marqué à la biotine à des concentrations allant de 20 ng / ul à 20 pg / pl (soit 100, à 0,1 ng oligo) comme une suite favorablerol à la membrane par pipetage. Utilisez un échantillon biotinylé, 4SU naïfs comme contrôle négatif.
- Air-sécher la membrane pendant 5 min.
- Incuber la membrane pendant 30 min à 40 ml avec un tampon de blocage à bascule.
- Incuber la membrane avec 10 ml de 1:1000 streptavidine-peroxydase de raifort pendant 15 min (5 ml de PBS + 5 ml SDS 20% + 10 pl streptavidine-peroxydase de raifort)
- Lavez membrane deux fois dans 40 ml de PBS + 10% SDS (20 ml PBS + 20 ml 20% SDS) pendant 5 min.
- Lavez membrane deux fois dans 40 ml de PBS + 1% SDS (38 ml PBS + 2 ml 20% SDS) pendant 5 min.
- Lavez membrane deux fois dans 40 ml de PBS + 0,1% SDS (40 ml PBS + 200 pi 20% SDS) pendant 5 min.
- Retirer un excès de liquide en plaçant la membrane entre deux serviettes en papier propre et en appuyant sur les fermement.
- Visualisez membrane-bound HRP en utilisant ECL selon les instructions du fabricant.
- Placer la membrane dans du papier / sac en plastique, enlever les bulles d'air et incuber pendant 2 min dans l'obscurité.
- Exposer la membrane àfilm 1 - 5 min.
5. Séparation de l'ARN étiquetés et non étiquetés en utilisant des billes magnétiques revêtues de streptavidine
- La chaleur du tampon de lavage (3 ml par exemple) à 65 ° C dans un bain d'eau.
- Préparer une solution fraîche mM dithiothréitol 100 (DTT) dans nucléase H 2 O. Le faire par décantation de 15 à 30 mg de poudre de TNT dans un tube propre de 50 ml Falcon placé sur l'ultra-fine échelle. Peser et ajouter la quantité requise de nucléase H 2 O.
- Heat biotinylé échantillons d'ARN à 65 ° C pendant 10 min pour dénaturer et immédiatement sur la glace.
- Lieu μMacs colonnes dans le support magnétique. Nous vous recommandons de ne pas traiter plus de 12 échantillons à la fois (6 - 8 échantillons sont optimales).
- Miltenyi colonnes pré-s'équilibrer avec la température ambiante 1 ml de tampon de lavage. Cela prendra environ 15 minutes.
- Pendant ce temps, ajouter 100 ul de billes de streptavidine à 50 - 100 ul d'ARN biotinylé. Incuber à température ambiante pendant 15 min avec rotation. < li> Si l'une des colonnes n'a pas engagé de vidange maintenant ce qui peut être facilité en appuyant doucement sur le haut de la colonne avec un doigt ganté. Une fois que le flux a commencé les colonnes drainent facilement.
- Appliquer les ARN / perles pour les colonnes. Jeter l'effluent à travers, sauf si vous voulez récupérer la fraction d'ARN sans étiquette (voir section 7).
- Laver trois fois avec 0,9 ml de 65 ° C tampon de lavage (1 ml pointes de pipette rétrécissent quand pipetage tampons à 65 ° C).
- Laver trois fois avec 0,9 température ambiante ml du tampon de lavage.
- Pipette 700 ul de tampon RLT (RNeasy Kit de nettoyage de MinElute, Qiagen) dans de nouveaux tubes de 2 ml et de les placer sous les colonnes.
- On élue l'ARN nouvellement transcrites dans le tampon RLT en ajoutant 100 ul de 100 mM DTT aux colonnes.
- Effectuer un deuxième tour élution 3 min plus tard dans le même tube en ajoutant un autre 100 pi de DTT 100 mM.
6. La récupération de l'ARN récemment transcrits
ontenu "> Continuer avec le nettoyage de MinElute RNeasy (Qiagen) protocole suivant les instructions du fabricant. Elute dans 25 pi H 2 concentrations d'ARN Mesure O. nucléase aide d'un spectrophotomètre Nanodrop. Pour éviter d'avoir à décongeler et recongeler ARN avant de soumettre à un dosage à haut débit, nous vous recommandons la préparation d'ADNc immédiatement après l'ARN nouvellement transcrit est purifié. Utiliser 2,5 pi de l'ARN nouvellement transcrites dans 20 pi de mélange de synthèse d'ADNc pour la synthèse d'ADNc en suivant les instructions du fabricant. effectuer des contrôles qRT-PCR en utilisant 1 : 10 dilutions du mélange d'ADNc magasin ARN à -80 ° C..7. Récupération de vierge, Unbound ARN (Facultatif)
Dans le cas où l'ARN non lié doit être récupéré; recueillir et combiner l'écoulement traversant (après addition de la solution de billes ARN-streptavidine pour les colonnes) et du premier lavage pour la précipitation subséquente. Habituellement, il est suffisante pour précipiter seulement 50% de l'ARN non lié comme this contiendra> 80% de la matière de départ.
- Ajouter un volume égal d'isopropanol (pas de sel doit être ajoutée comme le tampon de lavage contenant déjà 1 M de NaCl).
- Centrifuger à 20000 g pendant 20 min à 4 ° C. Rejeter le surnageant.
- Ajouter un volume égal d'éthanol à 75%, centrifuger à 20000 g pendant 10 min à 4 ° C, éliminer le surnageant.
- Centrifuger brièvement et enlever des résidus d'éthanol avec 200 pipette ul.
- Centrifuger brièvement et enlever des résidus d'éthanol avec 20 pipette ul.
- Ne laissez pas l'ARN à sécher. Remettre en suspension dans 100 ul H 2 O. Bien mélanger par pipetage de haut en bas 5 - 6 fois. Incuber à 65 ° C pendant 10 min avec agitation et transférer directement à la glace.
- Vérifiez la qualité de l'ARN par analyse électrophorétique d'exclure dégradation de l'ARN.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
1. Matériau de départ et rendements attendus
Après 1 heure (h) de 4SU-exposition ARN nouvellement transcrit représente environ 1-4% de l'ARN cellulaire total. Ce sera plus faible dans les cellules de la croissance arrêtés car ils ne synthétisent l'ARN pour tenir compte de la croissance / réplication cellulaire. Lors de l'étiquetage pendant 1 heure, nous vous conseillons de commencer le dosage avec 60 à 80 ug d'ARN total. A partir de moins de 30 ug de l'ensemble des résultats d'ARN dans des pastilles de petits ARN qui sont difficiles à voir après l'étape de biotinylation et peut donc être facilement perdu. Les niveaux d'ARN entrée peut être augmentée à autant que 150 mg pour des durées très courtes de l'étiquetage (par exemple 5 - 10 min). Lorsque la durée de l'ARN étiquetage est ramenée de 1 h à 5 min de la contribution de séquences introniques de courte durée dans les nouveaux transcrits d'ARN augmentation de ~ 60% à ~ 80% 9. Comme introns sont sensiblement plus longues par rapport aux séquences de codage, ainsi que 5'-et 3'-UTR, la quantité de transcrit nouvellementARN, qui peut être purifié suivant à court ou même ultra-court 4SU-tagging, ne baisse pas linéaire. En tant que tel, nous avons obtenu> 0,5% de l'ARN total après 5 min de 4SU-tagging dans les lignes de lymphocytes B humains non adhérentes 9. Il convient toutefois de noter que la concentration élevée des durées 4SU et un peu plus de l'étiquetage peut être nécessaire pour atteindre des taux d'incorporation 4SU similaires dans les cellules adhérentes. Alors que même un taux 4SU-incorporation faible permettra capture efficace et la purification de grandes transcriptions, uridine-riche, très court transcription à faible teneur en uridine (par exemple miARN) sont susceptibles d'échapper à la purification, même en utilisant des concentrations 4SU élevées (> 1 mm). Dans les fibroblastes murins NIH-3T3, 1 h de 200 um exposition 4SU ARN marqué récemment transcrits avec environ un 4SU résidu par 50 - 100 nucléotides (nt) 5. Cela devrait permettre une récupération très efficace de transcriptions> 500 - 1000 nt de longueur. En conséquence, nous avons observé une seule taille de transcription mineurepartialité lors de l'étiquetage pendant 1 heure en utilisant 200 um 4SU dans les deux fibroblastes murins et des cellules B humaines 7. Alors que 1 h de 200 4SU pM n'a pas entraîné de modifications importantes dans les niveaux de transcription cellulaire dans les fibroblastes murins, l'exposition prolongée des cellules à ≥ 200 um 4SU fait suite à un déficit de croissance mesurable dans les 24 heures (données non publiées). Par conséquent, tant la durée de l'étiquetage et de la 4SU-concentration travailleurs doivent être minimisées pour éviter les effets extra-utérines ou toxiques. Un moyen facile de déterminer la 4SU-concentration minimale requise pour une récupération efficace de l'ARN nouvellement transcrit est de purifier l'ARN nouvellement transcrite à la suite 4SU-étiquetage avec des concentrations croissantes de 4SU (par exemple de 50 à 1600 um). Comme le montrent les figures 2A et 2B, la récupération de l'ARN transcrits nouvellement marqués pendant 1 heure dans des fibroblastes humains primaires ont considérablement augmenté de 50 à 200 uM 4SU mais alors commencé à se stabiliser.
2. DotÉpongez Quantification de constitution 4SU (facultatif)
Dans certains cas, il peut être intéressant de mesurer la quantité d'incorporation 4SU dans l'ARN total. C'est mieux fait par analyse dot blot sur l'ARN biotinylé en utilisant un conjugué streptavidine. En raison de sa nature chimique iodoacétyle-biotine est plus réactif à thiol-groupes que la biotine HPDP entraînant la biotinylation de pratiquement tous les résidus 4SU de l'ARN nouvellement transcrit. Il est important de noter que, comme la biotine HDPD, iodoacétyle-biotine n'est pas soluble dans l'eau et est donc éliminé de manière efficace par extraction au chloroforme comme effectuée pour la biotine HPDP. Par conséquent, les conditions et les concentrations de réaction identiques peuvent être utilisés comme lors de l'utilisation de biotine HPDP. Cependant, iodoacétyle-biotine n'est pas réversible. Il ne peut donc pas être utilisé pour la purification de l'ARN nouvellement transcrites dans les approches fondées sur la colonne. Bien que l'utilisation de iodoacétyle-biotine permet de quantifier 4SU-constitution, les mesures basées sur la biotine HPDP considèrent à la fois4SU-incorporation et l'efficacité de biotinylation. Employant les deux réactifs de biotinylation au même échantillon permet la mesure de l'efficacité de biotinylation de l'ARN incorporé 4SU. efficacité Biotinylation de biotine-HPDP pour 4SU-ARN marqué semble être environ trois fois inférieure à celle de iodoacétyle-biotine qui indique que seulement environ un sur trois 4SU résidus dans l'ARN nouvellement transcrit est réellement biotinylé par la biotine HPDP (Figure 3). En comparant les échantillons de l'intensité de signal à la commande oligo ADN biotinylé, les densités de biotinylation peuvent être mesurées. Pour la plupart des lignées cellulaires de mammifères un signal positif devrait être encore détectable dans 10 ng d'ARN biotinylé après 1 h de 200 étiquetage 4SU iM. Un signal de fond faible est habituellement détectable pour la plus forte concentration (1 mg) de l'ARN non marqué.
3. Purification d'ARN récemment transcrits
La récupération de l'ARN nouvellement transcrit est très quantative. Si vous avez commencé avec la même concentration d'ARN vous pouvez vous attendre à obtenir les mêmes quantités d'ARN nouvellement transcrites pour tous les échantillons. Comme de nombreux tests colonne à base de collection de l'ARN nouvellement transcrit en utilisant le kit de MinElute RNeasy peut entraîner l'absorption supplémentaire de 230 à 260 nm (présence de détergents issus des tampons de lavage) qui peuvent interférer avec la DO 260 mesures. Cela se voit dans une moindre mesure pour l'utilisation d'un tube collecteur de 2 ml pour chaque étape de centrifugation. Néanmoins, les mesures de DO déraisonnablement élevés (> 2 fois plus que les autres échantillons) doivent être considérés avec précaution, surtout si OD 260/280 ratios sont <1.7. Pour les analyses en aval, il est donc souvent préférable d'utiliser la même quantité de volume d'ARN de modèle pour tous les échantillons. Dans les cas où les rendements des ARN marqués sont inférieurs chèque attendu des signes de dégradation de l'ARN par analyse électrophorétique. ARN transcrits nouvellement contient des quantités significativement plus importantes des grands, des transcriptions non épissés avec les bandes d'ARNr typiques étant beaucoup moins important (Figure 4).
4. La quantification de l'ARN récemment transcrits
Enfin, les taux d'incorporation de 4SU de l'ARN nouvellement transcrites peuvent être directement quantifiés par analyse spectrophotométrique basé sur le maximum d'absorption à 330 nm 4SU et la DO 330/260 Ratio 5,18. Cela nécessite> 3 pg d'ARN marqués sont concentrées dans un petit volume (10 - 20 pi) par isopropanol / précipitation à l'éthanol. Pour éviter de perdre le petit culot d'ARN co-précipitation avec 30 pg de glycogène sans nucléase (Fermentas, # R0551) doit être effectuée. Un pic supplémentaire est visible à 330 nm reflétant le taux d'incorporation de 4SU en ARN récemment transcrits (Figure 5).
/ Files/ftp_upload/50195/50195fig1highres.jpg "/>
Figure 1. . Principe de marquage métabolique avec la 4-thiouridine (4SU) 4SU est ajoutée aux cellules pour l'(5-120 min) requise temps suivie par la préparation d'ARN cellulaire total. Après biotinylation thiol-spécifique, l'ARN cellulaire total est séparé en 4SU marqués, ARN nouvellement transcrits d'ARN et vierge, pré-existantes à l'aide de billes magnétiques recouvertes de streptavidine. ARN nouvellement transcrit est récupéré à partir des billes à l'aide d'un agent réducteur qui clive les liaisons disulfures qui relient les ARN transcrits récemment aux billes. Cliquez ici pour agrandir la figure .
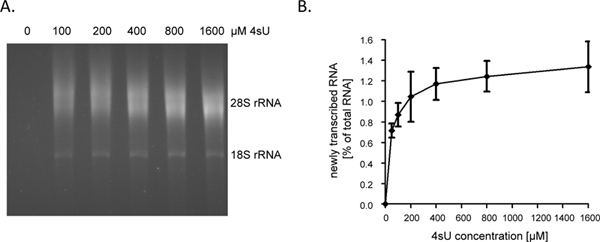
Figure 2. La récupération de l'ARN nouvellement transcrit suite des concentrations croissantes de 4SU. (A) fibroblastes de prépuce humain primaires (HFF) ont été incubées avec 100, 200, 400, 800 ou 1600 pM de 4SU. ARN récemment transcrits ont été purifiés à partir de 50 ug d'ARN cellulaire total et soumis à une analyse électrophorétique. Comme prévu, une augmentation dépendante de la concentration en ARN transcrits nouvellement récupérés a été observée qui a commencé à se stabiliser à des concentrations plus élevées. (B) Les montants de l'ARN purifié récemment transcrits ont été quantifiés en utilisant le logiciel ImageJ 1.45s. Les données combinées de quatre expériences indépendantes sur les quantités d'ARN transcrits nouvellement récupérés suite à différentes concentrations de 4SU-étiquetage allant de soit de 50 à 800 uM 4SU (n = 2) ou 100 -. 1,600 uM 4SU (n = 2) sont présentés Cliquez ici pour agrandir la figure .
upload/50195/50195fig3.jpg "alt =" Figure 3 "fo: content-width =" 4.5in "fo: src =" / files/ftp_upload/50195/50195fig3highres.jpg "/>
Figure 3. Estimation de constitution 4SU dans l'ARN total 4SU marqués en utilisant une analyse dot blot. L'ARN total a été isolé à partir de fibroblastes murins NIH-3T3 ou des fibroblastes de prépuce humain (HFF) incubées avec 200 uM 4SU pendant une heure. Pas 4SU a été ajouté à un plat comme contrôle négatif. Pour HFF deux inhibées par contact (n = cellules non-croissance) et des cellules de culture (y) ont été inclus. L'ARN a été isolé en utilisant le réactif Trizol et ensuite conjugué à la biotine-HPDP ou iodoacétyle-biotine. Concentration de chaque échantillon a été ajusté à 200 ng / pl et 5 pi de cette dilution (soit 1 pg d'ARN), ainsi que trois dilutions de 10 fois subséquentes (soit 100, 10 et 1 ng d'ARN, respectivement), étaient tous repéré sur un morceau de membrane Zeta. 5 pl de dilutions marqué à la biotine oligo ADN ont été placés sur la membrane comme contrôles positifs au concentratles ions allant de 20 ng / pl bas à 20 pg / ul (soit 100, à 0,1 ng, respectivement). Densité biotine a été sondé en utilisant un conjugué à la peroxydase de raifort streptavidine.
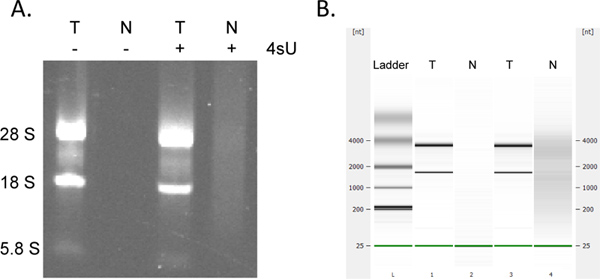
Figure 4. Analyse électrophorétique de l'ARN nouvellement transcrites et totale. L'ARN total (T) et de l'ARN nouvellement transcrit (N) préparé à partir de fibroblastes murins NIH-3T3 cultivées tant en présence qu'en l'absence de 500 um 4SU pendant 1 heure a été analysé par électrophorèse sur gel (A) et (dans le même ordre), en utilisant la Bioanalyser Agilent (B). Pas ARN a été récupéré sans traitement 4SU des cellules. Purifié l'ARN nouvellement transcrit contient une plus grande quantité d'ARNm de haut poids moléculaire et ARNr beaucoup moins mature que totaleARN comme notable entre le 28S, 18S, 5.8S et bandes d'ARNr. Cliquez ici pour agrandir la figure .
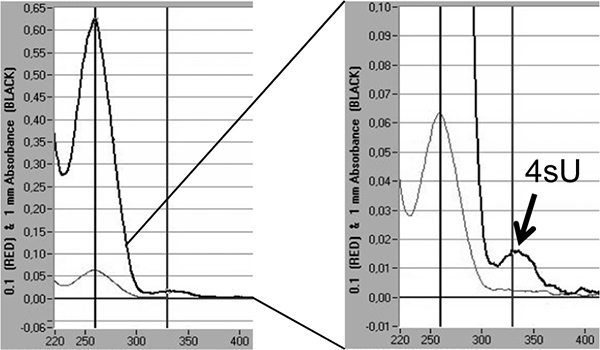
Figure 5. Quantification de l'incorporation dans l'ARN 4SU nouvellement transcrites par analyse spectrophotométrique. Des ARN transcrits nouvellement purifiés à partir de 2 x 100 ug d'ARN total après 1 h de 200 uM 4SU dans murins fibroblastes NIH-3T3. ARN récemment transcrits a été précipité avec de l'isopropanol / éthanol après addition de 30 pg de glycogène sans nucléase. Une analyse par spectrophotométrie d'ARN transcrits nouvellement obtenues par un Nanodrop 1000 spectrophotomètre est affiché. Les lignes en gris clair représentent des mesures à 0,1 mm, tandis que les lignes en gris foncé épais représentent des mesures à colonne de liquide de 1 mm. Sur la droite, un agrandissement du pic d'extinction représentant til a incorporé 4SU des résidus est affiché. Sur la base du coefficient d'extinction de 4SU 18, les taux d'incorporation de 4SU peuvent être estimées.
| Durée de l'étiquetage [min] | 4SU concentration recommandée [iM] |
| 120 | 100-200 |
| 60 | 200 - 500 |
| 15-30 | 500 - 1000 |
| <10 | 500 - 2000 |
Tableau 1. Recommandé concentrations 4SU.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Le marquage métabolique de l'ARN transcrit nouvellement renforce considérablement la puissance des technologies à haut débit comme des puces et ARN-Seq en fournissant des modèles plus appropriés pour aborder la question biologique d'intérêt. Le présent protocole a subi une optimisation considérable. Il permet l'enrichissement> 1000 fois de l'ARN nouvellement transcrites et fournit des résultats hautement reproductibles.
Le dispositif expérimental d'une expérience 4SU-tagging est d'une importance cruciale que l'ARN nouvellement transcrites seront dépeindre l'activité transcriptionnelle en temps réel uniquement pendant la durée de l'exposition des cellules à 4SU. Si les mouvements de taux de transcription suite à un stimulus ont déjà disparu, ceux-ci seront manquées lors de l'analyse d'ARN nouvellement transcrites même si les changements dans les niveaux d'ARN totaux peuvent encore être détectable. Par conséquent, une bonne compréhension de la biologie sous-jacente est important de définir le dispositif expérimental ainsi que la période optimale otemps de f pour l'exposition 4SU. Ci-dessous, nous fournissons des recommandations et des moyens pour éviter les pièges les plus courants pour les étapes les plus cruciales.
Préparation des solutions mères et des articles en matière plastique
Toutes les solutions mères doivent être préparées avec de l'eau sans nucléase. L'utilisation en interne purifié de l'eau déminéralisée peut entraîner des problèmes si l'eau contient des agents réducteurs. Dans un cas, cela s'est traduit par la perte complète de tous les ARN marqué. Par conséquent, nous vous recommandons fortement d'acheter pré-faites NaCl sans nucléase, Tris-Cl, EDTA, citrate de sodium et de l'eau. Assurer des conditions sans nucléase à tout moment. Le diméthylformamide (DMF) dissout certaines matières plastiques. Nous avons constaté que l'aide de 25 ml de culture cellulaire pipettes en plastique pour transférer DMF de ses actions bouteille de verre pour tubes Falcon de 50 ml pour préparer la solution de biotine HPDP était suffisante pour réduire sensiblement les rendements de l'ARN nouvellement transcrit à partir de l'ensemble de dosage. Fait intéressant, cela n'a pas d'incidence négative sur le biotinylefficacité ation (tel que testé par dot blot) mais a entraîné une réduction de 75 à> la perte de 90% de l'ARN nouvellement transcrites qui pourrait être récupérée à partir des billes. La perte a été plus prononcée lorsque la durée de l'étiquetage a été réduit de 60 à 30 min ou moins. Très probablement, une substance élue des pipettes en plastique par le DMF partiellement détruit le revêtement des billes de streptavidine. Par conséquent, l'utilisation de matières plastiques ne sont pas connus pour être compatible avec DMF doit être évitée par tous les moyens. Pour les mêmes raisons, des grattoirs de cellules ne doivent pas être utilisés pour améliorer la récupération des échantillons TRIzol à partir de plaques de culture cellulaire. Il est intéressant de noter que les substances présumées élues à partir de matières plastiques par le DMF ou Trizol ont apparemment été ni éliminés par extraction au chloroforme, ni isopropanol / précipitation à l'éthanol.
La culture cellulaire
La densité cellulaire sur les plaques est d'une importance cruciale. Dans une expérience où les cellules semblaient être un peu trop confluent (90 -100%), nous avons traité fibroblastes murins NIH-3T3 pendant 30 min avec 100 U / ml d'interféron (IFN) α ou γ. Dans les cellules confluentes moins encore 15 min de traitement par IFN déjà donné lieu à un 5 - à induction 8 fois des gènes comme IRF1 ou SOCS3 5. Avec les cellules étant légèrement l'analyse des microréseaux trop confluent n'a montré aucune induction de gènes IFN-inductibles, même pour les inductible plus rapidement les gènes comme IRF1 ou SOCS3. Par conséquent, la densité cellulaire est un facteur crucial pour les expériences 4SU-étiquetage et les plaques de culture cellulaire devrait être examinée avec soin avant de commencer l'étiquetage.
4SU est un ribonucléoside photo-activable et de l'ARN 4SU contenant est réticulé de manière efficace à des protéines après exposition à la source de lumière à 365 nm. 4SU cellules traitées doivent être cultivées dans l'obscurité et l'exposition à la lumière doit être évitée. Après l'élimination des protéines cellulaires par Trizol isolement de l'ARN, ce risque est sensiblement réduit.
<p class = "jove_content"> 4SU n'est pas incorporé dans l'ADN cellulaire. Il convient toutefois de noter que l'ARN total seront encore contenir de petites quantités d'ADN cellulaire. Lors de l'utilisation 4SU-tagging et l'analyse Q-RT-PCR pour étudier l'expression des gènes viraux dans l'infection à cytomégalovirus nous avons jugé nécessaire d'inclure une étape DNaseI digest dans le protocole pour éliminer les génomes viraux concatémériques 19. Ce n'est probablement pas nécessaire lors de l'utilisation des protocoles en aval qui ne sont pas sensibles à la présence de l'ADN.Taux d'incorporation 4SU et la concentration optimale 4SU
4SU est facilement absorbé par les cellules présentant des niveaux intra-et extra-cellulaires les plus susceptibles d'équilibrage en moins d'une minute 9,16. Les taux d'absorption et l'intégration de 4SU sont concentration-dépendante. Par conséquent, la concentration 4SU peut être facilement ajustée en fonction de la durée employée de l'étiquetage. Tableau 1 fournit des conseils sur les concentrations 4SU dans relation de la durée de l'étiquetage en fonction de notre meilleure expérience personnelle. Pour 1 heure de l'étiquetage 4SU dans les cellules de mammifères, 200 4SU uM seront suffisantes pour la plupart des applications qui en découlent dans environ un résidu 4SU par 50 à 100 nucléotides de l'ARN nouvellement transcrites dans les fibroblastes.
Au cours des deux dernières années, nous avons appliqué 4SU-tagging à un large éventail de types de cellules d'origine humaine et murine y compris les fibroblastes, cellules endothéliales, cellules épithéliales, les cellules du stroma de la moelle osseuse, les macrophages et les lymphocytes T. En outre, les cellules de drosophile et le xénope ont été utilisés avec succès. Dans toutes ces expériences, l'incorporation 4SU a été jugée très efficace nécessitant des ajustements minimes de concentration 4SU pour les différents types de cellules. Lorsque la mise en place de la méthode pour les nouveaux types de cellules, nous vous recommandons de marquer les cellules avec l'augmentation 4SU-concentrations (par exemple allant de 50 à 1600 M) et d'analyser la relation entre purifiée nouvellement transcrit RNA aux 4SU-concentrations appliquées (voir Figure 2A / B). Le 4SU-concentration à laquelle la quantité de l'ARN purifié récemment transcrits entre un plateau devrait être choisie.
Dans le cas où, le contact des cellules inhibées très confluentes sont utilisés, nous recommandons d'utiliser des concentrations 4SU légèrement plus élevées (par exemple 500 au lieu de 200 uM) pour assurer l'incorporation 4SU efficace. En outre, dans les cas où la capture de très courtes transcriptions nouvellement transcrites (<200 nt) est d'un intérêt particulier, la concentration 4SU peut aussi être nécessaire d'augmenter. Cela ne devrait pas être combiné avec des temps d'étiquetage prolongées (par exemple> 1 heure) afin d'éviter les effets extra-utérines ou de toxicité. Enfin, nous avons constaté que l'utilisation d'un trop petit volume de milieu de culture cellulaire peut réduire l'efficacité de l'incorporation 4SU. Nous vous recommandons donc d'utiliser 5 ml ou 10 ml de milieu par 10 cm ou 15 cm plat, respectivement.
Préparation de l'ARN cellulaire total
Pour la réussite de ce protocole, il est crucial d'obtenir des ARN cellulaires totaux propres, sans RNase. Utiliser 5 ml Trizol par 15 cm plat produit ARN propre, exempt de nucléases. Nous vous recommandons d'utiliser le protocole Trizol modifié par Chomczynski et al. 20. Tout d'abord, il est mieux adapté pour isoler de grandes quantités d'ARN (> 100 mg) que les résultats améliorés de force centrifuge dans pellets fermes qui sont plus faciles à manipuler pendant les étapes de lavage. Cependant, cela nécessite l'utilisation de tubes et adaptateurs en polypropylène spéciales comme les 15 ml tubes Falcon de laboratoire réguliers ne survivent pas plus de 6,000 × g. Deuxièmement, elle améliore l'élimination de l'ADN et des glycoprotéines. Cela devient particulièrement évident lors de la préparation d'ARN à partir d'organes ou de tissus. Troisièmement, il ne limite pas le montant maximum de l'ARN total qui peuvent être isolées. Même si nous avons également constaté méthodes d'isolement d'ARN colonne à base (par exemple RNeasy) pour fournir des ARN de qualité convenable, colonnes standards are seulement capable de capturer jusqu'à 100 ng d'ARN total limitant ainsi la quantité de matière de départ. Enfin, par élimination de l'éthanol restant deux fois avec une pipette, le séchage de l'ARN pour éliminer l'éthanol résiduel n'est plus nécessaire. Ceci élimine le risque de sur-séchage de l'ARN, ce qui peut être difficile à dissoudre à nouveau par la suite. En principe, 4SU-tagging est applicable in vivo, par exemple par injection intraveineuse de souris. Cependant, nous avons constaté que l'ARN pureté représente un problème majeur nécessitant la purification des transcriptions polyA avant de purification d'ARN nouvellement transcrit (données non publiées).
Biotinylation et l'élimination de la biotine non liée
Biotine-HPDP est de 100% thiol spécifique et forme une liaison disulfure entre le résidu de biotine et thiol-ARN marqué molécules nouvellement transcrites. efficacité Biotinylation de l'ARN 4SU marqué est d'environ 30%, tel que déterminé par analyse dot blot 5. Comme biotine-HPDP n'est pas soluble dans l'eauil peut être éliminée efficacement par extraction au chloroforme. Bien qu'une seule étape d'extraction de chloroforme est suffisante pour éliminer la grande majorité de la biotine non liée nous répétons régulièrement cette étape pour assurer une élimination complète. Pour réduire les pertes d'ARN au cours de l'étape d'extraction chloroforme 2 ml à verrouillage de phase gel des tubes lourds (Eppendorf) peut être utilisé en suivant les instructions du fabricant. Habituellement, nous utilisons des tubes à verrouillage de phase seulement pour la deuxième étape d'extraction de chloroforme que les volumes de modèle de la première étape sont souvent trop élevés pour être directement compatible avec ces tubes. Après l'élimination des non lié à la biotine HPDP, l'ARN est récupéré par l'isopropanol / précipitation à l'éthanol. Il est important de noter que les kits commerciaux colonne basés à récupérer l'ARN biotinylé (par exemple RNeasy de QIAGEN) ne devraient pas être utilisés car ils contiennent des agents réducteurs dans les tampons fournis, qui clivent la liaison disulfure et supprimer la biotine à partir de l'ARN nouvellement transcrit .
Purification de nouveauly ARN transcrit
Ne pas ajouter plus de 100 ARN biotinylé pi à 100 pi de billes de streptavidine. Ajout d'un volume inférieur est préférable. Cependant, le même volume d'ARN doit être ajoutée pour tous les échantillons. Réglez le volume d'entrée ARN (entre les échantillons) que vous ajoutez aux billes de streptavidine en ajoutant simplement le volume requis de 1x TE aux billes. Un moyen facile de faire des frais sans nucléase 100 mM DTT est de décanter une quantité suffisante de poudre DTT dans un tube falcon placé sur une échelle ultra-sensible, puis ajouter la quantité requise de nucléase H 2 O pour générer 100 mM DTT (64,8 pi d'eau par 1 mg TNT).
Pendant le développement de 4SU-tagging, nous avons testé billes de streptavidine de divers fournisseurs. Un certain nombre d'entre eux a généré de grandes quantités de fond. Par conséquent, nous vous recommandons fortement d'utiliser les billes de streptavidine Miltenyi que, jusqu'ici, nous n'avons jamais eu aucun problème avec le report de l'ARN non marquées à partir de tissu cLes échantillons d'ARN ulture dérivés. De cette façon, aussi peu que 150 ng d'ARN marqué peut être spécifiquement purifiée à partir de 150 pg d'ARN biotinylé (dans 100 pi d'eau) en utilisant 100 pi de billes de streptavidine. L'équilibrage des perles avec le tampon d'équilibrage fourni avec les billes peut être réalisée et peut légèrement accroître les taux de capture 13.
Des contrôles de qualité
Nous vous recommandons d'effectuer des contrôles Q-RT-PCR sur l'ARN nouvellement transcrites avant de le soumettre pour analyse à haut débit. Il peut s'agir de quantification de plusieurs gènes de référence connus pour être régulés différemment dans le cadre expérimental donné. Dans les cas où 4SU-tagging est utilisée pour étudier les taux de désintégration d'ARN, nous vous recommandons de quantifier la transcription de courte durée (par exemple myc, fos) et une longue durée un (par exemple GAPDH) dans l'ARN total et nouvellement transcrit. Le taux d'ARN nouvellement transcrites / total devrait être sensiblement plus élevé (~ 5 - à 10 fois)les transcriptions de courte durée. Sur la base de l'ARN demi-vie d'un gène de référence, ARN demi-vies peuvent être déterminées. Si les trois fractions d'ARN (l'ARN total, l'ARN nouvellement transcrites et de l'ARN pré-existant sans étiquette) sont analysées pour quatre ou plusieurs gènes, la normalisation des différents sous-ensembles d'ARN peut être effectuée par analyse de régression linéaire et les scores de contrôle de la qualité peut être déterminée comme décrit 7 , 21.
Pour l'analyse Q-RT-PCR, nous recommandons d'utiliser 2,5 ul d'ARN marquées dans 20 ul mélange de synthèse d'ADNc. A titre de comparaison optimale des résultats q-RT-PCR geler l'ADNc dans des aliquotes de 5 ul avant la première utilisation. Décongeler les tubes juste avant utilisation, ajouter 45 ul de H 2 O et sous réserve de 5 pi des dilutions à des analyses q-RT-PCR. Cela améliore considérablement la comparabilité entre les différentes courses PCR.
Les échantillons d'ARN nouvellement transcrites doivent être vérifiées pour des signes de dégradation de l'ARN en utilisant le Bioanalyser Agilent avant de les soumettre àanalyse à haut débit (puces à ADN ou ARN-Seq). Il convient toutefois de noter que les bandes supplémentaires sont parfois observées par le Bioanalyser Agilent. La signification biologique de cette demeure incertaine. Comme l'ARN nouvellement transcrit contient de l'ARN ribosomal beaucoup moins, ces échantillons ne parviennent pas à l'occasion des contrôles de qualité de l'Bioanalyser Agilent. Si cela n'est pas dû à des échantillons de dégradation de l'ARN visibles de qualité acceptable sont généralement très bien faire l'objet d'analyse à haut débit.
Compatibilité de l'ARN nouvellement transcrit avec des analyses en aval
ARN transcrits nouvellement contient beaucoup plus d'ARNm de l'ARN total. Ceci est principalement dû à l'augmentation des quantités de séquences d'introns de l'ARN nouvellement transcrites qui augmentent lorsque la durée de 4SU-tagging est raccourcie. Par conséquent, nous ne menons pas régulièrement l'épuisement des ARNr dans des échantillons d'ARN nouvellement transcrit comme ceci nécessite de grandes quantités de matière première tout en PROVIDING plutôt peu de gain (~ double) en non-ARNr lit. Enfin, il reste à noter que le plus grand pourcentage de non épissés, les transcriptions de haut poids moléculaire présents dans l'ARN nouvellement transcrites peut exiger fragmentation supplémentaire lors de la préparation de banques d'ADNc pour le séquençage de prochaine génération. Les résultats de l'étape de fragmentation de taille devraient donc être de qualité soigneusement contrôlées.
La normalisation des données pour l'ARN mesures demi-vie
L'approche standard pour normaliser les données expérimentales pour les mesures de demi-vie d'ARN est de normaliser toutes les données à l'ARN demi-vie d'un gène maintien de la maison bien caractérisé ou l'ARN demi-vie médiane dans un type cellulaire donné déterminé dans les expériences précédentes. Dans les cellules de mammifères, ce dernier se trouve dans la plage de 5 à 10 h 6,7. Bien que cette approche fonctionne aussi très bien pour les mesures 4SU basés, d'autres moyens pour la normalisation sont nécessaires si l'ARN demi-vie médiane n'est pas connu ou s'il MAy encore être affecté par des changements dans le système cellulaire à l'étude, par exemple par le knock-out d'une voie de désintégration ARN. 4SU-tagging offre une façon unique d'estimation de l'ARN demi-vie médiane basée sur l'analyse des trois fractions d'ARN, soit d'ARN cellulaire total, l'ARN nouvellement transcrit, et l'ARN pré-existantes non étiquetés. Comme l'ARN cellulaire total est séparé en deux dernières fractions d'ARN d'un modèle de régression linéaire simple peut être utilisé pour normaliser les trois fractions d'ARN à l'autre et de déterminer l'ARN demi-vie médiane 7,16. Un logiciel est disponible en ligne pour effectuer ces analyses 22.
Capture inefficace des transcriptions à faible teneur en uridine peut affecter les mesures de demi-vie d'ARN résultant artificiellement faibles ratios nouvellement transcrites / ARN total et les demi-vies prolongées ARN. L'ampleur de ce problème peut être évaluée en traçant des demi-vies d'ARN ou de journaux (nouvellement transcrit / ratios ARN total) contre le uridicontenu au NE de toutes les transcriptions 7,15. Cela fournit également un bon contrôle de la qualité afin d'évaluer les différences de taux 4SU-intégration entre les différents échantillons ou des conditions. Dans les cas où l'on observe une corrélation substantielle au contenu uridine cela peut être corrigé par la bioinformatique moyens 15. Toutefois, il convient de noter que la contribution des transcrits matures dans l'ARN nouvellement transcrit ne peut pas être facilement différencié du beaucoup plus grand et donc beaucoup plus riches en précurseurs uridine. Sauf si la cinétique de transformation de la transcription donnée sont connus (dont ils ne sont généralement pas) simplement pour corriger une faible teneur en uridine (capture inefficace) peut gravement fausser ARN demi-vies. En tant que tel, nous avons récemment trouvé le traitement de la plupart des snoRNAs humains d'être très inefficace 9. Si nous avions corrigé les ratios d'ARN nouvellement transcrites / total de la teneur en uridine bas de la relativement faible (70 à 300 nt) snoRNAs, cela aurait résulté en un très court snoRNA demi-lives (<5 min) avec de nombreux ratios ARN nouvellement transcrites / total de plus de 100%. Par conséquent, nous ne recommandons pas généralement la correction de faible teneur en uridine lors de la mesure d'ARN demi-vies.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Les auteurs déclarent qu'ils n'ont aucun intérêt financier concurrents.
Acknowledgments
Nous tenons à remercier Amie Regan pour la lecture attentive du manuscrit. Ce travail a été soutenu par NGFN plus de subvention # 01GS0801, MRC bourse subvention G1002523 et NHSBT subvention WP11-05 à LD et DFG subvention FR2938/1-1 au CCF
Materials
| Name | Company | Catalog Number | Comments |
| 4-thiouridine | Carbosynth | T4509 | Prepare 50 mM stock in sterile H2O, store at -20 °C in aliquots of 50-500 μl, discard unused reagent, do not refreeze. |
| Trizol | Invitrogen | 15596026 (100 ml), 15596018 (200 ml) | WARNING - CORROSIVE and HAZARDOUS TO HEALTH! Ensure immediate access to Phenol antidote (PEG-Methanol); Store at 4 °C. |
| Chloroform | Sigma | 372978 | WARNING - HAZARDOUS TO HEALTH |
| Isopropanol | Sigma | 650447 | |
| Sodium citrate, nuclease-free | Sigma | C8532 | Prepare 1.6 M stock solution using nuclease-free water. |
| 5M nuclease-free NaCl | Sigma | 71386 | Stock solution |
| Nuclease-free H2O | Sigma | W4502 | Make 1 ml aliquots in nuclease-free tubes. |
| RNA precipitation buffer | 1.2 M NaCl, 0.8 M sodium citrate in nuclease free water. Prepare in advance under strictly nuclease-free conditions. Store at room temperature in 50 ml falcon tubes. | ||
| Ethanol | Sigma | 459844 | Use with nuclease-free water to prepare 80% ethanol, store at -20 °C. |
| 1 M nuclease-free Tris Cl, pH 7.5 | Lonza | 51237 | Stock solution |
| 500 mM nuclease-free EDTA, pH 8.0 | Invitrogen | 15575-020 | Stock solution |
| 10x Biotinylation Buffer (BB) | 100 mM Tris pH 7.4, 10 mM EDTA in nuclease-free water, make aliquots of 1 ml. | ||
| Dimethylformamide (DMF) | Sigma | D4551 | |
| EZ-Link biotin-HPDP | Pierce | 21341 | Prepare 1 mg/ml stock solution by dissolving 50 mg biotin-HPDP in 50 ml DMF. Gentle warming enhances solubilisation. Store at 4 °C in aliquots of 1 ml. |
| Phase Lock Gel Heavy tubes 2.0 ml | Eppendorf | 0032 005.152 | Optional for the chloroform extraction step. |
| Zeta membrane | BIORAD | 162-0153 | |
| 10x Dot blot binding buffer | 100 mM NaOH, 10 mM EDTA | ||
| Biotin-oligo | 5'-biotin, 25 nucleotides, any sequence | ||
| Sodium dodecyl sulphate | Fisher | BPE9738 | For 100 ml 20% stock solution, add 20 g SDS to 80 ml PBS pH 7-8 and adjust volume to 100 ml. Keep all high-percentage SDS solutions above 20 °C. Warm the solutions slightly should SDS precipitate. |
| EZ-Link Iodoacetyl-LC-Biotin | Pierce | 21333 | Prepare 1 mg/ml stock solution by dissolving 50 mg iodoacetyl-biotin in 50 ml DMF. Gentle warming enhances solubilisation. Store at 4 °C in aliquots of 1 ml. Generates irreversible, thiol-specific biotinylation. |
| Phosphate buffer saline | Gibco | 10010-015 | |
| Dot blot blocking buffer | Mix 20 ml 20% SDS with 20 ml 1 x PBS pH 7-8 and add EDTA to the final concentration of 1 mM. | ||
| Streptavidin-horseradish peroxidase | Vector Laboratories | SA5004 | Store at -20 °C. Mix 10 ml 20% SDS with 10 ml 1 x PBS. Add 20 μl Streptavidin-HRP before use. |
| ECL reagent | GE Healthcare | RNP2109 | Use following the manufacturer's instructions. |
| Super RX, X-RA Film, 18x24 cm | Fujifilm | 47410 19236 | |
| μMacs Streptavidin Kit | Miltenyi | 130-074-101 | Store the beads at 4 °C. |
| Tween 20 | Sigma | P1379 | |
| Washing buffer | 100 mM Tris pH 7.4, 10 mM EDTA, 1 M NaCl, 0.1% Tween 20 in nuclease-free H2O. | ||
| Dithiothreitol (DTT) | Sigma | 43817 | Prepare as 100 mM DTT in nuclease-free H2O, always prepare fresh before use. |
| RNeasy MinElute Kit | Qiagen | 74204 | Store columns at 4 °C, remaining components of the kit at room temperature. |
| 1.5 ml screw-top polypropylene tubes | Sarstedt | 72.692.005 | Compatible with Dimethylformamide |
| 2.0 ml screw-top polypropylene tubes | Sarstedt | 72.694.005 | Compatible with Dimethylformamide |
| 15 ml tubes | BD Falcon | 352096 | Compatible with Dimethylformamide |
| 50 ml tubes | BD Falcon | 352070 | Compatible with Dimethylformamide |
| All solutions/reagents should be stored at room temperature unless otherwise specified. | |||
| Equipment | |||
| UV/VIS spectrophotometer | Thermo Scientific | NanoDrop 1000 | Or equivalent. Use low volume (1-2 μl) for measurements of low RNA concentrations to avoid excessive sample loss. |
| Polypropylene 15 ml centrifuge tubes | VWR International | 525-0153 | In contrast to standard 15 ml tubes, these tolerate up to 15,000 × g |
| High-speed centrifuge | Beckman Coulter | Avanti J-25 | Or equivalent equipment capable of reaching 13,000×g |
| High-speed rotor | Beckman Coulter | JLA-16250 | Or equivalent equipment capable of reaching 13,000×g |
| Adaptors for 15 ml tubes | Laborgeräte Beranek | 356964 | Or equivalent equipment capable of reaching 13,000×g |
| Refrigerated table-top centrifuge | Eppendorf | 5430 R | Or equivalent. |
| Thermomixer | Eppendorf | Thermomixer compact | Or equivalent. |
| Magnetic stand | Miltenyi Biotec | 130-042-109 | One stand holds 8 μMacs columns. |
| Waterbath | Grant | SUB Aqua 5 | Or equivalent. |
| Ultra-fine scale | A&D | GR-202 | Or equivalent. |
| E-Gel iBase Power System | Invitrogen | G6400UK | For RNA gels; or equivalent. |
| E-Gel EX 1% agarose precast gels | Invitrogen | G4020-01 | For RNA gels; or equivalent. |
References
- Brown, P. O., Botstein, D. Exploring the new world of the genome with DNA microarrays. Nature Genetics. 21, 33-37 (1999).
- Mortazavi, A., Williams, B. A., Mccue, K., Schaeffer, L., Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nature Methods. 5, 621-628 (2008).
- Nagalakshmi, U., Wang, Z., et al. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science. 320, 1344-1349 (2008).
- Wilhelm, B. T., Marguerat, S., et al. Dynamic repertoire of a eukaryotic transcriptome surveyed at single-nucleotide resolution. Nature. 453, 1239-U1239 (2008).
- Dölken, L., Ruzsics, Z., et al. High-resolution gene expression profiling for simultaneous kinetic parameter analysis of RNA synthesis and decay. RNA. 14, 1959-1972 (2008).
- Yang, E., van Nimwegen, E., et al. Decay rates of human mRNAs: correlation with functional characteristics and sequence attributes. Genome Res. 13, 1863-1872 (2003).
- Friedel, C. C., Dölken, L., Ruzsics, Z., Koszinowski, U. H., Zimmer, R. Conserved principles of mammalian transcriptional regulation revealed by RNA half-life. Nucleic Acids Res. 37, e115 (2009).
- Nilsen, T. W., Graveley, B. R. Expansion of the eukaryotic proteome by alternative splicing. Nature. 463, 457-463 (2010).
- Windhager, L., Bonfert, T., et al. Ultra short and progressive 4sU-tagging reveals key characteristics of RNA processing at nucleotide resolution. Genome Res. , (2012).
- Melvin, W. T., Milne, H. B., Slater, A. A., Allen, H. J., Keir, H. M. Incorporation of 6-thioguanosine and 4-thiouridine into RNA. Application to isolation of newly synthesised RNA by affinity chromatography. Eur. J Biochem. 92, 373-379 (1978).
- Cleary, M. D., Meiering, C. D., Jan, E., Guymon, R., Boothroyd, J. C. Biosynthetic labeling of RNA with uracil phosphoribosyltransferase allows cell-specific microarray analysis of mRNA synthesis and decay. Nature Biotechnology. 23, 232-237 (2005).
- Kenzelmann, M., Maertens, S., et al. Microarray analysis of newly synthesized RNA in cells and animals. Proc. Natl. Acad. Sci. U.S.A. 104, 6164-6169 (2007).
- Schwanhäusser, B., Busse, D., et al. Global quantification of mammalian gene expression control. Nature. 473, 337-342 (2011).
- Rabani, M., Levin, J. Z., et al. Metabolic labeling of RNA uncovers principles of RNA production and degradation dynamics in mammalian cells. Nat. Biotechnol. , (2011).
- Miller, M. R., Robinson, K. J., Cleary, M. D., Doe, C. Q. TU-tagging: cell type-specific RNA isolation from intact complex tissues. Nat. Methods. 6, 439-441 (2009).
- Miller, C., Schwalb, B., et al. Dynamic transcriptome analysis measures rates of mRNA synthesis and decay in yeast. Mol. Syst. Biol. 7, 458 (2011).
- Weintz, G., Olsen, J. V., et al. The phosphoproteome of toll-like receptor-activated macrophages. Mol. Syst. Biol. 6, 371 (2010).
- Lipsett, M. N. The isolation of 4-thiouridylic acid from the soluble ribonucleic acid of Escherichia coli. Journal of Biological Chemistry. 240, 3975-3978 (1965).
- Marcinowski, L., Liedschreiber, M., et al. Real-time Transcriptional Profiling of Cellular and Viral Gene Expression during Lytic Cytomegalovirus Infection. PLoS Pathog. 8, e1002908 (2012).
- Chomczynski, P., Mackey, K. Short technical reports. Modification of the TRI reagent procedure for isolation of RNA from polysaccharide- and proteoglycan-rich sources. Biotechniques. 19, 942-945 (1995).
- Friedel, C. C., Dölken, L. Metabolic tagging and purification of nascent RNA: implications for transcriptomics. Mol. Biosyst. 5, 1271-1278 (2009).
- Friedel, C. C., Kaufmann, S., Dölken, L., Zimmer, R. HALO - A Java framework for precise transcript half-life determination. Bioinformatics. , (2010).
