

Summary
전체 세포 RNA 단기 RNA 합성과 붕괴의 변화뿐만 아니라 RNA 처리 속도론을 연구 가난한 템플릿을 제공합니다. 여기에, 우리는 4 thiouridine에 새로 전사 RNA의 대사 라벨을 설명하는 이러한 한계를 극복 할 수 있도록 새로 전사 RNA의 싸이 특정 biotinylation 및 정제에 의해 따랐다.
Abstract
전체 - 사체 마이크로 어레이 차세대 시퀀싱의 개발은 세포의 유전자 발현의 복잡성에 대한 우리의 이해를 혁명하고 있습니다. 참여 분자 메커니즘에 대한 이해와 함께, 기본 역학의 정확한 측정은 점점 중요 해지고있다. 여기에 이러한 강력한 방법론 템플릿 샘플들이 연구 즉, 전체 세포 RNA의 고유 특성으로 인해 주요 한계에 직면하고 있습니다. 대부분의 경우 전체 세포 RNA의 변화는 충분한 해상도를 기본 분자 사건과 반응 속도를 나타내는 하나 너무 느리게 혹은 너무 빨리 발생합니다. 또한, RNA 합성, 처리 및 부패의 변화의 기여도는 쉽게 구별되지 않습니다.
우리는 최근 이러한 한계를 극복하기 위해 고해상도 유전자 발현 프로파일을 개발했습니다. 우리의 접근 방식은 4 thiouri에 새로 전사 RNA의 대사 라벨에 근거식사는 (따라서도 4SU를 붙이기로 함) 티올 특정 biotinylation와 스트렙 타비 딘 - 코팅 자석 구슬을 사용하여 새로 전사 RNA의 엄격한 정화가 따랐다. 그것은 척추 동물, 초파리, 효모 등 미생물의 넓은 범위에 적용 할 수 있습니다. 우리는 성공적으로, 전사 인자의 활동을 실시간으로 속도를 연구 RNA 반감기의 정확한 측정을 제공하며, RNA 처리 속도론에 새로운 통찰력을 얻을 수 4SU - 태그를 적용했다. 마지막으로, 전산 모델링은 기본 분자 메커니즘의 통합, 포괄적 인 분석을 생성하기 위해 사용될 수있다.
Introduction
유전자 발현 프로파일 링은 세포 프로세스와 관련된 복잡한 상호 작용 네트워크를 연구하는 데 중요한 도구입니다. mRNA의 풍요 로움에 대한 연구는 일반적으로 기본 분자 메커니즘에 대한 기본적인 통찰력을 얻기 위해 선택의 방법이었다. 전체 - 사체 마이크로 어레이 1, RNA (RNA-SEQ) 2-4 최근에는 차세대 시퀀싱의 개발이 방법을 연료. 이 기술은 세포의 유전자 발현의 복잡성에 대한 우리의 이해를 혁명을 가지고 있지만, 그들은 그들의 템플릿 샘플의 고유 특성 즉, 전체 세포 RNA로 인해 큰 한계에 직면하고 있습니다. 총 RNA 수준에서 첫째, 단기 변화는 전사 속도의 변화와 일치하지만, RNA 해당 증명서의 반감기에 근본적으로 의존하지 않는다. 전사 인자에 대한 짧은 증명서, 예를 들어, 인코딩의 다섯 배 유도, 총 RNA에서 쉽게 감지 할 수있을 것입니다 반면시간 내에, 대사 효소 수명이 긴 증명서, 예를 들어, 인코딩 같은 유도가 거의 보이지 않는 상태로 유지됩니다. 또,이 셧다운 (> 1,000 배 아래로 조절) RNA 오시간의 반감기는 평균 유전자의 전사 속도 완료는 단순히 단지 두 가지에 의해 감소하기 위해 총 RNA 수준에 대한 다섯 시간 소요됩니다 . 따라서 총 RNA의 분석은 규제 기능 5와 전사 인자와 유전자 인코딩 중 많은 짧은 성적의 최대 규제의 검출을 선호. 또한, 규제의 진정한 운동 폭포가 가려 및 기본 신호 이벤트는 보조 구별 할 수 없습니다. 모두 차례로 하류 생물 정보학 분석에 상당한 편차가 발생할 수 있습니다. 둘째, 총 RNA 수준의 변화는 RNA 합성 또는 붕괴의 변화에 기인 할 수 없습니다. 후자의 측정 transcripti을 차단하는 세포 침입 방법, 예를 들면 필요actinomycin D 6 시간이 지남에 지속적인 RNA 붕괴의 확장 된 모니터링을 사용합니다. 5 포유류 세포에서 평균 mRNA의 반감기 - 10 시간 5,7, 대부분의 유전자의 mRNA 수준은 두 가지 전사 체포 몇 시간에 따라보다 감소 할 것이다. 이 오히려 작은 차이는 기본 수학 방정식의 지수 특성으로 인해 세포의 유전자의 대부분 mRNA의 반감기 심하게 부정확 한 측정의 결과. 마지막으로, 동시에 전체 세포 RNA의 RNA-SEQ는 우리의 유전자의 절반 정도가 대안 접합 이벤트 8, 기본 역학뿐만 아니라 제대로 이해 남아 RNA 가공의 조직 및 상황 별 규제 지침 동적 메커니즘이 적용됩니다 것으로 나타났습니다. 또한, RNA의 기여, 특히 비 코딩 RNAs를 위해 결정되어야 유지, 차등 유전자 발현을 처리. 모두 이러한 제한에 대한 주요 장애물을 나타냅니다기본 분자 메커니즘의 생물 정보학 운동 모델링.
우리는 최근에 이러한 문제에게 5,7,9을 극복하기 위해,라고 고해상도 유전자 발현 프로파일 링 방법을 개발했다. 그것은 4 thiouridine (4SU 태깅), 자연적으로 발생 딘 유도체를 사용하여 새로 전사 RNA의 대사 라벨에 따라 세포 성장과 유전자 발현의 최소한의 간섭 (그림 1 참조) 5를 새로 전사 전사에 직접 액세스를 제공합니다 10-12. 급속한 흡수, 4SU - 인산을 인산화, 그리고 새로 전사 RNA에 결합의 4SU 결과 진핵 세포의 노출. 전체 세포 RNA의 분리 후, 4SU - 라벨 RNA 분획은 티올, 특히 비오틴하고 새로 전사 RNA 사이의 이황화 결합을 생성로 만들었다이다. '전체 세포 RNA'는 다음 정량 ( '새로 전사')과 레이블 ( '사전 existin 표시로 구분 될 수있다스트렙 타비 딘 - 코팅 자석 구슬을 사용하여 고순도 G ') RNA. 마지막으로, 라벨 RNA는 단순히 이황화 결합을 쪼개서 환원제 (예를 들면 디티 올 트레이 톨)을 추가하고 구슬에서 새로 전사 RNA를 풀어 구슬에서 복구됩니다.
새로 전사 RNA는 4SU 노출의 기간 동안 모든 유전자의 전사 활성을 보여줍니다. 분의 척도에 4SU 태깅 따라서 진핵 생물의 유전자 발현의 스냅 사진과 다운 스트림 생물 정보학 분석 (예 : 프로모터 분석)를위한 이상적인 템플릿을 제공합니다. 정상 상태를 가정 할 수 경우의 비율 새로 전사 / 전체 새로 베껴 / 레이블이와 레이블이없는 / 총 RNA 정확한 RNA 반감기 7,13에 대한 비 침습 액세스를 제공합니다. 또, 4SU - 태깅으로 작은 5로 분 (5 분 4SU-RNA) 후 정제가 새로 전사 RNA를주의하는 것이 중요하다 15, 60 분 4SU-RNA 미만이다.RNA-SEQ와 함께 하나의 실험 설정에서 4SU를 붙이고는 매우 짧고 점차적으로 더 이상 모두를 수행 할 때, RNA 프로세싱의 반응 속도는 염기 해상도 9시에 공개된다. 마지막으로, 전산 모델링과 함께 새로 베껴 총 RNA 시간 코스 분석은 RNA 합성과 부패 14의 통합 분석을 할 수 있습니다.
결론적으로,이 방법은 진핵 세포의 RNA 합성, 처리 및 분해의 역 동성을 직접 분석 할 수 있습니다. 그것은 포유 동물, 곤충 (초파리), 양서류 (Xenopus의), 효모 5,15,16 포함한 모든 주요 모델 생물에 적용됩니다. 그것은 마이크로 어레이 분석 5,17, RNA-SEQ 9,13,14과 직접 호환 및 생체 12,15에 적용됩니다. 여기에, 우리는 세부 방법론, 라벨 분리, 배양 포유 동물 세포에서 새로 전사 RNA를 정화. 또한, 적합한 한쌍에게있는알 문제와 함정을 설명합니다.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. 4 thiouridine과 신진 대사 라벨링
실험 설정 / 일정 세부 계획을 만들어 세포 배양에 4SU를 추가 할 때 샘플을 채취 할 때 예를 들어. 각 조건 사이에 적어도 5 분 계획. 한 번에 하나의 조건의 세포를 처리합니다. 최대 처리 할 수 있습니다. 3 - 주어진 시간에 5 접시. 온도와 CO 2 농도의 변화를 최소화하기 위해 가능한 한 빨리 전지를 처리합니다. 이 세포 단백질 4SU - 라벨 RNA의 가교 될 수 있으므로 4SU가 추가 된 후 밝은 빛에 세포를 노출시키지 마십시오.
라벨의 시작
- 해동 4 thiouridine (4SU) 단지 멸균 팔콘 튜브에 각 조건에 대한 4SU의 사용과 피펫 필요한 양 전.
- 요리 떨어져 세포 배양 매체의 필요한 금액 (10cm 접시 당 5 ML)를 타고 4SU 함유 팔콘 튜브에 첨가하고 잘 섞는다. 제거하고 요리의 나머지 매체를 폐기하십시오. <리> 요리에 4SU 함유 배지를 다시 적용합니다.
라벨의 끝
- 세포에서 세포 배양액을 제거합니다. 각 플레이트에 TRIZOL의 5 ML을 추가합니다. 여러 시간 지점 또는 조건을 포함 복잡한 실험을 위해,이 단계가 가장 두 사람이 중간 TRIZOL을 추가하고 해물을 수확 기타를 제거 하나에 의해 이루어집니다.
- 완전한 세포 용해 실온에서 5 분 알을 품다.
- 추가 TRIZOL으로 조심스럽게 접시를 헹굴 10 ㎖ 피펫을 사용합니다. 이 완전한 세포 용해 및 샘플 복구 제품. 피부 나 눈에 접촉 가져올 때 TRIZOL 매우 위험한 상태로 취급주의! 손 페놀 화상 (예를 들면 폴리에틸렌 글리콜 300 또는 산업 메틸화 영혼 (70:30) 400)에 대한 해독제가있다. 폴리 프로필렌 튜브에 샘플을 전송합니다. 표준 팔콘 튜브)이 높은 G의 힘에 저항하지 않도록주의하시기 바랍니다. 샘플은 내가 총 RNA 때까지 적어도 한 달 동안 -20 ° C에 저장할 수 있습니다들 준비.
2. 수정 TRIZOL 프로토콜을 사용하여 RNA 준비
- 1 ML 클로로포름 (ML TRIZOL 당 0.2 ML) 추가하고 15 초 동안 격렬하게 흔들어. 3 분 - 2 실온에서 알을 품다.
- 4시 15 분 13,000 × g에서 원심 분리기 ° C.
- 새로운 15 ML의 폴리 프로필렌 튜브에 수성 상위 단계 (RNA 포함) 전송합니다.
- 추가 ½ RNA 침전 버퍼와 이소프로판올 모두의 반응 볼륨 (3 ML 상층에 예를 들어, 1.5 ML RNA 침전 버퍼와 1.5 ML 이소프로판올 추가).
- 잘 섞는다. 10 분 실온에서 알을 품다.
- 4에서 10 분간 13,000 × g에서 원심 분리기 ° C. 상층 액을 버린다.
- 간단히 스핀 다운 (5,000 × g에서 30 초) 200 μL 피펫 잔류 이소프로판올을 제거합니다.
- 75 % 에탄올의 동일한 볼륨을 추가하고 펠렛 분리 될 때까지 튜브를 흔들어. residua이 같은 여러 개의 작은 조각으로 파손되지 않도록하는 것은 만들 수 있습니다 제거L의 에탄올 어렵다.
- 4에서 10 분간 13,000 × g에서 원심 분리기 ° C. 상층 액을 버린다.
- 간단히 RNA를 회전하고 200 μL 피펫으로 남아있는 에탄올을 제거합니다. 20 μL 피펫으로 남아있는 에탄올 단계를 제거를 반복합니다. 이 두 단계 후, 펠렛의 더 건조가 수행되지 않아야합니다.
- 100 μg 예상 RNA 수율 당 H 2 O의 100 μl를 추가하고 5를 위아래로 pipetting에 의해 잘 섞는다 - 6 번 RNA를 용해에 도움.
- 용해와 65 가열하여 RNA를 변성 ° C 10 분 (뿌리)에 대한 즉시 얼음에 배치합니다.을
- 제조업체의 지침에 따라, Nanodrop 분광 광도계를 사용하여 260 nm에서 농도를 RNA 측정합니다. 이 RNA는 적어도 한 달 동안 -80 ° C에 저장할 수 있습니다.
3. 새로 전사 RNA의 싸이 특정 Biotinylation
- 60로 시작 - 전체 세포 RNA 80 μg합니다.
- 반응에 레이블을 구성합니다. 다음의 피펫순서 (당 μg의 RNA)
- 1 μL 10X Biotinylation 버퍼
- 7 μL RNA (핵산 무료 H 2 O에 희석 1 μg의 RNA를 포함)
- 2 μL 비오틴 HPDP (1 MG / ML DMF)
항상 비오틴 HPDP 마지막으로 추가하고 pipetting하여 즉시 섞는다. 비오틴은 침전물 경우에, DMF 콘텐츠는 40 %의 최종 농도를 증가시킬 수있다.
- 회전 1.5 시간 동안 실온에서 알을 품다.
- 클로로포름의 동일한 볼륨을 추가합니다. 적극적으로 섞는다. 단계 분리하기 시작하고 거품이 사라 시작할 때까지 삼분 - 2 품어.
- 4 ℃에서 5 분간 20,000 × g에서 원심 분리 조심스럽게 새로운 튜브로 위 수성 단계를 전송합니다.
- 한 단계 3.4 및 3.5를 반복합니다. 당신은 RNA의 손실을 줄이기 위해 2 ㎖ 위상 잠금 젤 무거운 관이 단계를 수행 할 수 있습니다.
- RNA 강수량 : 추가 1 / 10 5 M NaCl을의 부피와 같은 부피의물 단계로 이소프로판올.
- 4 ℃에서 20 분간 20,000 × g에서 원심 분리 상층 액을 버린다.
- 상층 액을 버리고, 4시 10 분 ° C에 대해 20,000 75 % 에탄올, 원심 분리기의 동일한 볼륨 × g을 추가합니다.
- 간단히 스핀 200 μL 피펫 잔류 에탄올을 제거합니다.
- 간단히 스핀 20 μL 피펫 잔류 에탄올을 제거합니다.
- RNA 건조 할 수 없습니다. 50에 다시 일시 중지합니다 - 100 μL H 2 O (1 μg 입력 RNA 당 ~ 1 μL). 6 번 - 5 아래로 피펫에 의해 잘 섞는다.
- RNA 저하를 제외 electrophoretical 분석 RNA의 품질을 확인합니다.
4. 4SU - 정관 점 오점 분석 (선택 사항)
4SU 법인은 쉽게 바이오틴 RNA의 점 blot 분석에 의해 결정하실 수 있습니다. 이 문제 해결과 바이오틴 DNA 올리고 컨트롤을 기준으로 4SU 편입 비율의 추정을 허용하는 선택적 단계입니다. 이 분석 우리 연구에 대한단계 3.2 4SU - 라벨 RNA의 biotinylation 대신에 바이오틴 HPDP의 iodoacetyl-비오틴 사용 ecommend. 4SU-RNA의 돌이킬 수없는 biotinylation이 결과. 따라서 열 기반의 방법 (예 RNeasy) 바이오틴 RNA의 매우 적은 양 (예를 들어, 5 μg)의 복구에 사용할 수 있습니다. RNA는이 분석에 적합합니다 비오틴 - HPDP를 사용하여로 만들었다하지만, 그 결과 신호는 (그림 3) 약한 신호 대 잡음 비율 덜 유리한 것입니다.
- 섹션 1과 2에 설명 된대로 전체 세포 RNA의 4SU-라벨 및 격리를위한 프로토콜을 따르십시오.
- iodoacetyl - 비오틴과 비오틴 - HPDP 교체 3 장에서 설명한 완전히 과도한 iodoacetyl-비오틴 잔류 물을 제거하기 위해 두 클로로포름 추출을 수행 할 때 Biotinylate RNA 4SU - 라벨.
- 설명이나 사건 RNA의 소량 (에 (예를 들어, RNeasy) 컬럼 기반 접근 방법을 사용하는 것과 이소 프로필 알코올 / 에탄올 침전 바이오틴 RNA를 복구 <10 μg) 사용됩니다.
- 10 분 동안 흔들 nuclease가없는 물 제타 막 품어.
- 핵산이없는 물에서 막 가지고 두 개의 깨끗한 종이 타월 단단히 눌러 중간에 막 배치하여 과도한 액체를 제거합니다. 공기 건조 5 분 막하는 것이 더 좋은 점 발생합니다.
- 각 샘플은 차가운 얼음 점 오점 바인딩 버퍼 (10 MM NaOH를 1 mM의 EDTA)를 사용하여 200 μL / NG RNA의 20 μl를 준비합니다. pipetting하여 제타 막이 희석의 5 μL (RNA 즉 1 μg)뿐만 아니라 세 이후에 10 배 희석 (각각 즉, 100, 10, 1 NG RNA를) 적용 할 수 있습니다. 피펫 팁의 빈 선반을 피펫은 고르게 분포 간격을 제공하기 위해 사용될 수있다. 또한, 제조업체의 지침에 따라 점 오점 장치를 사용합니다.
- 긍정적 연속으로 20 일부터 μL / NG 20 PG / μL (즉, 100-0.1 NG 올리고)에 이르기까지 농도에서 비오틴 - 라벨 DNA 올리고의 5 μl를 적용pipetting하여 막 ROL. 대조군으로 바이오틴, 4SU - 순진 샘플을 사용합니다.
- 공기 건조 5 분 막.
- 락 40 ML 차단 버퍼에 30 분 동안 막 품어.
- 15 분 (5 ML PBS가 + 5 ML 20 % SDS + 10 ㎕의 스트렙 타비 딘 - 고추 냉이 과산화 효소)에 대한 1:1,000 스트렙 타비 딘 - 고추 냉이 퍼 옥시 데이즈 10 mL로 막을 배양
- 5 분 40 ML PBS + 10 % SDS (20 ML PBS가 + 20ml를 20 % SDS)에 두 번 막 씻으십시오.
- 40 ML PBS로 두 번 막 씻어 5 분 + 1 % SDS (38 ML PBS가 + 2 ML 20 % SDS).
- 40 ML PBS로 두 번 막 씻어 5 분 + 0.1 % SDS (40 ML PBS가 + 200 ㎕의 20 % SDS).
- 두 깨끗한 종이 타월 사이에 막 배치하고 단단히 그들을 눌러 과도한 액체를 제거합니다.
- 제조업체의 지침에 따라 ECL을 사용하는 멤브레인 바인딩 HRP을 시각화.
- 플라스틱 호일 / 가방에 막 배치, 공기 방울을 제거하고 어둠 속에서 2 분 동안 품어.
- 막에 노출1 막 - 5 분.
5. 스트렙 타비 딘 - 코팅 자석 구슬을 사용하여 레이블과 레이블없는 RNA의 분리
- 열 ° C의 물을 욕조에 65 (샘플 당 3 mL)에 버퍼를 세척.
- nuclease가없는 H 2 O에 신선한 100 MM의 디티 올 트레이 톨 (DTT)를 준비 초 미세 규모에 배치 깨끗한 50 ML 팔콘 튜브에 DTT 분말의 30 MG - 15 경사 분리하여이를 수행. nuclease가없는 H 2 O의 필요한 양을 달아 추가
- 열 ° C 10 변성하는 분, 얼음에 즉시 장소를 65 RNA 샘플로 만들었다.
- 장소는 마그네틱 스탠드에 열을 μMacs. 우리는 한 번에 12 개 이상의 샘플 (- 8 샘플이 최적이다 6) 처리를하지 않는 것이 좋습니다.
- 1 ML 실온 버퍼를 세척으로 미리 평형 소개 Miltenyi 열. 이 약 15 분 소요됩니다.
- 한편, 50 스트렙 타비 딘 구슬 100 μL를 추가합니다 - 바이오틴 RNA의 100 μl를. 회전 15 분 실온에서 알을 품다. < 리> 열 중 하나가 지금까지 배출 시작되지 않은 경우이를 부드럽게 장갑을 낀 손가락으로 열 상단을 눌러 의해 촉진 될 수있다. 일단 흐름은 열이 쉽게 배출하기 시작했다.
- 열에 RNA / 비즈를 적용합니다. 흐름을 통해 당신은 레이블이없는 RNA 분획 (7 항 참조)를 복구하려는 경우가 아니면 폐기하십시오.
- 0.9 65 ML ° C 세척 버퍼 (65 ° C에서 버퍼를 피펫 팅 할 때 1 ML 피펫 팁 축소).로 3 회 세척
- 0.9 ML 실온 버퍼 세탁 3 회 씻는다.
- 피펫 700 μL 버퍼 새로운 2 ㎖ 튜브에 RLT (RNeasy MinElute 정리 키트 퀴 아젠)와 열 아래에 배치합니다.
- 열을 100 MM의 DTT의 100 μl를 추가하여 RLT 버퍼에 새로 전사 RNA를 용출.
- 100 MM의 DTT의 또 다른 100 μl를 추가하여 나중에 동일한 튜브에 두 번째 용출 라운드 3 분을 수행합니다.
6. 새로 전사 RNA 복구
ontent는 "> 제조 업체의 지침에 따라 RNeasy MinElute 정리 (Qiagen의) 프로토콜을 계속 진행합니다. 25 μL nuclease가없는 H 2 O 측정 RNA 농도에서 용출이 Nanodrop 분광 광도계를 사용하여. 제출하기 전에 RNA를 해동하고 다시 동결하지 않으려면 그것은 높은 처리량 분석에, 우리는 즉시 새로 전사 RNA 후 cDNA를 준비하는 것이 좋습니다이 정화됩니다. 제조업체의 지침에 따라 cDNA를 합성 20 μL의 cDNA 합성 믹스 새로 전사 RNA의 2.5 μl를 사용합니다. 1을 사용 QRT-PCR 컨트롤을 수행 : cDNA를 혼합 10 희석 -80에서 보관 RNA ° C..7. 레이블이없는, 언 바운드 RNA (선택 사항) 복구
경우에 언 바운드 RNA 복구 할 필요가; 흐름을 통해 (열을 RNA-스트렙 타비 딘 비즈 솔루션을 추가 한 후) 이후의 강수량에 대한 첫 번째 세척을 수집하고 결합한다. 보통 생로 언 바운드 RNA의 50 %가 침전 충분하다s는 출발 물질의> 80 %를 포함합니다.
- 이소프로판올의 동일한 볼륨을 추가 (아무런 소금 세척 버퍼가 이미 1 M의 NaCl을 포함으로 추가 할 필요가 없습니다).
- 4 ℃에서 20 분간 20,000 × g에서 원심 분리 상층 액을 버린다.
- 상층 액을 버리고, 4시 10 분 ° C에 대해 20,000 75 % 에탄올, 원심 분리기의 동일한 볼륨 × g을 추가합니다.
- 간단히 스핀 200 μL 피펫 잔류 에탄올을 제거합니다.
- 간단히 스핀 20 μL 피펫 잔류 에탄올을 제거합니다.
- RNA 건조 할 수 없습니다. 100 μL H 2 O에 resuspend을 6 번 - 5 아래로 피펫에 의해 잘 섞는다. 동요와 함께 10 분 동안 65 ° C에서 알을 품다과 얼음에 직접 전송할 수 있습니다.
- RNA 저하를 제외 electrophoretical 분석 RNA의 품질을 확인합니다.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
1. 원료 및 예상 수익률
4SU 노출 1 시간 (시간)에 따라 새로 전사 RNA는 약 1 대표 - 전체 세포 RNA의 4 %를. 그들은 더 이상 세포 성장 / 복제 계정에 RNA를 합성하지 않기 때문에이 성장 체포 세포에서 낮은 것입니다. 총 RNA의 80 μg - 1 시간 동안 붙일 경우, 우리는 60와 분석을 시작하는 것이 좋습니다. biotinylation 단계 이후 확인하기 어려운 작은 RNA 펠렛 총 RNA 결과보다 30 μg으로 시작하기 때문에 쉽게 손실 될 수 있습니다. 입력 RNA 수준은 라벨의 매우 짧은 기간 (- 10 분 예를 들면 5)를위한 많은 150으로 μg 증가 될 수 있습니다. RNA 라벨의 지속 시간은 1 시간에 5 분 ~ 60 %에서 ~ 80 % 9 새로 전사 RNA 증가의 짧은 인트론 서열의 기여에서 단축됩니다. 인트론은 상당히 긴 시퀀스를 코딩뿐만 아니라, 5'-및 3'-UTRs 비교되기 때문에, 양이 새로 베꼈단락 또는 매우 짧은 4SU를 붙이는 다음과 같은 정화 할 수있는 RNA는 선형 적으로 삭제하지 않습니다. 따라서, 우리는 비 부착 인간 B 세포 라인 9 4SU - 태그의 5 분 후 총 RNA의> 0.5 %를 얻었다. 그것은, 그러나, 라벨 4SU 약간 더 긴 기간의 높은 농도가 자기편 세포에서 유사한 4SU 설립 속도를 달성하는 데 필요한 수 있다는 것을 주목해야한다. 심지어 낮은 4SU - 정관 속도가 허용되지만 대형 딘 풍부한 성적 효율적으로 캡처 및 정화, 낮은 딘 함량이 매우 짧은 증명서 (예를 들어, miRNAs는)은 높은 4SU 농도 (> 1 ㎜)를 사용하는 경우에도 정화를 벗어날 가능성이 있습니다. 5 100 세포핵 (NT) - NIH-3T3 생쥐 섬유 아세포에서 200 μM 4SU 노출의 1 시간은 50 4SU 약 잔류 새로 전사 RNA를 표시. 길이 1000 NT -이 증명서> 500 매우 효율적인 복구를 허용해야합니다. 따라서, 우리는 작은 사본의 크기를 관찰생쥐 섬유 아세포와 인간 B 세포 7 모두에서 200 μM의 4SU를 사용하여 1 시간 동안 라벨 편견. 200 μM의 4SU의 1 시간은 생쥐 섬유 아세포, ≥ 200 μM의 4SU에 세포의 장기간 노출에 세포 성적 수준에 어떤 중요한 변화가 발생하지 않았지만 24 시간 (미발표 자료)에서 측정 성장 적자의 결과를 않습니다. 따라서 라벨의 기간과 4SU 농도 고용 모두 자궁외 또는 독성을 피하기 위해 최소화되어야한다. 새로 전사 RNA의 효율적인 복구에 필요한 4SU 농도 최소화를 결정하는 쉬운 방법은 4SU의 농도 증가 (- 1.5 μM 예 : 50)로 4SU 라벨링 다음 새로 전사 RNA를 정화하는 것입니다. 로 그림에 표시된 2A와 2B, 인간의 기본 섬유 아세포에서 1 시간 동안 표시 새로 전사 RNA의 회복은 50 4SU 200 μM로 크게 증가하지만 고원에 시작했다.
2. 점4SU 법인 (옵션)의 정량화를 몰아
어떤 경우에는 총 RNA의 4SU 법인의 양을 측정하기 위해 관심이있을 수 있습니다. 이 가장 스트렙 타비 딘 복합체를 사용하여 바이오틴 RNA에 점 blot 분석에 의해 이루어집니다. 화학적 특성으로 인해 iodoacetyl - 비오틴 새로 전사 RNA의 거의 모든 4SU 잔류 biotinylation의 결과 비오틴 HPDP보다 티올 그룹에 더 반응이다. 그것은 비오틴 HDPD 같은 iodoacetyl - 비오틴은 수용성하지 않으며, 따라서 효율적으로 바이오틴 HPDP에 대해 수행 클로로포름 추출에 의해 제거됩니다 점에 유의하는 것이 중요합니다. 따라서 동일한 반응 조건 및 농도는 비오틴 - HPDP를 사용하는 경우로 고용 할 수 있습니다. 그러나 iodoacetyl - 비오틴은 되돌릴 수 없습니다. 그것은 따라서 열 기반의 접근 방식에 새로 전사 RNA의 정제에 사용할 수 없습니다. iodoacetyl - 비오틴의 사용 4SU-결합을 정량화 할 수 있지만, 비오틴 - HPDP 기반의 측정을 모두 고려4SU - 정관 및 biotinylation 효율성. 동일한 샘플에 두 biotinylation 시약을 채용하는 RNA 함유 4SU의 biotinylation의 효율성을 측정 할 수 있습니다. 4SU - 라벨 RNA에 대한 비오틴 - HPDP의 Biotinylation 효율은 새로 전사 RNA에서만 약 1 4SU 세의 잔류가 실제로 비오틴 HPDP (그림 3)에 의해로 만들었다을 나타내는 iodoacetyl-비오틴보다 약 3 배 적은 것으로 보인다. 바이오틴 제어 DNA 올리고 샘플과 신호 강도를 비교하여, biotinylation 밀도를 측정 할 수 있습니다. 대부분의 포유 동물 세포 라인에 대한 긍정적 인 신호는 여전히 200 μM 4SU 라벨 1 시간을 다음과 바이오틴 RNA의 10 NG에 감지해야한다. 약한 배경 신호 레이블 RNA의 높은 농도 (1 μg) 보통 감지합니다.
3. 새로 전사 RNA의 정제
새로 전사 RNA의 복구는 매우 콴입니다titative. 당신은 같은 RNA의 농도를 시작하면 모든 샘플 새롭게 전사 RNA의 동일한 금액을 얻을 것으로 예상 할 수있다. OD 260 측정을 방해 할 수있는 260 나노 미터 (세척 버퍼에서 파생 된 세제의 존재) - 많은 컬럼 기반의 분석처럼, RNeasy MinElute 키트를 사용하여 새로 전사 RNA의 컬렉션 230 추가 흡수 될 수 있습니다. 각 원심 분리 단계에 신선한 2 ML 수집 관을 사용하는 경우이 정도는 덜 볼 수 있습니다. 그럼에도 불구하고, 모든 부당하게 높은 OD 측정은 (다른 샘플보다> 2 배 이상) OD 280분의 260 비율이 <1.7 경우 특히주의 고려되어야한다. 다운 스트림 분석을 위해 모든 샘플 템플릿 RNA 볼륨의 동일한 금액을 사용하는 것이 있으므로 좋습니다. 라벨 RNA의 수율 electrophoretical 분석에 의한 RNA 분해의 징후 예상 수표보다 낮은 경우. 새로 전사 RNA는 상당히 큰 금액을 포함 일반적인 rRNA의 밴드 (그림 4) 훨씬 덜 눈에 띄는되는 대형, unspliced 성적표.
4. 새로 전사 RNA의 정량
마지막으로, 새로 전사 RNA의 4SU의 결합 속도는 주로 330 nm에서 4SU의 흡수 최대 값과 OD 260분의 330 비율 5-18에 따라 분광 분석으로 정량화 할 수있다. 이소 프로필 알코올 / 에탄올 침전에 의해 -이 작은 양 (20 ㎕의 10)에 집중 표지 RNA의> 3 μg을 필요로합니다. nuclease가없는 글리코겐의 30 μg (Fermentas, # R0551)와 작은 RNA 펠렛 공침의 손실을 방지하려면 수행해야합니다. 추가 피크 새로 전사 RNA (그림 5)에 4SU의 결합 비율을 반영하는 330 nm에서 볼 수 있습니다.
/ files/ftp_upload/50195/50195fig1highres.jpg "/>
그림 1. . 전체 세포 RNA의 준비 다음 시간 - 4 thiouridine (4SU)와 신진 대사 라벨의 원리 4SU는 필수 (120 분 5)에 대한 세포에 추가됩니다. 싸이 특정 biotinylation에 따라 총 세포 RNA는 스트렙 타비 딘 - 코팅 자석 구슬을 사용 4SU - 라벨, 새로 전사 RNA와 레이블, 기존의 RNA로 구분됩니다. 새로 전사 RNA는 구슬로 새로 전사 RNA를 연결 이황화 결합을 클리브 환원제를 사용하여 구슬에서 복구됩니다. 큰 그림을 보려면 여기를 클릭하십시오 .
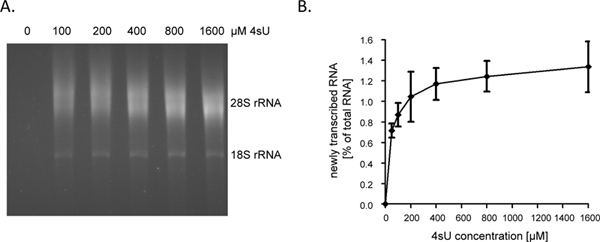
그림 2. 다음과 같은 새로 전사 RNA 복구 4SU의 농도 증가. (A) 인간의 기본 포피 섬유 아세포 (HFF)가 100으로 배양 하였다, 4SU 200, 400, 800 또는 1,600 μM. 새로 전사 RNA는 50 μg 전체 세포 RNA의 정제 electrophoretical 분석을 실시 하였다. 예상대로 회복 새로 전사 RNA의 농도에 따라 증가는 높은 농도에서 고원에 시작한 관찰되었다. (B) 새로 전사 RNA 정제 된 금액은 ImageJ에 1.45s 소프트웨어를 사용하여 정량 하였다. 800 μM 4SU은 (N = 2) 또는 100 - - 새로 전사 RNA의 양에 4 개의 독립적 인 실험의 결합 된 데이터가 50도에 이르기까지 4SU 라벨링의 다른 농도에 따라 복구. 1,600 μM 4SU (N = 2) 표시됩니다는 여기를 클릭하십시오 더 큰 그림을 볼 수 있습니다 .
upload/50195/50195fig3.jpg "고도 ="그림 3 "FO : 콘텐츠 너비 ="4.5in "FO : SRC ="/ files/ftp_upload/50195/50195fig3highres.jpg "/>
그림 3. 점 오점 분석을 사용하여 4SU - 라벨 총 RNA의 4SU 법인의 추정. 총 RNA는 한 시간 4SU 200 μM로 배양 NIH-3T3 생쥐 섬유 아세포 또는 인간의 포피 섬유 아세포 (HFF)에서 분리되었다. 더 4SU는 대조군으로 한 접시에 추가되지 않았습니다. HFF 모두에 접촉 금지 (N은 = 비 성장 세포) 성장 세포 (Y)가 포함되었다. RNA는 TRIZOL 시약 및 비오틴 HPDP 또는 iodoacetyl - 바이오틴을 연속적으로 결합을 사용하여 분리 하였다. 각 샘플의 농도는이 희석 200 NG / μL 5 μL (RNA 즉 1 μg)뿐만 아니라, 세 이후에 10 배 희석 (각각 즉, 100, 10, 1 NG RNA)로 조정 모든 하였다되었다 제타 막 조각에 발견했다. 비오틴 - 라벨 DNA 올리고 5 ㎕의 희석은 정광에 긍정적 인 컨트롤로 막에 배치μL / NG 아래 20 ~ 20 PG / μL (각각 즉, 100-0.1 NG)에 이르기까지 이온. 비오틴 밀도는 스트렙 타비 딘 - 고추 냉이 과산화 효소 복합체를 사용하여 탐색 하였다.
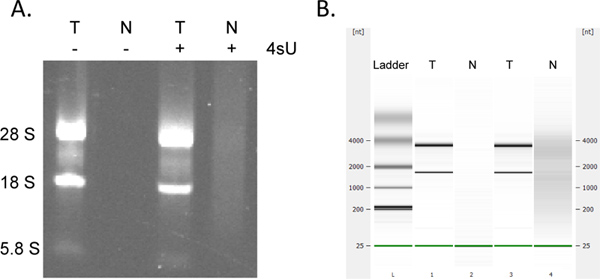
그림 4. 새로 베꼈 총 RNA의 Electrophoretical 분석. 총 RNA (T)와 새로 전사 RNA (N)은 1 시간이 아가 로스 겔 전기 영동으로 분석 하였다 위해 500 μM 4SU의 존재와 부재에 모두 배양 쥐 NIH-3T3 섬유 아세포 (A)에서 준비 와 (같은 순서로) 애질런트 Bioanalyser (B)를 사용하여. 어떤 RNA는 세포의 4SU 치료없이 회복되지 않았다. 새로 전사 RNA를 정제하여 고 분자량의 mRNA의 큰 금액과 총보다 훨씬 덜 성숙 rRNAs를 포함28S, 18S, 5.8S rRNA의와 밴드 사이의 주목할만한으로 RNA. 큰 그림을 보려면 여기를 클릭하십시오 .
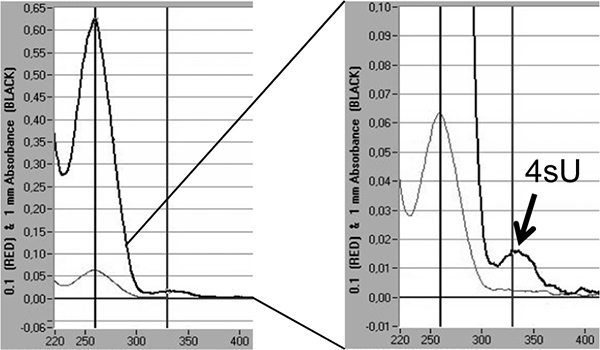
그림 5. 분광 분석에 의해 새로 전사 RNA의 4SU 설립 정량화. 쥐 NIH-3T3 섬유 아세포의 4SU 200 μM의 1 시간 후 2 × 100 μg 총 RNA의 정제 새로 전사 RNA. 새로 전사 RNA는 핵산이없는 글리코겐의 30 μg을 추가 한 후 이소 프로필 알코올 / 에탄올로 침전시켰다. Nanodrop 분광 1000 분광 광도계에 의해 얻어진 새로 전사 RNA의 분광 분석이 표시됩니다. 두꺼운 어두운 회색 선이 1mm 유체 열을 측정을 나타내는 동안 밝은 회색 선은 0.1 mm에서 측정을 나타냅니다. t를 나타내는 멸종의 피크의 오른쪽에, 배율그는 4SU - 잔류가 표시됩니다 통합. 4SU 18의 공동 효율적으로 멸종 위기에 따라 4SU의 결합 비율은 추정 할 수있다.
| 라벨 [분] 기간 | 추천 4SU 농도 [μM] |
| 120 | 100-200 |
| 60 | 200-500 |
| 15 - 30 | 500-1000 |
| <10 | 500 - 2000 |
표 1. 4SU 농도를 권장합니다.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
새로 전사 RNA 대사 라벨이 실질적으로 관심의 생물학적 문제를 해결하기 위해 더 적합한 템플릿을 제공하여 마이크로 어레이 및 RNA-SEQ와 같은 높은 처리량 기술의 힘을 강화한다. 본 프로토콜은 광범위한 최적화를 받았다. 그것은 새로 전사 RNA의> 1,000 배 농축을 허용하고 재현성이 높은 결과를 제공합니다.
새로 전사 RNA는 4SU에 세포의 노출 시간 동안 실시간으로 전사 활동을 묘사하므로 4SU 태깅 실험의 실험 설계는 매우 중요합니다. 자극에 따라 전사 요금의 실제 변화는 이미 가라 경우 총 RNA 수치의 변화는 여전히 감지 할 수 있더라도 새로 전사 RNA를 분석 할 때, 이러한 누락됩니다. 따라서, 기본 생물학의 좋은 이해는 실험 장치뿐 아니라 최적의 기간 (을)를 정의하는 것이 중요합니다4SU 노출 F 시간. 아래, 우리는 추천과 가장 중요한 단계는 일반적인 함정을 피할 수있는 방법을 제공합니다.
주식 솔루션 및 플라스틱 제품의 제조
모든 주식 솔루션은 nuclease가없는 물을 사용하여 준비해야합니다. 물이 환원제가 포함 된 경우 증류수를 정제 사내에서 사용하면 문제가 발생할 수 있습니다. 한 경우,이 레이블이있는 모든 RNA의 전체 손실 발생. 따라서, 우리는 강하게 nuclease가없는 염화나트륨, 트리스-CL, EDTA, 구연산 나트륨과 물을 미리 만들어 구입을 권장합니다. 항상 nuclease가없는 조건을 확인하십시오. 디메틸 포름 아미드 (DMF)는 일부 플라스틱 물질을 용해. 우리는 비오틴 - HPDP 재고 솔루션을 준비하는 50 ML 팔콘 튜브에 자사의 주식 유리 병에서 DMF를 전송하는 25 ML 세포 배양 플라스틱 피펫을 사용하여 실질적으로 전체 분석에서 새로 전사 RNA의 수율을 줄이기 위해 충분한 것으로 나타났습니다. 흥미롭게도,이 부정적인 biotinyl에 영향을 미치지 않았다ATION 효율 (같은 점 얼룩에 의해 시험)하지만 구슬에서 복구 할 수있는 새로 전사 RNA의 75> 90 % 손실 발생. 라벨의 지속 시간이 60에서 30 분 이하로 감소되었을 때 손실이 가장 뚜렷했다. 대부분의 경우, DMF에 의해 플라스틱 피펫에서 용출 물질이 부분적으로 스트렙 타비 딘 구슬의 코팅을 파괴했다. 따라서, DMF와 호환하지 않은 것으로 알려져 플라스틱 재료의 사용은 반드시 피해야한다. 같은 이유로, 세포 스크레이퍼는 세포 배양 접시에서 TRIZOL 샘플의 복구를 강화하는 데 사용할 수 없습니다. 그것은 DMF 또는 TRIZOL에 의해 플라스틱에서 용출 추정되는 물질이 분명히 둘 클로로포름 추출이나 이소 프로필 알코올 / 에탄올 침전에 의해 제거되지 않았다는 점에 주목하라.
세포 배양
접시에 세포 밀도가 매우 중요합니다. 세포가 (90 조금 너무 합류 것으로 나타났다 하나의 실험 -100 %), 우리는 인터페론의 100 U / mL로 30 분 동안 NIH-3T3 생쥐 섬유 아세포 치료 (IFN) α 또는 γ. 적은 합류 세포에서 IFN 치료도 15 분에 이미 5 결과 - IRF1 또는 socs3 5와 같은 유전자의 8 배 유도합니다. 세포가 약간 존재와도 합류 마이크로 어레이 분석은 가장 빠르게 IRF1 또는 socs3 같은 유전자를 유도이라도 IFN-유도 유전자의 유도를 보여주지 않았다. 따라서, 세포 밀도가 중요한 4SU - 라벨 실험과 모든 세포 배양 플레이트 요인 신중하게 라벨을 시작하기 전에 검사해야합니다.
4SU는 photoactivatable ribonucleoside과 4SU을 포함하는 RNA를 효율적으로 365 nm의 광원에 노출 된 후 단백질 가교 있습니다. 4SU 처리 세포는 피해야한다 어둡고 밝은 빛에 노출 배양해야한다. TRIZOL RNA 분리에 의한 세포 단백질의 제거 후 이러한 위험이 크게 감소됩니다.
<P 클래스 = "jove_content는"> 4SU은 세포의 DNA에 포함되지 않습니다. 그것은, 그러나, 총 RNA는 여전히 세포 DNA의 소량이 포함됩니다 주목해야한다. 4SU - 태그를 사용하여 Q-RT-PCR 분석 사이토 메갈로 바이러스 감염에서 바이러스 유전자 발현을 연구 할 때 우리는 concatemeric 바이러스 게놈에게 19 제거하는 프로토콜 DNaseI 다이제스트 단계를 더 포함하는 것이 필요 발견했다. DNA의 존재에 민감하지 않은 다운 스트림 프로토콜을 사용하는 경우 이것은 아마도 필요하지 않습니다.4SU 법인 속도와 최적의 4SU 농도
4SU은 쉽게 내 및 세포 외 농도가 가장 가능성이 분 9,16 이하에서 equilibrating와 세포에 의해 수행됩니다. 4SU의 흡수 및 결합 요금은 농도 의존적이다. 따라서 4SU 농도는 편리하게 라벨의 고용 기간에 따라 조정될 수있다. 표 1은 상대의 4SU 농도에 대한 조언을 제공합니다우리의 가장 개인적인 경험을 바탕으로 라벨의 기간에 ATION. 포유 동물 세포에서 4SU 라벨 1 시간, 200 μM의 4SU은 섬유 아세포에서 새로 전사 RNA의 50 ~ 100 뉴클레오티드 당 약 4SU 잔류의 결과로 대부분의 응용 프로그램에 충분합니다.
지난 몇 년 동안, 우리는 섬유 아세포, 내피 세포, 상피 세포, 골수 기질 세포, 대식 세포와 T-세포를 포함한 인간과 쥐 기원의 세포 유형의 넓은 범위에 4SU-태그를 적용했습니다. 또한, 초파리 및 Xenopus의의 세포가 성공적으로 사용되었다. 이 실험의 모든에 4SU 법인이 다른 세포 유형에 대한 4SU 농도 최소한의 조정을 필요로하는 매우 효율적인 것으로 나타났다. 새로운 세포 유형에 대한 메소드를 설정할 때, 우리는 4SU-농도 (예를 들어 50에서 1.5 μM까지) 증가와 라벨 세포에 추천 새롭게 전사 정제의 관계를 분석 할 R적용 4SU-농도 NA (그림 참조 2A / B). 정제 새로 전사 RNA의 양이 고원를 입력하는 4SU 농도를 선택해야합니다.
매우 합류, 접촉 억제 세포를 사용하는 경우에, 우리는 효율적으로 4SU 설립을 위해 약간 높은 4SU 농도 (예를 들어 500 대신에 200 μM)를 사용하는 것이 좋습니다. 또한, 매우 짧은 새로 전사 성적 캡처 (<200 NT)이 특히 관심의 경우, 4SU 농도도 증가해야 할 수 있습니다. 이 자궁외 효과 나 독성을 방지하기 위해 장기간의 표시 시간 (예> 1 시간)과 결합 할 수 없습니다. 마지막으로, 우리는 너무 작다에게 세포 배양 매체 볼륨을 사용하여 4SU 법인의 효율성을 줄일 수 있다는 것을 발견했다. 그러므로 우리는 각각 5 ML 또는 10 cm 또는 15cm 접시 당 배지 10 ㎖를 사용하는 것이 좋습니다.
전체 세포 RNA의 준비
이 프로토콜의 성공을 위해 청결, RNase가없는 전체 세포 RNA를 얻기 위해 매우 중요합니다. 15cm 접시 당 5 ML의 TRIZOL를 사용하여 핵산을 무료로 깨끗한 RNA를 생성합니다. 우리는 Chomczynski 등에 의해 수정 TRIZOL 프로토콜을 사용하는 것이 좋습니다. 20. 첫째, 더 나은 세척 단계에서 쉽게 처리 할 수있다 확고한 펠릿의 향상된 원심력 결과로 RNA의 대량 (> 100 μg)을 분리 적합합니다. 일반 15 ML 실험실 팔콘 튜브 이상 6,000 × g에서 생존하지 않는 그러나,이 특수 폴리 프로필렌 튜브 및 어댑터의 사용을 필요로합니다. 둘째, DNA 및 당 단백질의 제거를 향상시킵니다. 장기 나 조직에서 RNA를 준비 할 때 특히 명백하게된다. 셋째, 그것은 분리 될 수 총 RNA의 최대 크기를 제한하지 않습니다. 우리는 또한 발견하지만 열 기반의 RNA 분리 방법 (예 RNeasy) 적당한 품질의 RNA 표준 열 AR을 제공합니다전자 만하여 출발 물질의 양을 제한하는 총 RNA의 100 μg까지 캡처 할 수. 마지막으로, 잔류 에탄올을 제거하려면, 피펫 두 번, 나머지 에탄올을 제거하는 RNA의 건조가 더 이상 필요하지 않습니다. 이 후 다시 분해하기 어려울 수 있습니다 RNA를 통해 건조의 위험을 제거합니다. 원칙적으로, 4SU 태깅은 생쥐의 정맥 주사에 의해, 예를 들면 생체 내에서 적용됩니다. 그러나, 우리는 RNA의 순도가 새로 전사 RNA (미발표 자료)의 정화하기 전에 polyA 성적의 정화를 필요로하는 중요한 문제를 나타냅니다 지적했다.
Biotinylation 및 언 바운드 비오틴의 제거
비오틴 HPDP 100 % 티올 특정이며 비오틴 잔류 티올 - 라벨 RNA 새로 전사 분자 사이의 이황화 결합을 형성한다. 4SU - 라벨 RNA의 Biotinylation 효율 점 blot 분석 5의 결정에 따라 약 30 %이다. 비오틴 - HPDP는 수용성 아니므로그것은 효율적으로 클로로포름 추출에 의해 제거 할 수 있습니다. 하나의 클로로포름 추출 단계는 언 바운드 비오틴의 대부분을 제거하기에 충분 동안 우리는 정기적으로 제거를 완료하기 위해이 단계를 반복합니다. 클로로포름 추출 단계 2 ㎖ 위상 잠금 젤 무거운 튜브 (에펜 도르프) 동안 RNA 손실을 줄이기 위해 제조 업체의 지침에 따라 사용할 수 있습니다. 첫 번째 단계의 템플릿 볼륨이 종종 이러한 튜브와 직접 호환 될 너무 높은만큼 보통 우리는 두 번째 클로로포름 추출 단계에 대한 위상 잠금 튜브를 사용합니다. 언 바운드 비오틴 HPDP 제거 후, RNA는 이소 프로필 알코올 / 에탄올 침전에 의해 복구됩니다. 그것은 그들이 제공된 버퍼에있는 다니엘 이황화 결합을 환원제 포함하고 새로 전사 RNA에서 비오틴을 제거로 사용해서는 안 바이오틴 RNA를 (QIAGEN에서 예를 들어, RNeasy)를 복구하는 상용 열 기반의 장비에 유의하는 것이 중요합니다 .
새로운 정제LY 전사 RNA
100 ㎕의 스트렙 타비 딘 비즈에 100 개 이상의 μL 바이오틴 RNA를 추가하지 마십시오. 적은 볼륨을 추가하는 것은 바람직하다. 그러나 RNA의 동일한 볼륨은 모든 샘플에 대해 추가해야합니다. 당신은 단순히 비즈 1X TE의 필요한 볼륨을 추가하여 스트렙 타비 딘 구슬에 추가 RNA 입력 볼륨 (샘플 사이)을 조정합니다. 신선한 nuclease가없는 100 mM의 DTT를 만들 수있는 쉬운 방법은 매우 민감한 규모에 배치 팔콘 튜브에 DTT 분말의 충분한 양을 가만히 따르다하고 100 MM의 DTT를 생성하는 핵산 무료 H 2 O의 필요한 양을 추가하는 것입니다 (1 MG DTT 당 64.8 μL 물).
4SU - 태그의 개발 과정에서 우리는 다양한 공급 업체로부터 스트렙 타비 딘 구슬을 테스트했습니다. 그들의 수는 배경의 많은 양의를 생성합니다. 따라서, 우리는 강하게 지금까지, 우리는 조직을 C에서 레이블이없는 RNA의 이월 어떤 문제를 경험 한 적이 있고,있는 Miltenyi 스트렙 타비 딘 비즈로 사용하는 것이 좋습니다ulture 유래 RNA 샘플. 이 방법에서는, 라벨 RNA의 한 작은 150으로 NG는 특히 스트렙 타비 딘 구슬 100 μL를 사용하여 150 μg 바이오틴 RNA (물 100 μL의)에서 정제 할 수있다. 비즈와 함께 제공되는 평형 버퍼 구슬의 평형을 수행 할 수 있으며, 약간 캡처 속도에게 13을 향상시킬 수 있습니다.
품질 관리
우리는 높은 처리량 분석을 위해 복종하기 전에 새로 전사 RNA에 Q-RT-PCR 컨트롤을 수행하는 것이 좋습니다. 이 차동 주어진 실험 설정에서 조절하는 것으로 알려진 몇 가지 참조 유전자의 정량을 포함 할 수 있습니다. 4SU - 태그는 RNA의 붕괴 속도를 연구하기 위해 사용되는 경우에, 우리는 짧은 증명서 (예를 들어, MYC, FOS) 총 새롭게 전사 모두 RNA의 장수 하나를 (예를 들어 GAPDH)을 정량화하는 것이 좋습니다. 새로 전사 / 총 RNA의 비율은 (- 10 배 ~ 5) 상당히 높아야한다짧은 성적표합니다. RNA 참조 유전자의 반감기에 따라 RNA 반감기를 결정할 수 있습니다. 세 RNA 분획은 (총 RNA 새로 전사 RNA와 레이블이없는 기존의 RNA) 네 개 이상의 유전자가 서로 다른 RNA 부분 집합의 정상화를 분석하는 경우 등 7 설명 결정할 수 선형 회귀 분석 및 품질 관리 점수에 의해 수행 될 수있다 21.
Q-RT-PCR 분석을 위해, 우리는 20 μL의 cDNA 합성 믹스에 표시된 RNA의 2.5 μl를 사용하는 것이 좋습니다. Q-RT-PCR 결과의 최적의 비교를 위해 처음 사용하기 전에 5 μL 일정량의 cDNA를 동결. 사용 직전에 해동 튜브, H 2 O 45 μL와 Q-RT-PCR 분석에 희석 제목 5 μl를 추가합니다. 이 유의 한 차이를 PCR의 실행 사이의 비교를 향상시킵니다.
새로 전사 RNA 샘플은 그들을 복종하기 전에 애질런트 Bioanalyser를 사용하여 RNA 저하의 징후를 확인해야합니다높은 처리량 분석 (마이크로 어레이 또는 RNA-SEQ). 그것은, 그러나, 추가 밴드 때때로 애질런트 Bioanalyser에 의해 관찰되는 것을주의해야한다. 이것의 생물학적 중요성은 불분명 남아 있습니다. 새로 전사 RNA가 훨씬 적은 리보솜 RNA가 들어 있으므로, 이러한 샘플은 때때로 애질런트 Bioanalyser 품질 컨트롤을 실패합니다. 이 허용 품질 표시 RNA 분해 시료에 의한하지 않은 경우 일반적으로 높은 처리량 분석을 실시하여야 정상입니다.
다운 스트림 분석과 새로 전사 RNA의 호환성
새로 전사 RNA는 총 RNA보다 훨씬 더 많은 발현이 포함되어 있습니다. 이 4SU - 태그의 기간이 단축 될 때 증가 새롭게 전사 RNA에서 인트론 서열의 대용량 주로 때문입니다. 이 원료의 큰 금액을 필요로하기 때문에, 우리는 정기적으로 새로 전사 RNA 샘플에서 rRNAs의 고갈을 수행하지 않는 동안하는 Provid비 rRNA의에서 ING 오히려 작은 (~ 이중) 이득 읽습니다. 마지막으로, 차세대 시퀀싱의 cDNA 라이브러리를 준비 할 때 새로 전사 RNA에 존재 unspliced, 고 분자량 성적의 큰 비율을 추가 조각화가 필요할 수 있습니다 주목해야 할 남아있다. 크기 분열 단계의 결과에 따라서 품질을주의 깊게 통제되어야한다.
RNA 반 라이브 측정 데이터를 정규화
RNA 반감기 측정을위한 실험 데이터를 표준화하는 표준 방법은 RNA 특징 잘 집 지키는 유전자의 반감기 또는 중간 RNA 이전 실험에서 결정된 특정 세포 유형의 반감기에 모든 데이터를 정상화하는 것입니다. 포유 동물 세포에서, 후자는 5 ~ 10 시간 6.7의 범위에있다. 이 방법은 또한 4SU 기반의 측정을 위해 아주 잘 작동하는 동안 중간 RNA 반감기가 엄마를 알고 있거나하면하지 않는 경우, 정상화를위한 다른 수단이 필요합니다Y 심지어 노크 아웃 RNA의 붕괴 통로에 의해 예를 들어, 연구중인 셀룰러 시스템의 변화에 의해 영향을받을. 4SU - 태그는 세 가지 RNA 분획, 즉 전체 세포 RNA, 새로 전사 RNA와 레이블이없는 기존의 RNA의 분석에 따라 중간 RNA 반감기를 측정하는 독특한 방법을 제공합니다. 전체 세포 RNA는 단순 선형 회귀 모델은 서로 다른 세 개의 RNA의 분수를 정상화하고 중간 RNA 반감기 7,16를 결정하기 위해 사용될 수있다 후자의 두 RNA 분획으로 분리되는. 소프트웨어 패키지는 이러한 분석에게 22을 수행 할 온라인으로 사용할 수 있습니다.
낮은 딘 함량 성적 비효율적 캡처 인위적으로 낮은 새로 베껴 / 총 RNA 비율과 장기 RNA 반감기의 결과 RNA 반감기 측정에 영향을 미칠 수 있습니다. 이 문제의 범위는 uridi에 RNA 반감기 또는 로그 (새로 베껴 / 총 RNA 비율) 플롯에 의해 평가 될 수있다모든 성적 7,15의 북동쪽 내용. 이것은 또한 다른 샘플이나 조건 사이 4SU - 정관 속도의 차이를 평가하기 위해 좋은 품질 제어를 제공합니다. 딘 콘텐츠에 상당한 상관 관계가 관찰되는 경우에이 생물 정보학는 15을 의미하여 위해 수정 될 수 있습니다. 그러나 새로 전사 RNA의 성숙 성적의 기여를 쉽게 훨씬 크고 따라서 더 많은 딘 풍부한 전구체 구별 할 수없는 주목해야한다. 주어진 증명서의 처리 속도론은 (그들이 일반적으로하지 않습니다) 단지 (비효율적 캡처) 낮은 딘의 컨텐츠에 대해 정정 크게 RNA 반감기를 왜곡 수있는 알려진하지 않는 한. 따라서, 우리는 최근 9 매우 비효율적 대부분의 인간 snoRNAs 처리를 발견했다. 우리는 오히려 작은 낮은 딘 콘텐츠 (70-300 NT)에 대한 새로 전사 / 총 RNA 비율을 수정 한 경우 snoRNAs을,이 매우 짧은 snoRNA 반 거실로 이어질 것100 %를 초과하는 다수의 새로 베껴 / 총 RNA의 비율이 ES (<5 분). 그러므로, 우리는 일반적으로 RNA 반감기를 측정 할 때 낮은 딘의 콘텐츠를 수정하지 않는 것이 좋습니다.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
저자는 그들이 더 경쟁 재정적 이익이 없다는 것을 선언합니다.
Acknowledgments
우리는 원고의주의 읽기 에이미 레이건에게 감사의 말씀을 전합니다. 이 작품은 LD와 CCF에 DFG 부여 FR2938/1-1에 NGFN 플러스 부여 # 01GS0801, MRC 교제를 부여 G1002523 및 NHSBT 부여 WP11-05에 의해 지원되었다
Materials
| Name | Company | Catalog Number | Comments |
| 4-thiouridine | Carbosynth | T4509 | Prepare 50 mM stock in sterile H2O, store at -20 °C in aliquots of 50-500 μl, discard unused reagent, do not refreeze. |
| Trizol | Invitrogen | 15596026 (100 ml), 15596018 (200 ml) | WARNING - CORROSIVE and HAZARDOUS TO HEALTH! Ensure immediate access to Phenol antidote (PEG-Methanol); Store at 4 °C. |
| Chloroform | Sigma | 372978 | WARNING - HAZARDOUS TO HEALTH |
| Isopropanol | Sigma | 650447 | |
| Sodium citrate, nuclease-free | Sigma | C8532 | Prepare 1.6 M stock solution using nuclease-free water. |
| 5M nuclease-free NaCl | Sigma | 71386 | Stock solution |
| Nuclease-free H2O | Sigma | W4502 | Make 1 ml aliquots in nuclease-free tubes. |
| RNA precipitation buffer | 1.2 M NaCl, 0.8 M sodium citrate in nuclease free water. Prepare in advance under strictly nuclease-free conditions. Store at room temperature in 50 ml falcon tubes. | ||
| Ethanol | Sigma | 459844 | Use with nuclease-free water to prepare 80% ethanol, store at -20 °C. |
| 1 M nuclease-free Tris Cl, pH 7.5 | Lonza | 51237 | Stock solution |
| 500 mM nuclease-free EDTA, pH 8.0 | Invitrogen | 15575-020 | Stock solution |
| 10x Biotinylation Buffer (BB) | 100 mM Tris pH 7.4, 10 mM EDTA in nuclease-free water, make aliquots of 1 ml. | ||
| Dimethylformamide (DMF) | Sigma | D4551 | |
| EZ-Link biotin-HPDP | Pierce | 21341 | Prepare 1 mg/ml stock solution by dissolving 50 mg biotin-HPDP in 50 ml DMF. Gentle warming enhances solubilisation. Store at 4 °C in aliquots of 1 ml. |
| Phase Lock Gel Heavy tubes 2.0 ml | Eppendorf | 0032 005.152 | Optional for the chloroform extraction step. |
| Zeta membrane | BIORAD | 162-0153 | |
| 10x Dot blot binding buffer | 100 mM NaOH, 10 mM EDTA | ||
| Biotin-oligo | 5'-biotin, 25 nucleotides, any sequence | ||
| Sodium dodecyl sulphate | Fisher | BPE9738 | For 100 ml 20% stock solution, add 20 g SDS to 80 ml PBS pH 7-8 and adjust volume to 100 ml. Keep all high-percentage SDS solutions above 20 °C. Warm the solutions slightly should SDS precipitate. |
| EZ-Link Iodoacetyl-LC-Biotin | Pierce | 21333 | Prepare 1 mg/ml stock solution by dissolving 50 mg iodoacetyl-biotin in 50 ml DMF. Gentle warming enhances solubilisation. Store at 4 °C in aliquots of 1 ml. Generates irreversible, thiol-specific biotinylation. |
| Phosphate buffer saline | Gibco | 10010-015 | |
| Dot blot blocking buffer | Mix 20 ml 20% SDS with 20 ml 1 x PBS pH 7-8 and add EDTA to the final concentration of 1 mM. | ||
| Streptavidin-horseradish peroxidase | Vector Laboratories | SA5004 | Store at -20 °C. Mix 10 ml 20% SDS with 10 ml 1 x PBS. Add 20 μl Streptavidin-HRP before use. |
| ECL reagent | GE Healthcare | RNP2109 | Use following the manufacturer's instructions. |
| Super RX, X-RA Film, 18x24 cm | Fujifilm | 47410 19236 | |
| μMacs Streptavidin Kit | Miltenyi | 130-074-101 | Store the beads at 4 °C. |
| Tween 20 | Sigma | P1379 | |
| Washing buffer | 100 mM Tris pH 7.4, 10 mM EDTA, 1 M NaCl, 0.1% Tween 20 in nuclease-free H2O. | ||
| Dithiothreitol (DTT) | Sigma | 43817 | Prepare as 100 mM DTT in nuclease-free H2O, always prepare fresh before use. |
| RNeasy MinElute Kit | Qiagen | 74204 | Store columns at 4 °C, remaining components of the kit at room temperature. |
| 1.5 ml screw-top polypropylene tubes | Sarstedt | 72.692.005 | Compatible with Dimethylformamide |
| 2.0 ml screw-top polypropylene tubes | Sarstedt | 72.694.005 | Compatible with Dimethylformamide |
| 15 ml tubes | BD Falcon | 352096 | Compatible with Dimethylformamide |
| 50 ml tubes | BD Falcon | 352070 | Compatible with Dimethylformamide |
| All solutions/reagents should be stored at room temperature unless otherwise specified. | |||
| Equipment | |||
| UV/VIS spectrophotometer | Thermo Scientific | NanoDrop 1000 | Or equivalent. Use low volume (1-2 μl) for measurements of low RNA concentrations to avoid excessive sample loss. |
| Polypropylene 15 ml centrifuge tubes | VWR International | 525-0153 | In contrast to standard 15 ml tubes, these tolerate up to 15,000 × g |
| High-speed centrifuge | Beckman Coulter | Avanti J-25 | Or equivalent equipment capable of reaching 13,000×g |
| High-speed rotor | Beckman Coulter | JLA-16250 | Or equivalent equipment capable of reaching 13,000×g |
| Adaptors for 15 ml tubes | Laborgeräte Beranek | 356964 | Or equivalent equipment capable of reaching 13,000×g |
| Refrigerated table-top centrifuge | Eppendorf | 5430 R | Or equivalent. |
| Thermomixer | Eppendorf | Thermomixer compact | Or equivalent. |
| Magnetic stand | Miltenyi Biotec | 130-042-109 | One stand holds 8 μMacs columns. |
| Waterbath | Grant | SUB Aqua 5 | Or equivalent. |
| Ultra-fine scale | A&D | GR-202 | Or equivalent. |
| E-Gel iBase Power System | Invitrogen | G6400UK | For RNA gels; or equivalent. |
| E-Gel EX 1% agarose precast gels | Invitrogen | G4020-01 | For RNA gels; or equivalent. |
References
- Brown, P. O., Botstein, D. Exploring the new world of the genome with DNA microarrays. Nature Genetics. 21, 33-37 (1999).
- Mortazavi, A., Williams, B. A., Mccue, K., Schaeffer, L., Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nature Methods. 5, 621-628 (2008).
- Nagalakshmi, U., Wang, Z., et al. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science. 320, 1344-1349 (2008).
- Wilhelm, B. T., Marguerat, S., et al. Dynamic repertoire of a eukaryotic transcriptome surveyed at single-nucleotide resolution. Nature. 453, 1239-U1239 (2008).
- Dölken, L., Ruzsics, Z., et al. High-resolution gene expression profiling for simultaneous kinetic parameter analysis of RNA synthesis and decay. RNA. 14, 1959-1972 (2008).
- Yang, E., van Nimwegen, E., et al. Decay rates of human mRNAs: correlation with functional characteristics and sequence attributes. Genome Res. 13, 1863-1872 (2003).
- Friedel, C. C., Dölken, L., Ruzsics, Z., Koszinowski, U. H., Zimmer, R. Conserved principles of mammalian transcriptional regulation revealed by RNA half-life. Nucleic Acids Res. 37, e115 (2009).
- Nilsen, T. W., Graveley, B. R. Expansion of the eukaryotic proteome by alternative splicing. Nature. 463, 457-463 (2010).
- Windhager, L., Bonfert, T., et al. Ultra short and progressive 4sU-tagging reveals key characteristics of RNA processing at nucleotide resolution. Genome Res. , (2012).
- Melvin, W. T., Milne, H. B., Slater, A. A., Allen, H. J., Keir, H. M. Incorporation of 6-thioguanosine and 4-thiouridine into RNA. Application to isolation of newly synthesised RNA by affinity chromatography. Eur. J Biochem. 92, 373-379 (1978).
- Cleary, M. D., Meiering, C. D., Jan, E., Guymon, R., Boothroyd, J. C. Biosynthetic labeling of RNA with uracil phosphoribosyltransferase allows cell-specific microarray analysis of mRNA synthesis and decay. Nature Biotechnology. 23, 232-237 (2005).
- Kenzelmann, M., Maertens, S., et al. Microarray analysis of newly synthesized RNA in cells and animals. Proc. Natl. Acad. Sci. U.S.A. 104, 6164-6169 (2007).
- Schwanhäusser, B., Busse, D., et al. Global quantification of mammalian gene expression control. Nature. 473, 337-342 (2011).
- Rabani, M., Levin, J. Z., et al. Metabolic labeling of RNA uncovers principles of RNA production and degradation dynamics in mammalian cells. Nat. Biotechnol. , (2011).
- Miller, M. R., Robinson, K. J., Cleary, M. D., Doe, C. Q. TU-tagging: cell type-specific RNA isolation from intact complex tissues. Nat. Methods. 6, 439-441 (2009).
- Miller, C., Schwalb, B., et al. Dynamic transcriptome analysis measures rates of mRNA synthesis and decay in yeast. Mol. Syst. Biol. 7, 458 (2011).
- Weintz, G., Olsen, J. V., et al. The phosphoproteome of toll-like receptor-activated macrophages. Mol. Syst. Biol. 6, 371 (2010).
- Lipsett, M. N. The isolation of 4-thiouridylic acid from the soluble ribonucleic acid of Escherichia coli. Journal of Biological Chemistry. 240, 3975-3978 (1965).
- Marcinowski, L., Liedschreiber, M., et al. Real-time Transcriptional Profiling of Cellular and Viral Gene Expression during Lytic Cytomegalovirus Infection. PLoS Pathog. 8, e1002908 (2012).
- Chomczynski, P., Mackey, K. Short technical reports. Modification of the TRI reagent procedure for isolation of RNA from polysaccharide- and proteoglycan-rich sources. Biotechniques. 19, 942-945 (1995).
- Friedel, C. C., Dölken, L. Metabolic tagging and purification of nascent RNA: implications for transcriptomics. Mol. Biosyst. 5, 1271-1278 (2009).
- Friedel, C. C., Kaufmann, S., Dölken, L., Zimmer, R. HALO - A Java framework for precise transcript half-life determination. Bioinformatics. , (2010).
