

Summary
Total cellulær RNA gir et dårlig mal for å studere kortvarige endringer i RNA-syntese og nedbrytning, så vel som kinetikken til RNA prosessering. Her beskriver vi metabolsk merking av nylig transkriberte RNA med 4-thiouridine etterfulgt av tiol-spesifikk biotinylering og rensing av nylig transkriberte RNA tillater å overvinne disse begrensningene.
Abstract
Utvikling av hel-transkriptom mikromatriser og neste generasjons sekvensering har revolusjonert vår forståelse av kompleksiteten i mobilnettet genuttrykk. Sammen med en bedre forståelse av de involverte molekylære mekanismer, har nøyaktige målinger av de underliggende kinetikk blitt stadig viktigere. Her disse kraftige metoder overfor store begrensninger på grunn av iboende egenskapene til malen prøvene de studerer, dvs. totalt cellulære RNA. I mange tilfeller vil endringene i total cellulær RNA forekommer enten er for sakte eller for fort for å representere de underliggende molekylære hendelser og deres kinetikk med tilstrekkelig oppløsning. I tillegg er bidraget fra endringer i RNA syntese, bearbeiding, og forfallet ikke lett differensiert.
Vi har nylig utviklet høy oppløsning genuttrykk profilering for å overvinne disse begrensningene. Vår tilnærming er basert på metabolsk merking av nylig transkribert RNA med 4-thiourispis (dermed også referert til som 4SU-merking) etterfulgt av grundig rensing med ny-transkriberte RNA ved hjelp av tiol-spesifikk biotinylering og streptavidin-belagte magnetiske kuler. Det er aktuelt for et bredt spekter av organismer inkludert virveldyr, Drosophila, og gjær. Vi vellykket anvendt 4SU-tagging å studere real-time kinetikk transkripsjonsfaktor aktiviteter, gir presise målinger av RNA halveringstider, og få ny innsikt i kinetikken av RNA prosessering. Endelig kan beregningsmessig modellering bli anvendt for å generere et integrert, omfattende analyse av de underliggende molekylære mekanismer.
Introduction
Genuttrykk profilering er et viktig verktøy som brukes til å studere cellulære prosesser og tilhørende kompleks interaksjon nettverk. Studier av mRNA-overflod har typisk vært metoden for valg å skaffe grunnleggende innsikt inn i de underliggende molekylære mekanismer. Utvikling av hel-transkriptom mikromatriser en og, mer nylig, neste generasjons sekvensering av RNA (RNA-seq) 2-4 drevet denne tilnærmingen. Mens disse teknologiene har revolusjonert vår forståelse av kompleksiteten i mobilnettet genuttrykk, står de overfor store begrensninger på grunn av iboende egenskapene til deres mal sample, dvs. total mobilnettet RNA. Først kortsiktige endringer i total RNA-nivåer samsvarer ikke endringer i transkripsjon priser, men er iboende avhengig RNA halveringstid av de respektive transkripsjoner. Mens en fivefold induksjon av en kortvarig transkript, f.eks koding for en transkripsjonsfaktor, vil det bli mulig å påvise i total RNAinnen en time, den samme induksjon av en langlivet transkripsjon, for eksempel som koder for et metabolsk enzym, vil forbli praktisk talt usynlig. I tillegg til og med en fullstendig stengning (> 1000 ganger nedregulering) i transkripsjon frekvensen av en gjennomsnittlig genet med en RNA halveringstid på fem timer vil bare ta fem timer til sin samlede RNA-nivåer for å redusere med bare todelt . Derfor favoriserer analyse av total RNA påvisning av oppregulering av kortlivede transkripter, hvorav mange koder for transkripsjonsfaktorer og gener med regulatoriske funksjoner 5. I tillegg er den sanne kinetiske kaskade av regulering formørket og primære signaleringshendelser kan ikke skilles fra sekundære. Både i sin tur kan føre til betydelig skjevhet i nedstrøms bioinformatikk analyser. Sekund, kan endringer i total RNA-nivåer ikke tilskrives endringer i RNA syntese eller forfall. Målinger av sistnevnte krever celle invasive metoder, f.eks blokkerer transcriptiom bruk Actinomycin D 6 og forlenget overvåkning av pågående RNA-nedbrytning over tid. Med en gjennomsnittlig mRNA halveringstid i mammalieceller av 5 - 10 hr 5,7, vil mRNA nivåer av de fleste gener har bare redusert med mindre enn todelt etter flere timer med transkripsjonell arrest. Disse ganske små forskjeller resultere i grovt upresise målinger av mRNA halveringstider for de fleste cellulære gener på grunn av den eksponentielle i den underliggende matematiske ligninger. Til slutt, mens RNA-seq av total mobilnettet RNA avslørte at omtrent halvparten av genene våre er underlagt alternativ spleising hendelser åtte, de underliggende kinetikk samt de dynamiske mekanismer guiding vev-og kontekst-spesifikk regulering av RNA prosessering fortsatt dårlig forstått. I tillegg er bidraget av RNA prosessering til differensial genekspresjon, spesielt for ikke-kodende RNA, gjenstår å bli bestemt. Til sammen disse begrensningene representerer store hindringer forbioinformatiske kinetisk modellering av de underliggende molekylære mekanismer.
Vi har nylig utviklet en tilnærming, kalt høy oppløsning genuttrykk profilering, for å overvinne disse problemene 5,7,9. Den er basert på metabolsk merking av nylig transkriberte RNA ved hjelp av 4-thiouridine (4SU-tagging), et naturlig forekommende uridin derivat, og gir direkte tilgang til nylig transkriberte utskrifter med minimal innblanding i cellevekst og genuttrykk (se figur 1) 5, 10-12. Eksponering av eukaryote celler til 4SU resultater i sin raske opptak, fosforylering til 4SU-trifosfat, og innlemmelse i nylig transkriberte RNA. Etter isolering av total cellulær RNA, er det 4SU-merkede RNA fraksjon tiol-spesifikt biotinylert generere en disulfidbinding mellom biotin og de nylig transkriberte RNA. 'Total mobilnettet RNA "kan da bli kvantitativt delt inn merket (' nylig transkribert ') og umerket (' pre-existing ') RNA med høy renhet ved hjelp av streptavidin-belagte magnetiske kuler. Endelig er merket RNA utvinnes fra kulene ved å legge et reduksjonsmiddel (for eksempel ditiotreitol) spalte disulfidbinding og frigivelsen av nylig transkriberte RNA fra kulene.
Nylig transkribert RNA skildrer transkripsjonell aktiviteten til hvert gen i løpet av tidsrammen for 4SU eksponering. 4SU-tagging på tidsskalaen minutter gir dermed et øyeblikksbilde bilde av eukaryote genuttrykk og en ideell mal for ned-stream bioinformatiske analyser (f.eks promoter analyse). I tilfeller der steady-state kan antas, blir forholdet mellom nylig transkribert / totalt, nylig transkribert / umerket og umerkede / total RNA gi ikke-invasiv tilgang til presise RNA halveringstider 7,13. I tillegg er det viktig å merke seg at ny-transkriberte RNA renset etter så lite som 5 minutter av 4SU-merking (5 min 4SU-RNA) er yngre enn 15 og 60 min 4SU-RNA.Når du utfører både ultra-korte og gradvis lengre 4SU-tagging i en enkelt eksperimentell innstilling kombinert med RNA-seq, er kinetikken av RNA prosessering avslørt på nukleotid oppløsning ni. Til slutt, tid-retters analyser av nylig transkriberte og total RNA kombinert med beregningsorientert modellering tillate en integrerende analyse av RNA syntese og forfall 14..
Som konklusjon, tillater denne tilnærming for den direkte analyse av dynamikken til RNA-syntese, behandling og nedbrytning i eukaryote celler. Det er aktuelt i alle større modell organismer inkludert pattedyr, insekter (Drosophila), amfibier (Xenopus), og gjær 5,15,16. Det er direkte kompatibelt med microarray analyse 5,17, RNA-seq 9,13,14, og gjelder in vivo 12,15. Her, vi detalj metodikken til å merke, isolere og rense nylig transkriberte RNA i dyrkede celler. I tillegg potenal problemer og fallgruver blir diskutert.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
En. Metabolsk Merking med 4-thiouridine
Lag en detaljert plan for den eksperimentelle oppsett / tidsplan, for eksempel når man skal legge til 4SU til cellekultur og når man skal høste prøvene. Plan i minst 5 min mellom hver betingelse. Bare behandle celler fra en tilstand av gangen. Håndtak maks. 3-5 retter på et gitt tidspunkt. Håndter celler så raskt som mulig for å minimalisere endringer i temperatur og CO 2 nivåer. Unngå å utsette cellene for sterkt lys etter 4SU legges da dette kan resultere i fornetning av 4SU-merket RNA til cellulære proteiner.
Starten av merking
- Tine 4-thiouridine (4SU) like før bruk og pipette nødvendige mengden 4SU for hver tilstand i en steril Falcon tube.
- Ta den nødvendige mengden av cellekulturmedium (5 ml per 10 cm parabol) av rettene og legge til 4SU-holdig Falcon rør og bland godt. Fjern og kast de resterende medium fra retter. <li> Bruk 4SU-holdig medium tilbake til rettene.
Slutten av merking
- Fjern cellekulturmedium fra cellene. Tilsett 5 ml av Trizol til hver plate. For kompliserte eksperimenter inkludert flere tidspunkter eller betingelser, er dette trinnet best gjøres av to personer, én fjerne medium, den andre legger Trizol og høsting lysatet.
- Inkuber i 5 min ved romtemperatur for fullstendig cellelyse.
- Bruk en 10 ml pipette å skylle platen forsiktig med den ekstra Trizol. Dette hjelpemidler komplett cellelyse og prøve utvinning. Handle with care som Trizol er ekstremt farlig når komme i kontakt med hud eller øyne! Har motgift for fenol brannsår på hånden (f.eks Polyethylene glycol 300 eller 400 i industrielle denaturert sprit (70:30)). Overfør prøvene til polypropylenrør. Vær oppmerksom på at standard Falcon rør ikke motstå disse høye g krefter). Prøver kan bli lagret ved -20 ° C i minst én måned inntil total RNA jegs forberedt.
2. RNA Forberedelser Bruk Forandringer Trizol Protocol
- Tilsett 1 ml kloroform (0,2 ml per ml Trizol) og ristes kraftig i 15 sek. Inkuber ved romtemperatur i 2 - 3 min.
- Sentrifuger ved 13000 x g i 15 minutter ved 4 ° C.
- Overfør vandige øvre fase (inneholdende RNA) til et nytt 15 ml polypropylenrør.
- Legg? Den reaksjonsvolum på begge RNA nedbør buffer og isopropanol (for eksempel til 3 ml supernatant Tilsett 1,5 ml RNA-buffer nedbør og 1,5 ml isopropanol).
- Bland godt. Inkuber ved romtemperatur i 10 min.
- Sentrifuger ved 13000 x g i 10 minutter ved 4 ° C. Kast supernatanten.
- Spinn ned en kort stund (5000 × g for 30 sek) og fjerne rester av isopropanol med 200 mL pipette.
- Legg et likt volum av 75% etanol og riste røret til pellet løsner. Unngå å bryte den i mange små biter som dette kan gjøre fjerning av restl etanol vanskelig.
- Sentrifuger ved 13000 x g i 10 minutter ved 4 ° C. Kast supernatanten.
- Spinn ned RNA kort og fjerne gjenværende etanol med en 200 mL pipette. Gjenta trinn og fjerne gjenværende etanol med en 20 mL pipette. Etter disse to trinnene, skal ingen ytterligere tørking av pellets utføres.
- Tilsett 100 ul av H 2 O per 100 mikrogram forventet RNA yield og bland godt med pipettering opp og ned 5-6 ganger for å hjelpe til med å løse opp RNA.
- Oppløs og denaturere RNA ved oppvarming til 65 ° C i 10 min (shaker) og umiddelbart plassere på is.
- Måle RNA konsentrasjon på 260 nm ved hjelp av en NanoDrop spektrofotometer, etter produsentens anvisninger. Dette RNA kan lagres ved -80 ° C i minst én måned.
3. Tiol-spesifikk biotinylering med ny-transkriberte RNA
- Begynn med 60 - 80 mikrogram total cellulære RNA.
- Utgjør merking reaksjon. Pipette i følgenderekkefølge (per mikrogram RNA):
- 1 mL 10x biotinylering Buffer
- 7 mL RNA (som inneholder ett mikrogram RNA fortynnet i nukleasefritt H 2 O)
- 2 ul biotin-HPDP (1 mg / mL DMF)
Alltid legge til biotin-HPDP siste og bland umiddelbart ved pipettering. I tilfelle biotin utfelles, kan DMF innhold økes til en sluttkonsentrasjon på 40%.
- Inkuber ved romtemperatur i 1,5 time med rotasjon.
- Legg et likt volum kloroform. Bland kraftig. Inkuber i 2 - 3 minutter før fasene begynner å skille og bobler begynner å forsvinne.
- Sentrifuger ved 20000 x g i 5 min ved 4 ° C. Nøye overføre den øvre vannfase i et nytt rør.
- Gjenta trinn 3,4 og 3,5 gang. Det kan være lurt å utføre dette trinnet i 2 ml Phase Lock Gel Tunge rør for å redusere tap av RNA.
- RNA ventet: legg til 1/10 volum av 5 M NaCl, og et like stort volumisopropanol til vannfasen.
- Sentrifuger ved 20000 x g i 20 min ved 4 ° C. Kast supernatanten.
- Legg et likt volum av 75% etanol, sentrifuger ved 20 000 x g i 10 minutter ved 4 ° C, supernatanten kastes.
- Spinn kort og fjerne rester av etanol med 200 mL pipette.
- Spinn kort og fjerne rester av etanol med 20 mL pipette.
- Ikke la RNA tørke. Re-suspendere den i 50 - 100 mL H 2 O (~ 1 mL per 1 mikrogram innspill RNA). Bland godt med pipettering opp og ned 5-6 ganger.
- Sjekk RNA kvalitet ved electrophoretical analyse for å utelukke RNA degradering.
4. Dot Blot Analysis of 4SU-inkorporering (valgfritt)
4SU inkorporering kan lett bestemmes ved punkt-blot-analyse av biotinylerte RNA. Dette er et valgfritt trinn som gjør feilsøking og estimering av 4SU inkorporering priser i forhold til en biotinylated DNA oligo kontroll. For denne analysen vi recommend hjelp jodacetyl-biotin i stedet for biotin-HPDP for biotinylering av 4SU-merket RNA i trinn 3.2. Dette resulterer i en irreversibel biotinylering av 4SU-RNA. Derfor kolonne-baserte metoder (f.eks RNeasy) kan anvendes for gjenvinning av langt mindre mengder av biotinylert RNA (for eksempel 5 ug). Mens RNA biotinylert ved hjelp av biotin-HPDP er også egnet for denne analyse, er det resulterende signalet svakere, og det signal-støy-forholdet er mindre gunstige (figur 3).
- Følg protokoll for 4SU-merking og isolering av totalt cellulære RNA som beskrevet i punkt 1 og 2.
- Biotinylate 4SU-merket RNA som beskrevet i pkt. 3 erstatte biotin-HPDP med jodacetyl-biotin og utføre to kloroform ekstraksjon å fjerne overdreven jodacetyl-biotin rester.
- Gjenopprette biotinylerte RNA av isopropanol / etanol nedbør som beskrevet eller bruke en kolonne tilnærming (f.eks RNeasy) i tilfelle små mengder RNA (<10 mikrogram) Blir brukt.
- Inkuber Zeta membranen i nuclease-fritt vann med gående i 10 min.
- Ta membranen ut av nuclease-fritt vann og fjerne overdreven væsker ved å plassere membranen i mellom to ren papirhåndklær og trykke hardt. Lufttørkende membranen i 5 min vil resultere i finere prikker.
- For hver prøve forberede 20fil 200 ng / ul RNA ved hjelp av punkt-blot iskald bindingsbuffer (10 mM NaOH, 1 mM EDTA). Påfør 5 ul av denne fortynningen (dvs. 1 ug av RNA) samt tre påfølgende 10-doble fortynninger (dvs. 100, 10 og 1 ng RNA, respektivt) til en Zeta membranen ved pipettering. Pipettering gjennom et tomt rack av pipettespisser kan brukes til å gi jevnt fordelt avstand. Alternativt kan du bruke en prikk blot apparat i henhold til produsentens instruksjoner.
- Påfør 5 pl av biotin-merket DNA oligo ved konsentrasjoner varierende fra 20 ng / mL til 20 pg / mL (dvs. 100 til 0,1 ng oligo) som en positiv fortsrol til membranen ved pipettering. Bruk en biotinylated, 4SU-naive prøven som negativ kontroll.
- Lufttørk membranen i 5 min.
- Inkuber membranen i 30 minutter i 40 ml blokkeringsbuffer med vuggende.
- Inkuber membran med 10 ml 1:1.000 streptavidin-pepperrot peroksidase i 15 minutter (5 ml PBS + 5 ml 20% SDS + 10 ul streptavidin-pepperrot peroksidase)
- Vask membran to ganger i 40 ml PBS + 10% SDS (20 ml PBS + 20 ml 20% SDS) i 5 min.
- Vask membran to ganger i 40 ml PBS + 1% SDS (38 ml PBS + 2 ml 20% SDS) i 5 min.
- Vask membran to ganger i 40 ml PBS + 0,1% SDS (40 ml PBS + 200 ul 20% SDS) i 5 min.
- Fjern overdreven væske ved å plassere membran i mellom to rene papirhåndklær og trykke på dem fast.
- Visualisere membran-bundet HRP bruker ECL henhold til produsentens anvisninger.
- Plasser membranen i plastfolie / pose, fjerne luftbobler og inkuber i 2 min i mørket.
- Expose membranfilm til 1-5 min.
5. Separasjon av Merket og uten merking RNA Bruke Streptavidin-belagte Magnet perler
- Varme vaskebuffer (3 ml per prøve) til 65 ° C i et vannbad.
- Forbered fersk 100 mM ditiotreitol (DTT) i nukleasefritt H 2 O. Gjøre det ved dekantering 15 - 30 mg DTT pulver i en ren 50 ml Falcon rør plasseres på det ultrafine skala. Veie og legge nødvendige mengden av nukleasefritt H 2 O.
- Heat biotinylert RNA prøver til 65 ° C i 10 min for å denaturere og straks sted på is.
- Sted μMacs kolonner i den magnetiske stativet. Vi anbefaler ikke å behandle mer enn 12 prøver på en gang (6 - 8 prøver er optimal).
- Pre-likevekt Miltenyi søyler med 1 ml romtemperatur vaske buffer. Dette vil ta ca 15 min.
- Samtidig tilsettes 100 pl av streptavidin perler til 50 - 100 ul av biotinylerte RNA. Inkuber ved romtemperatur i 15 min med rotasjon. < li> Dersom noen av kolonnene ikke har innledet ved drenering nå dette kan lettes ved lett trykk i toppen av kolonnen med en behansket finger. Når strømmen har startet kolonnene renne lett.
- Påfør RNA / perler til kolonnene. Kast gjennomstrømning med mindre du ønsker å gjenopprette den umerkede RNA fraksjon (se kapittel 7).
- Vask tre ganger med 0,9 ml av 65 ° C vaskebuffer (1 ml pipettespisser krympe når pipettering buffere ved 65 ° C).
- Vask tre ganger med 0.9 ml vaskebuffer romtemperatur.
- Pipette 700 mL buffer RLT (RNeasy MinElute Opprydding Kit, Qiagen) inn i nye 2 ml rør og plassere dem under søylene.
- Eluere nylig transkriberte RNA til den RLT-buffer ved å tilsette 100 ul av 100 mM DTT til søylene.
- Utføre en andre eluering rund 3 min senere inn i samme rør ved å tilsette ytterligere 100 ul av 100 mM DTT.
6. Gjenoppretting av ny-transkriberte RNA
ontent "> Fortsett med RNeasy MinElute Cleanup (Qiagen) protokoll følge produsentens instruksjoner. Elute i 25 mL nukleasefritt H 2 O. Mål RNA konsentrasjoner ved hjelp av en NanoDrop spektrofotometer. For å unngå behovet for å tine og fryses RNA før du sender den til en high-throughput analysen, anbefaler vi forbereder cDNA umiddelbart etter den nylig transkribert RNA er renset. Bruk 2,5 mL av de nylig transkriberte RNA i 20 mL cDNA syntese mix for cDNA syntese følge produsentens instruksjoner. Utfør QRT-PCR-kontroller med en : 10 fortynninger av cDNA-blanding butikk RNA ved -80 ° C..7. Gjenoppretting av uten merking, Unbound RNA (valgfritt)
I tilfelle den ubundne RNA må utvinnes, samle og kombinere den gjennomstrømningskanalen (etter tilsetting av RNA-streptavidin perler løsning til kolonnene) og den første vask for etterfølgende utfelling. Vanligvis er det tilstrekkelig å utfelle bare 50% av den ubundne RNA som this vil inneholde> 80% av utgangsmaterialet.
- Legg et likt volum isopropanol (ingen salt som må tilsettes som vaskebuffer allerede inneholder 1 M NaCl).
- Sentrifuger ved 20000 x g i 20 min ved 4 ° C. Kast supernatanten.
- Legg et likt volum av 75% etanol, sentrifuger ved 20 000 x g i 10 minutter ved 4 ° C, supernatanten kastes.
- Spinn kort og fjerne rester av etanol med 200 mL pipette.
- Spinn kort og fjerne rester av etanol med 20 mL pipette.
- Ikke la RNA tørke. Gjensuspender det i 100 mL H 2 O. Bland godt med pipettering opp og ned 5-6 ganger. Inkuber ved 65 ° C i 10 min med risting og overfører direkte til is.
- Sjekk RNA kvalitet ved electrophoretical analyse for å utelukke RNA degradering.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
En. Starter Material og Forventet Rentene
Etter en time (t) av 4SU-eksponering nylig transkribert RNA representerer om lag 1-4% av totalt cellulære RNA. Dette vil være lavere i vekststimulerende arrestert celler som de ikke lenger syntetisere RNA for å ta hensyn til cellevekst / replikasjon. Ved merking for en hr, anbefaler vi starter analysen med 60 - 80 mikrogram total RNA. Fra og med mindre enn 30 mikrogram av total RNA resulterer i små RNA-pellets som er vanskelig å se etter biotinylering trinnet og således lett kan gå tapt. Input RNA-nivåer kan økes til så mye som 150 mikrogram for svært korte varigheten av merking (f.eks 5 - 10 min). Når varigheten av RNA merking er forkortet fra 1 time til 5 min bidraget av kortlivede Intronic sekvenser i nylig transkribert RNA øker fra ~ 60% til ~ 80% 9. Som introner er vesentlig lenger enn for kodende sekvenser, så vel som 5'-og 3'-UTRs, mengden av nylig transkriberteRNA, som kan renses etter kort-eller til og med ultra-kort 4SU-tagging, faller ikke lineært. Som sådan, erholdt vi> 0,5% av total RNA etter 5 min av 4SU-merking i ikke-adherente humane B-cellelinjer 9. Det bør imidlertid bemerkes at høyere konsentrasjon av 4SU og litt lengre varigheter av merking kan være nødvendig for å oppnå lignende 4SU innlemmelse satsene i adherente celler. Mens selv en lav 4SU-inkorporasjonsraten vil tillate effektiv fangst og rensing av store, uridin-rik transkripsjoner, veldig kort karakterutskrift med lav uridin innhold (f.eks miRNAs) er sannsynlig å unnslippe rensing selv når du bruker høye 4SU konsentrasjoner (> 1 mm). I NIH-3T3 murine fibroblaster, merket 1 time på 200 mikrometer 4SU eksponering nylig transkriberte RNA med om lag en 4SU rester per 50-100 nukleotider (nt) 5. Dette bør tillate svært effektiv utvinning av vitnemål> 500 - 1000 nt i lengde. Følgelig, vi bare observert en mindre avskrift størrelseskjevhet når merking i 1 time ved anvendelse av 200 uM 4SU i både murine fibroblaster og humane B-celler 7. Mens en hr av 200 mikrometer 4SU ikke medføre vesentlige endringer i cellulære transkripsjoner nivåer i murine fibroblaster, langvarig eksponering av celler til ≥ 200 mikrometer 4SU ikke resulterer i en målbar vekst underskudd innen 24 timer (upubliserte data). Derfor bør både varigheten av merkingen og den anvendte 4SU-konsentrasjonen bli minimalisert for å unngå ektopisk eller toksiske effekter. En enkel måte å bestemme minimal 4SU-konsentrasjonen som kreves for en effektiv utvinning av nylig transkribert RNA er å rense nylig transkriberte RNA etter 4SU-merking med økende konsentrasjoner av 4SU (f.eks 50-1600 mm). Som vist på figurene 2A og 2B, gjenvinning av nylig transkriberte RNA-merket i 1 time i primære humane fibroblaster økes vesentlig 50-200 uM 4SU men deretter begynte å flate ut.
2. DotBlot Kvantifisering av 4SU innlemmelse (valgfritt)
I noen tilfeller kan det være av interesse å måle mengden av 4SU inkorporering i total RNA. Dette gjøres best ved dot blot analyse på biotinylerte RNA ved hjelp av en streptavidin konjugat. På grunn av sin kjemiske natur jodacetyl-biotin er mer reaktiv til tiol-gruppene enn biotin-HPDP resulterer i biotinylering av praktisk talt alle 4SU rester i nylig transkribert RNA. Det er viktig å bemerke at, som biotin-HDPD, er jodacetyl-biotin ikke vannoppløselig, og er således effektivt fjernet ved kloroformekstraksjon som utført for biotin-HPDP. Derfor kan identiske reaksjonsbetingelser og konsentrasjoner som anvendes ved bruk av biotin-HPDP. Imidlertid er jodacetyl-biotin ikke reversibel. Det kan således ikke benyttes for rensing av ny-transkriberte RNA i kolonne tilnærminger. Mens bruk av jodacetyl-biotin gjør det mulig å kvantifisere 4SU-innlemmelse, biotin-HPDP baserte målinger vurdere både4SU-innlemmelse og biotinylering effektivitet. Tilsatt de to biotinylering reagenser til samme prøve tillater måling av biotinylering effektiviteten av RNA-innarbeidet 4SU. Biotinylering effektiviteten av biotin-HPDP for 4SU-merkede RNA synes å være omtrent tre ganger mindre enn for jodacetyl-biotin som indikerer at bare omtrent én av tre 4SU rester i nylig transkriberte RNA er faktisk biotinylert med biotin-HPDP (figur 3). Ved å sammenligne signalet SAMPLE intensiteter med den biotinylerte DNA-oligo-kontroll, kan biotinylering tettheter måles. For de fleste pattedyr cellelinjer et positivt signal bør fortsatt være synlig i 10 ng av biotinylerte RNA etter en time på 200 mikrometer 4SU merking. En svak bakgrunn signal er påviselig for den høyeste konsentrasjonen (1 pg) av umerket RNA.
3. Rensing av ny-transkriberte RNA
Gjenoppretting av nylig transkribert RNA er svært kvantitative. Hvis du startet med samme RNA konsentrasjonen du kan forvente å oppnå de samme mengder nylig transkriberte RNA for alle prøvene. Som mange søyle-baserte analyser, kan innsamling av nylig transkriberte RNA ved bruk av RNeasy-sett MinElute resultere i ytterligere absorpsjon ved 230-260 nm (nærvær av vaskemidler som stammer fra vaskingen buffere) som kan interferere med OD 260 målinger. Dette er sett i mindre grad ved bruk av en frisk 2 ml samling rør for hvert sentrifugeringstrinnet. Likevel bør noen urimelig høye OD målinger (> 2 ganger høyere enn andre prøver) vurderes med forsiktighet, spesielt hvis OD 260/280 forholdstall er <1.7. For down-stream analyser er det derfor ofte best å bruke samme mengde mal RNA volum for alle prøvene. I de tilfeller hvor avkastningen av merket RNA er lavere enn forventet sjekk for tegn på RNA degradering av electrophoretical analyse. Nylig transkribert RNA inneholder betydelig større mengder av store, unspliced transkripsjoner med de typiske rRNA band å bli mye mindre fremtredende (figur 4).
4. Kvantifisering av ny-transkriberte RNA
Endelig kan inkorporering priser av 4SU i nylig transkriberte RNA være direkte kvantifisert ved spektrofotometrisk analyse basert på absorpsjon maksimalt 4SU på 330 nm og OD 330/260 ratio 5,18. Dette krever> 3 mikrogram av merkede RNA konsentrert i et lite volum (10 - 20 ul) av isopropanol / etanol-utfelling. For å unngå å miste den lille RNA pellet co-nedbør med 30 mikrogram nukleasefritt glykogen (Fermentas, # R0551) bør utføres. En ekstra peak er synlig på 330 nm reflekterer inkorporering rate på 4SU inn nylig transkribert RNA (figur 5).
/ Files/ftp_upload/50195/50195fig1highres.jpg "/>
Figur 1. . Prinsippet om metabolsk merking med 4-thiouridine (4SU) 4SU legges til celler for den nødvendige (5-120 min) tid etterfulgt av utarbeidelse av totalt cellulære RNA. Etter tiol-spesifikk biotinylering, er total cellular RNA delt inn 4SU-merket, nylig transkriberte RNA, og umerkede, pre-eksisterende RNA bruker streptavidin-belagte magnetiske kuler. Nylig transkribert RNA er restituert fra perlene med et reduksjonsmiddel som kløyver de disulfidbindinger som forbinder nylig transkriberte RNA til perlene. Klikk her for å se større figur .
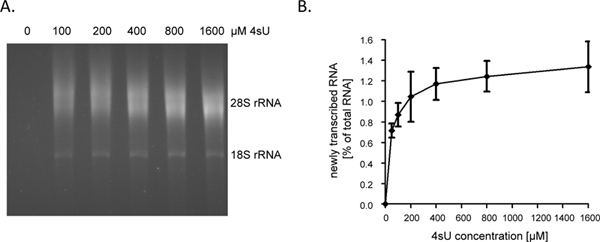
Figur 2. Gjenoppretting av nylig transkribert RNA etter økende konsentrasjoner av. 4SU (A) Primære humane forhud fibroblaster (HFF) ble inkubert med 100, 200, 400, 800 eller 1600 iM 4SU. Ny-transkriberte RNA ble renset fra 50 ug total cellulær RNA og underkastet electrophoretical analyse. Som forventet ble det en konsentrasjonsavhengig økning i gjenvunnet nylig transkribert RNA observert som startet å platå ved høyere konsentrasjoner. (B) Mengder av renset nylig transkriberte RNA ble kvantifisert ved hjelp ImageJ 1.45s programvare. Kombinerte data fra fire uavhengige eksperimenter på de mengder av ny-transkriberte RNA gjenopprettet etter ulike konsentrasjoner av 4SU-merking som spenner fra enten 50-800 mikrometer 4SU (n = 2) eller 100 -. 1600 mikrometer 4SU (n = 2) er vist Klikk her å se større figur .
upload/50195/50195fig3.jpg "alt =" Figur 3 "fo: content-width =" 4.5in "fo: src =" / files/ftp_upload/50195/50195fig3highres.jpg "/>
Figur 3. Estimering av 4SU inkorporering i 4SU-merket total RNA ved hjelp av punkt-blot-analyse. Total RNA ble isolert fra NIH-3T3-fibroblaster murine eller humane forhud fibroblaster (HFF) inkubert med 200 pM 4SU i én time. Ingen 4SU ble lagt til en parabol som negativ kontroll. For HFF både kontakten inhibert (N = ikke-voksende celler), og celler i vekst (y) ble inkludert. RNA ble isolert ved anvendelse av Trizol reagens og deretter konjugert til biotin-HPDP eller jodacetyl-biotin. Konsentrasjon av hver prøve ble justert til 200 ng / mL, og 5 ul av denne fortynningen (dvs. 1 ug av RNA), samt tre påfølgende 10-doble fortynninger (dvs. 100, 10 og 1 ng RNA, respektivt), ble alle flekket på et stykke Zeta membran. 5 mL fortynninger av biotin-merket DNA oligo ble plassert på membranen som positive kontroller på concentrationer som strekker seg fra 20 ng / mL ned til 20 pg / mL (dvs. 100 til 0,1 ng, henholdsvis). Biotin tetthet ble analysert ved hjelp av en streptavidin-pepperrot peroksidase konjugat.
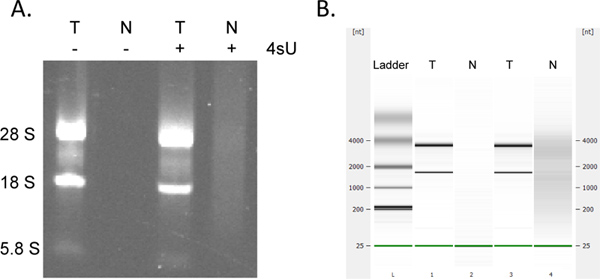
Figur 4. Electrophoretical analyse av nylig transkriberte og total RNA. Totalt RNA (T) og ny-transkriberte RNA (N) fremstilt fra muse-NIH-3T3-fibroblaster dyrket både i nærvær og fravær av 500 pM 4SU i 1 time ble analysert ved agarosegel-elektroforese (a) og (i samme rekkefølge) ved hjelp av Agilent Bioanalyser (B). Ingen RNA ble restituert uten 4SU behandling av celler. Renset nylig transkriberte RNA inneholder større mengder av høymolekylære mRNAer og betydelig mindre modne rRNAs enn totalRNA som bemerkelsesverdig mellom 28S, 18S, og 5.8S rRNA band. Klikk her for å se større figur .
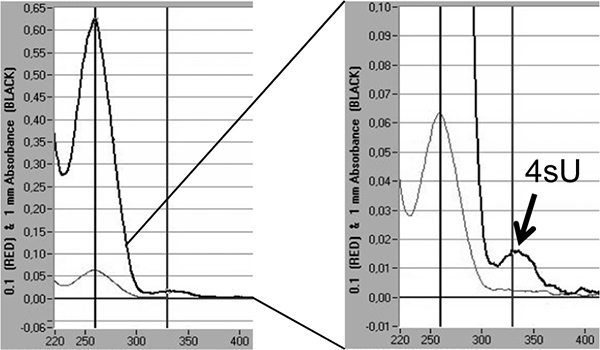
Figur 5. Kvantifisering av 4SU innlemmelse i nylig transkriberte RNA ved spektrofotometrisk analyse. Newly transkriberte RNA renset fra 2 x 100 mikrogram total RNA etter en time på 200 mikrometer 4SU i murine NIH-3T3 fibroblaster. Nylig transkriberte RNA ble fremskyndet med isopropanol / etanol etter tilsetting 30 mikrogram nukleasefritt glykogen. Spektrofotometrisk analyse av nylig transkriberte RNA innhentet av en NanoDrop 1000 spektrofotometer vises. De lysegrå linjer representerer målinger på 0,1 mm, mens de tykkere, mørk grå linjene representerer målinger på 1 mm væske kolonne. På høyre, en forstørrelse på toppen av utryddelse representerer than innlemmet 4SU-rester vises. Basert på utryddelse co-effektive av 4SU 18 innlemmelse priser av 4SU kan anslås.
| Varigheten av merking [min] | Anbefalt 4SU konsentrasjon [mM] |
| 120 | 100-200 |
| 60 | 200-500 |
| 15 - 30 | 500 - 1000 |
| <10 | 500 - 2000 |
Tabell 1. Anbefalt 4SU konsentrasjoner.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Metabolsk merking av nylig transkribert RNA forbedrer betydelig kraft av high-throughput teknologier som mikromatriser og RNA-seq ved å tilby mer egnede maler for å løse biologiske spørsmål av interesse. Denne protokoll gjennomgikk omfattende optimalisering. Den lar> 1000-fold berikelse av nylig transkriberte RNA og gir svært reproduserbare resultater.
Den eksperimentelle utformingen av en 4SU-tagging eksperimentet er av avgjørende betydning som nylig transkriberte RNA vil skildre sanntid transkripsjonell aktivitet bare i den tiden av eksponering av celler til 4SU. Hvis de faktiske endringer i transkripsjon priser etter en stimulans allerede har stilnet, vil disse bli savnet når analysere nylig transkriberte RNA selv om endringene i total RNA-nivåer kan fortsatt være synlig. Derfor er en god forståelse av den underliggende biologi viktig å definere den eksperimentelle oppsett samt den optimale perioder of tid for 4SU eksponering. Nedenfor gir vi anbefalinger og måter for å unngå vanlige fallgruver for de mest avgjørende skritt.
Utarbeidelse av lager løsninger og plast ware
Alle stamløsninger må være forberedt på å bruke nukleasefritt vann. Ved hjelp av in-house renset deionisert vann kan resultere i problemer dersom vannet inneholder reduksjonsmidler. I ett tilfelle førte dette i fullstendig tap av alle merkede RNA. Derfor anbefaler vi sterkt anbefale å kjøpe pre-laget nukleasefritt NaCl, Tris-Cl, EDTA, natriumsitrat og vann. Sikre nukleasefritt forholdene til enhver tid. Dimetylformamid (DMF) oppløser noen plastmaterialer. Vi har funnet at bruk av 25 ml cellekultur plastpipetter å overføre DMF fra sitt lager glassflaske til 50 ml Falcon-rør for å forberede den biotin-HPDP stamløsning var tilstrekkelig til vesentlig å redusere utbytter av nylig transkriberte RNA fra hele analysen. Interessant, gjorde dette ikke negativt påvirke biotinylasjon effektivitet (som testet ved dot blot), men resulterte i en 75 til> 90% tap av nylig transkriberte RNA som kunne utvinnes fra kulene. Tapet var mest uttalt når varigheten av merking ble reduseres fra 60 til 30 minutter eller mindre. Mest sannsynlig en substans elueres fra plastpipetter av DMF delvis ødelagt belegg av streptavidin perlene. Derfor bør bruken av plast-materialer som ikke er kjent for å være kompatibel med DMF unngås med alle midler. Av samme grunner bør celle skrapere ikke brukes til å forbedre utvinningen av TRIzol prøver fra cellekultur plater. Det er interessant å merke seg at de putative stoffene elueres fra plasten ved den DMF eller Trizol var tilsynelatende ingen fjernet ved kloroformekstraksjon nor isopropanol / etanol-utfelling.
Cellekultur
Celletetthet på platene er av avgjørende betydning. I ett eksperiment hvor cellene så ut til å være litt for konfluent (90 -100%), behandlet vi NIH-3T3-murine fibroblaster i 30 min med 100 U / ml av interferon (IFN) eller α γ. I mindre konfluente cellene selv 15 minutter av IFN-behandling allerede medfører en 5 - til 8-ganger induksjon av gener som irf1 eller socs3 5.. Med celler blir litt for konfluent microarray analyse viste ingen induksjon av IFN-inducible gener for selv den mest raskt induserbar gener som irf1 eller socs3. Derfor er celletetthet en avgjørende faktor for 4SU-merking eksperimenter og alle cellekultur plater bør undersøkes nøye før du starter merking.
4SU er en photoactivatable ribonukleosid og 4SU-holdig RNA er effektivt kryssbundet til proteiner etter eksponering til 365 nm lyskilde. 4SU-behandlede celler skal dyrkes i mørket og eksponering for sterkt lys bør unngås. Etter fjerning av cellulære proteiner ved Trizol RNA isolering denne risiko reduseres vesentlig.
<p class = "jove_content"> 4SU ikke er innlemmet i mobilnettet DNA. Det bør imidlertid bemerkes at total RNA vil fremdeles inneholde små mengder av cellulær DNA. Når du bruker 4SU-merking og q-RT-PCR analyse for å studere viral genuttrykk i cytomegalovirus infeksjon vi funnet det nødvendig å inkludere en DNaseI fordøye skritt i protokollen for å fjerne concatemeric virale genomer 19. Dette er sannsynligvis ikke nødvendig ved bruk av nedstrøms-protokoller som ikke er følsomme for tilstedeværelse av DNA.4SU inkorporering priser og optimal 4SU konsentrasjon
4SU er lett tas opp av celler med intra-og ekstra-cellulære nivåer mest sannsynlig equilibrating i løpet av mindre enn et minutt 9,16. Opptak og inkorporering priser av 4SU er konsentrasjonsavhengig. Derfor kan 4SU konsentrasjonen være beleilig justeres i henhold til de sysselsatte varigheten av merking. Tabell 1 gir råd om 4SU konsentrasjoner i relasjon til varigheten av merking basert på vår beste personlige erfaring. For en time av 4SU merking i mammalske celler, vil 200 mikrometer 4SU være tilstrekkelig for de fleste applikasjoner som resulterer i om lag en 4SU rester per 50 til 100 nukleotider i nylig transkriberte RNA i fibroblaster.
I de siste par årene har vi brukt 4SU-tagging til et bredt spekter av celletyper av menneskelige og murine opprinnelse, inkludert fibroblaster, endotelceller, epitelceller, benmarg stroma celler, makrofager og T-celler. I tillegg ble celler fra Drosophila og Xenopus hell brukes. I alle disse forsøkene ble 4SU inkorporering funnet å være svært effektiv som krever minimale justeringer i 4SU konsentrasjon for de forskjellige celletyper. Når du setter opp metoden for nye celletyper, vil vi anbefale å merke celler med økende 4SU-konsentrasjoner (f.eks strekker 50-1600 mm) og analysere forholdet mellom renset nylig transkribert RNA til de anvendte 4SU-konsentrasjoner (se figur 2A / B). Den 4SU-konsentrasjon hvorved mengden av renset nylig transkriberte RNA entrer et platå bør velges.
I tilfeller hvor høyt konfluerende, kontakt hemmet celler er brukt, vil vi anbefale å bruke litt høyere 4SU konsentrasjoner (f.eks 500 i stedet for 200 mm) for å sikre effektiv 4SU innlemmelse. I tillegg, i tilfeller der fangst av svært korte nylig transkriberte transkripsjoner (<200 nt) er av spesiell interesse, kan 4SU konsentrasjonen må også økes. Dette bør ikke kombineres med langvarig merking ganger (f. eks> 1 t) for å unngå ektopiske effekter eller toksisitet. Til slutt fant vi at bruk av for lite volum på cellekulturmedier kan redusere 4SU innlemmelse effektivitet. Vi anbefaler derfor å bruke 5 ml eller 10 ml medium per 10 cm eller 15 cm tallerken, henholdsvis.
Utarbeidelse av total cellular RNA
For å lykkes med denne protokollen er det avgjørende å få rene, RNase-frie totale cellulære RNA. Bruker 5 ml Trizol per 15 cm tallerken produserer rene RNA gratis nukleaser. Vi anbefaler å bruke den modifiserte Trizol protokollen ved Chomczynski et al. 20.. Først blir det bedre egnet til å isolere store mengder RNA (> 100 ug) som den forbedrede sentrifugalkraft resulterer i fastere pellets som er lettere å håndtere under vasketrinnene. Dette krever imidlertid bruk av spesielle polypropylen rør og adaptere som de vanlige 15 ml laboratorium Falcon rør ikke overleve mer enn 6000 × g. For det andre forbedrer det for fjerning av DNA og glykoproteiner. Dette blir spesielt tydelig når forbereder RNA fra organer eller vev. For det tredje, betyr det ikke begrense den maksimale mengden av total RNA som kan isoleres. Selv om vi også funnet kolonne-baserte RNA isolasjon metoder (f.eks RNeasy) for å gi RNA av egnet kvalitet, standard kolonner are bare i stand til å fange opp til 100 pg av total RNA og derved begrense mengden av utgangsmateriale. Endelig er ved å fjerne den gjenværende etanol to ganger med en pipette, tørking av RNA for å fjerne gjenværende etanol var ikke lenger nødvendig. Dette eliminerer risikoen for over-tørking av RNA, som kan være vanskelig å oppløse igjen etterpå. I prinsippet er 4SU-merking gjelder in vivo, for eksempel ved iv injeksjon av mus. Men, bemerket vi at RNA renhet representerer et stort problem som krever rensing av polyA transkripsjoner før rensing av nylig transkribert RNA (upubliserte data).
Biotinylering og fjerning av ikke-bundet biotin
Biotin-HPDP er 100% tiol-spesifikk og danner en disulfidbinding mellom biotin rest og tiol-merkede RNA nylig transkriberte molekyler. Biotinylering effektiviteten av 4SU-merket RNA er ca 30% som bestemmes av dot blot analyse fem. Som biotin-HPDP er ikke vannløseligdet kan fjernes effektivt ved kloroformekstraksjon. Mens en enkelt kloroformekstraksjon trinnet er tilstrekkelig til å fjerne det store flertallet av ubundet biotin vi regelmessig gjenta dette trinnet for å sikre fullstendig fjerning. For å redusere RNA tap under kloroformekstraksjon trinn 2 ml Phase Lock Gel Tunge rør (Eppendorf) kan brukes i henhold til produsentens instruksjoner. Vanligvis bruker vi de faselåst rør bare for den andre kloroformekstraksjon trinn som mal volumer av det første trinnet er ofte for høyt til å være direkte kompatibel med disse rør. Etter fjerning av ubundet biotin-HPDP, er RNA utvinnes av isopropanol / etanol nedbør. Det er viktig å merke seg at kommersielle kolonne-baserte kitt for å gjenopprette den biotinylerte RNA (f.eks RNeasy fra Qiagen) bør ikke brukes, da de inneholder reduksjonsmiddel i bufferne forutsatt, som spalte disulfidbinding og fjern biotin fra den nylig transkriberte RNA .
Rensing av nyly transkribert RNA
Ikke legg mer enn 100 mL biotinylerte RNA til 100 ul streptavidin perler. Legge til mindre volum er å foretrekke. Imidlertid bør det samme volum av RNA tilsettes for alle prøver. Juster RNA innspill volum (mellom samples) som du legger til streptavidin perler ved å legge det nødvendige volumet av 1x TE til perlene. En enkel måte å lage fersk nukleasefritt 100 mM DTT er å dekantere en tilstrekkelig mengde av DTT pulver i en falk tube plasseres på en ultra-sensitive skala og deretter legge den nødvendige mengden av nukleasefritt H 2 O for å generere 100 mM DTT (64.8 mL vann per 1 mg DTT).
Under utviklingen av 4SU-tagging testet vi streptavidin perler fra ulike leverandører. En rekke av dem genereres store mengder bakgrunn. Derfor anbefaler vi sterkt anbefale å bruke Miltenyi Streptavidin perler som, så langt har vi aldri opplevd noen problemer med overheng av umerkede RNA fra vev culture-deriverte RNA prøver. På denne måten, kan så lite som 150 ng av merkede RNA være spesielt renset fra 150 ug biotinylert RNA (i 100 ul vann) ved anvendelse av 100 pl av streptavidin perlene. Likevekt av perlene med likevektsbufferen leveres med perlene kan utføres og kan lett øke fangst priser 13.
Kvalitetskontroller
Vi anbefaler å utføre q-RT-PCR-kontroller på ny-transkriberte RNA før den utsettes for høy gjennomstrømming analyser. Dette kan omfatte kvantifisering av flere referanse gener kjent for å bli differensielt regulert i den gitte eksperimentelle omgivelser. I tilfeller der 4SU-merking er ansatt for å studere RNA dempefaktorer, vil vi anbefale å kvantifisere en kortvarig transkripsjon (f.eks myc, fos) og en langlivet en (f.eks GAPDH) i både totalt og nylig transkribert RNA. Forholdet mellom nylig transkriberte / totalt RNA bør være vesentlig høyere (~ 5 - til 10-ganger)for de kortvarige transkripsjoner. Basert på det RNA halveringstiden til et referanse-genet, kan RNA halveringstidene bestemmes. Ved alle de tre RNA-fraksjoner (total RNA, nylig transkriberte RNA og umerket pre-eksisterende RNA) blir analysert for fire eller flere gener, normalisering av de forskjellige delsettene RNA kan utføres ved lineær regresjonsanalyse og kvalitetskontroll resultatet kan bli bestemt som beskrevet 7 21..
For Q-RT-PCR analyse, anbefaler vi å bruke 2,5 mL av merket RNA i 20 mL cDNA syntese mix. For optimal sammenligning av q-RT-PCR resultater fryse cDNA i mengder på henholdsvis 5 ul før første bruk. Tine rørene like før bruk, tilsett 45 mL H 2 O og er underlagt 5 pl av fortynninger til Q-RT-PCR-analyser. Dette forbedrer betydelig sammenlignbarhet mellom ulike PCR-kjøringer.
Nylig transkriberte RNA prøver bør kontrolleres for tegn på RNA degradering ved hjelp av Agilent Bioanalyser før utsette dem forhigh-throughput analyser (mikromatriser eller RNA-seq). Det bør imidlertid bemerkes at ytterligere bånd blir noen ganger observert av Agilent Bioanalyser. Den biologiske betydningen av dette er uklar. Som nylig transkribert RNA inneholder betydelig mindre ribosomale RNA, disse prøvene feile de Agilent Bioanalyser kvalitetskontroller. Hvis dette ikke er synlige på grunn av RNA-degradering prøver av akseptabel kvalitet er vanligvis fine til å bli utsatt for high-throughput-analyse.
Kompatibiliteten nylig transkribert RNA med down-stream analyser
Nylig transkribert RNA inneholder vesentlig mer mRNA enn total RNA. Dette er hovedsakelig på grunn av større mengder Intronic sekvenser i nylig transkriberte RNA som øker når varigheten av 4SU-merking er forkortet. Derfor trenger vi ikke regelmessig gjennomføre uttømming av rRNAs fra nylig transkriberte RNA prøver da dette krever større mengder av utgangsmaterialet mens innsamlingsløsing heller lite (~ todelt) gevinst i ikke-rRNA leser. Til slutt gjenstår det å merke at jo større prosentandel av unspliced, høymolekylære transkripter stede i nylig transkriberte RNA kan kreve ytterligere fragmentering ved tilberedning cDNA-bibliotek og for neste generasjons sekvensering. Resultater av størrelsen fragmentering trinnet bør derfor være kvalitet kontrolleres nøye.
Data normalisering for RNA halv live-målinger
Standard måte å normalisere eksperimentelle data for RNA halv levetid målinger er å normalisere alle data til den RNA halveringstiden til et godt karakterisert hus-holder genet eller median RNA halveringstid i en gitt celletype forklart i de foregående eksperimenter. I pattedyrceller, ligger den sistnevnte i området fra 5 til 10 t 6,7. Mens denne tilnærmingen fungerer også ganske bra for 4SU-baserte målinger, er andre virkemidler for normalisering nødvendig hvis median RNA halveringstiden ikke er kjent eller hvis det may selv bli påvirket av endringer i den cellulære systemet som studeres, for eksempel ved knock-out av en RNA forfall veien. 4SU-tagging tilbyr en unik måte å beregne median RNA halveringstid basert på analyse av alle tre RNA fraksjoner, dvs. total mobilnettet RNA, nylig transkribert RNA, og umerkede pre-eksisterende RNA. Som total cellulær RNA separeres i de to sistnevnte RNA-fraksjoner en enkel lineær regresjonsmodell kan anvendes for å normalisere de tre RNA-fraksjonene til hverandre og bestemme medianen RNA halveringstid 7,16. En programvarepakke er tilgjengelig på nettet for å utføre disse analysene 22.
Ineffektiv fangst av vitnemål med lav uridin innhold kan påvirke RNA halveringstid målinger som resulterer i kunstig lave nylig transkriberte / total RNA forholdstall og langvarig RNA halveringstider. Omfanget av dette problemet kan bli vurdert ved å plotte RNA halveringstider eller logg (nylig transkribert / total RNA forholdstall) mot uridine innhold av alle vitnemål 7,15. Dette gir også en god kvalitetskontroll for å vurdere forskjeller i 4SU-inkorporering priser mellom ulike prøver eller betingelser. I tilfeller der en betydelig korrelasjon til uridin innhold er observert dette kan korrigeres for ved bioinformatiske betyr 15. Imidlertid bør det bemerkes at bidraget av modne transkripter i nylig transkriberte RNA ikke lett kan skilles fra den mye større og dermed mye mer uridin-rik forløpere. Med mindre behandlingen kinetikken i en gitt avskrift er kjent (som de er vanligvis ikke) bare korrigere for lav uridin innhold (ineffektiv fangst) kan grovt forvrenge RNA halveringstider. Som sådan, vi nylig funnet behandling av de fleste menneskelige snoRNAs å være svært ineffektive ni. Hvis vi hadde rettet den nylig transkriberte / total RNA forholdstall for den lave uridin innholdet i ganske liten (70-300 nt) snoRNAs, ville dette ha resultert i ekstremt kort snoRNA halvt lives (<5 minutter) med en rekke nylig transkriberte / totalt RNA forholdstall som overstiger 100%. Derfor har vi generelt ikke anbefale å korrigere for lav uridin innhold ved måling av RNA halveringstider.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Forfatterne erklærer at de har ingen konkurrerende finansielle interesser.
Acknowledgments
Vi vil gjerne takke Amie Regan for grundig lesning av manuskriptet. Dette arbeidet ble støttet av NGFN Plus stipend # 01GS0801, MRC stipendet G1002523 og NHSBT stipend WP11-05 til LD og DFG stipend FR2938/1-1 til CCF
Materials
| Name | Company | Catalog Number | Comments |
| 4-thiouridine | Carbosynth | T4509 | Prepare 50 mM stock in sterile H2O, store at -20 °C in aliquots of 50-500 μl, discard unused reagent, do not refreeze. |
| Trizol | Invitrogen | 15596026 (100 ml), 15596018 (200 ml) | WARNING - CORROSIVE and HAZARDOUS TO HEALTH! Ensure immediate access to Phenol antidote (PEG-Methanol); Store at 4 °C. |
| Chloroform | Sigma | 372978 | WARNING - HAZARDOUS TO HEALTH |
| Isopropanol | Sigma | 650447 | |
| Sodium citrate, nuclease-free | Sigma | C8532 | Prepare 1.6 M stock solution using nuclease-free water. |
| 5M nuclease-free NaCl | Sigma | 71386 | Stock solution |
| Nuclease-free H2O | Sigma | W4502 | Make 1 ml aliquots in nuclease-free tubes. |
| RNA precipitation buffer | 1.2 M NaCl, 0.8 M sodium citrate in nuclease free water. Prepare in advance under strictly nuclease-free conditions. Store at room temperature in 50 ml falcon tubes. | ||
| Ethanol | Sigma | 459844 | Use with nuclease-free water to prepare 80% ethanol, store at -20 °C. |
| 1 M nuclease-free Tris Cl, pH 7.5 | Lonza | 51237 | Stock solution |
| 500 mM nuclease-free EDTA, pH 8.0 | Invitrogen | 15575-020 | Stock solution |
| 10x Biotinylation Buffer (BB) | 100 mM Tris pH 7.4, 10 mM EDTA in nuclease-free water, make aliquots of 1 ml. | ||
| Dimethylformamide (DMF) | Sigma | D4551 | |
| EZ-Link biotin-HPDP | Pierce | 21341 | Prepare 1 mg/ml stock solution by dissolving 50 mg biotin-HPDP in 50 ml DMF. Gentle warming enhances solubilisation. Store at 4 °C in aliquots of 1 ml. |
| Phase Lock Gel Heavy tubes 2.0 ml | Eppendorf | 0032 005.152 | Optional for the chloroform extraction step. |
| Zeta membrane | BIORAD | 162-0153 | |
| 10x Dot blot binding buffer | 100 mM NaOH, 10 mM EDTA | ||
| Biotin-oligo | 5'-biotin, 25 nucleotides, any sequence | ||
| Sodium dodecyl sulphate | Fisher | BPE9738 | For 100 ml 20% stock solution, add 20 g SDS to 80 ml PBS pH 7-8 and adjust volume to 100 ml. Keep all high-percentage SDS solutions above 20 °C. Warm the solutions slightly should SDS precipitate. |
| EZ-Link Iodoacetyl-LC-Biotin | Pierce | 21333 | Prepare 1 mg/ml stock solution by dissolving 50 mg iodoacetyl-biotin in 50 ml DMF. Gentle warming enhances solubilisation. Store at 4 °C in aliquots of 1 ml. Generates irreversible, thiol-specific biotinylation. |
| Phosphate buffer saline | Gibco | 10010-015 | |
| Dot blot blocking buffer | Mix 20 ml 20% SDS with 20 ml 1 x PBS pH 7-8 and add EDTA to the final concentration of 1 mM. | ||
| Streptavidin-horseradish peroxidase | Vector Laboratories | SA5004 | Store at -20 °C. Mix 10 ml 20% SDS with 10 ml 1 x PBS. Add 20 μl Streptavidin-HRP before use. |
| ECL reagent | GE Healthcare | RNP2109 | Use following the manufacturer's instructions. |
| Super RX, X-RA Film, 18x24 cm | Fujifilm | 47410 19236 | |
| μMacs Streptavidin Kit | Miltenyi | 130-074-101 | Store the beads at 4 °C. |
| Tween 20 | Sigma | P1379 | |
| Washing buffer | 100 mM Tris pH 7.4, 10 mM EDTA, 1 M NaCl, 0.1% Tween 20 in nuclease-free H2O. | ||
| Dithiothreitol (DTT) | Sigma | 43817 | Prepare as 100 mM DTT in nuclease-free H2O, always prepare fresh before use. |
| RNeasy MinElute Kit | Qiagen | 74204 | Store columns at 4 °C, remaining components of the kit at room temperature. |
| 1.5 ml screw-top polypropylene tubes | Sarstedt | 72.692.005 | Compatible with Dimethylformamide |
| 2.0 ml screw-top polypropylene tubes | Sarstedt | 72.694.005 | Compatible with Dimethylformamide |
| 15 ml tubes | BD Falcon | 352096 | Compatible with Dimethylformamide |
| 50 ml tubes | BD Falcon | 352070 | Compatible with Dimethylformamide |
| All solutions/reagents should be stored at room temperature unless otherwise specified. | |||
| Equipment | |||
| UV/VIS spectrophotometer | Thermo Scientific | NanoDrop 1000 | Or equivalent. Use low volume (1-2 μl) for measurements of low RNA concentrations to avoid excessive sample loss. |
| Polypropylene 15 ml centrifuge tubes | VWR International | 525-0153 | In contrast to standard 15 ml tubes, these tolerate up to 15,000 × g |
| High-speed centrifuge | Beckman Coulter | Avanti J-25 | Or equivalent equipment capable of reaching 13,000×g |
| High-speed rotor | Beckman Coulter | JLA-16250 | Or equivalent equipment capable of reaching 13,000×g |
| Adaptors for 15 ml tubes | Laborgeräte Beranek | 356964 | Or equivalent equipment capable of reaching 13,000×g |
| Refrigerated table-top centrifuge | Eppendorf | 5430 R | Or equivalent. |
| Thermomixer | Eppendorf | Thermomixer compact | Or equivalent. |
| Magnetic stand | Miltenyi Biotec | 130-042-109 | One stand holds 8 μMacs columns. |
| Waterbath | Grant | SUB Aqua 5 | Or equivalent. |
| Ultra-fine scale | A&D | GR-202 | Or equivalent. |
| E-Gel iBase Power System | Invitrogen | G6400UK | For RNA gels; or equivalent. |
| E-Gel EX 1% agarose precast gels | Invitrogen | G4020-01 | For RNA gels; or equivalent. |
References
- Brown, P. O., Botstein, D. Exploring the new world of the genome with DNA microarrays. Nature Genetics. 21, 33-37 (1999).
- Mortazavi, A., Williams, B. A., Mccue, K., Schaeffer, L., Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nature Methods. 5, 621-628 (2008).
- Nagalakshmi, U., Wang, Z., et al. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science. 320, 1344-1349 (2008).
- Wilhelm, B. T., Marguerat, S., et al. Dynamic repertoire of a eukaryotic transcriptome surveyed at single-nucleotide resolution. Nature. 453, 1239-U1239 (2008).
- Dölken, L., Ruzsics, Z., et al. High-resolution gene expression profiling for simultaneous kinetic parameter analysis of RNA synthesis and decay. RNA. 14, 1959-1972 (2008).
- Yang, E., van Nimwegen, E., et al. Decay rates of human mRNAs: correlation with functional characteristics and sequence attributes. Genome Res. 13, 1863-1872 (2003).
- Friedel, C. C., Dölken, L., Ruzsics, Z., Koszinowski, U. H., Zimmer, R. Conserved principles of mammalian transcriptional regulation revealed by RNA half-life. Nucleic Acids Res. 37, e115 (2009).
- Nilsen, T. W., Graveley, B. R. Expansion of the eukaryotic proteome by alternative splicing. Nature. 463, 457-463 (2010).
- Windhager, L., Bonfert, T., et al. Ultra short and progressive 4sU-tagging reveals key characteristics of RNA processing at nucleotide resolution. Genome Res. , (2012).
- Melvin, W. T., Milne, H. B., Slater, A. A., Allen, H. J., Keir, H. M. Incorporation of 6-thioguanosine and 4-thiouridine into RNA. Application to isolation of newly synthesised RNA by affinity chromatography. Eur. J Biochem. 92, 373-379 (1978).
- Cleary, M. D., Meiering, C. D., Jan, E., Guymon, R., Boothroyd, J. C. Biosynthetic labeling of RNA with uracil phosphoribosyltransferase allows cell-specific microarray analysis of mRNA synthesis and decay. Nature Biotechnology. 23, 232-237 (2005).
- Kenzelmann, M., Maertens, S., et al. Microarray analysis of newly synthesized RNA in cells and animals. Proc. Natl. Acad. Sci. U.S.A. 104, 6164-6169 (2007).
- Schwanhäusser, B., Busse, D., et al. Global quantification of mammalian gene expression control. Nature. 473, 337-342 (2011).
- Rabani, M., Levin, J. Z., et al. Metabolic labeling of RNA uncovers principles of RNA production and degradation dynamics in mammalian cells. Nat. Biotechnol. , (2011).
- Miller, M. R., Robinson, K. J., Cleary, M. D., Doe, C. Q. TU-tagging: cell type-specific RNA isolation from intact complex tissues. Nat. Methods. 6, 439-441 (2009).
- Miller, C., Schwalb, B., et al. Dynamic transcriptome analysis measures rates of mRNA synthesis and decay in yeast. Mol. Syst. Biol. 7, 458 (2011).
- Weintz, G., Olsen, J. V., et al. The phosphoproteome of toll-like receptor-activated macrophages. Mol. Syst. Biol. 6, 371 (2010).
- Lipsett, M. N. The isolation of 4-thiouridylic acid from the soluble ribonucleic acid of Escherichia coli. Journal of Biological Chemistry. 240, 3975-3978 (1965).
- Marcinowski, L., Liedschreiber, M., et al. Real-time Transcriptional Profiling of Cellular and Viral Gene Expression during Lytic Cytomegalovirus Infection. PLoS Pathog. 8, e1002908 (2012).
- Chomczynski, P., Mackey, K. Short technical reports. Modification of the TRI reagent procedure for isolation of RNA from polysaccharide- and proteoglycan-rich sources. Biotechniques. 19, 942-945 (1995).
- Friedel, C. C., Dölken, L. Metabolic tagging and purification of nascent RNA: implications for transcriptomics. Mol. Biosyst. 5, 1271-1278 (2009).
- Friedel, C. C., Kaufmann, S., Dölken, L., Zimmer, R. HALO - A Java framework for precise transcript half-life determination. Bioinformatics. , (2010).
